

Background: FXYD proteins are auxiliary subunits of the Na+/K+ ATPase that modulate its kinetic properties.
Results: FXYD1 can be expressed in the plasma membrane only when coexpressed with Na+/K+ ATPase whereasFXYD7 may reach the membrane without the Na+/K+ ATPase.
Conclusion: O-glycosylation of FXYD7 in the Golgi is required for its surface expression.
Significance: FXYD7 and FXYD1 have very different trafficking behavior.
Keywords: Intracellular Trafficking, Membrane Transport, NAK-ATPase; O-glcnacylation, Oocyte, FXYD Proteins, Xenopus Oocytes
Abstract
FXYD proteins are a group of short single-span transmembrane proteins that interact with the Na+/K+ ATPase and modulate its kinetic properties. This study characterizes intracellular trafficking of two FXYD family members, FXYD1 (phospholemman (PLM)) and FXYD7. Surface expression of PLM in Xenopus oocytes requires coexpression with the Na+/K+ ATPase. On the other hand, the Na+/Ca2+ exchanger, another PLM-interacting protein could not drive it to the cell surface. The Na+/K+ ATPase-dependent surface expression of PLM could be facilitated by either a phosphorylation-mimicking mutation at Thr-69 or a truncation of three terminal arginine residues. Unlike PLM, FXYD7 could translocate to the cell surface of Xenopus oocytes independently of the coexpression of α1β1 Na+/K+ ATPase. The Na+/K+ ATPase-independent membrane translocation of FXYD7 requires O-glycosylation of at least two of three conserved threonines in its ectodomain. Subsequent experiments in mammalian cells confirmed the role of conserved extracellular threonine residues and demonstrated that FXYD7 protein, in which these have been mutated to alanine, is trapped in the endoplasmic reticulum and Golgi apparatus.
Introduction
FXYD is a family of seven single-span transmembrane proteins termed after the invariant extracellular motif Phe-XXX-Tyr-Asp (1). They were all shown to interact with the Na+/K+ ATPase and modulate its kinetic properties (for review, see Ref. 2, 3). Thus, a common notion is that FXYD proteins are tissue-specific regulators, or auxiliary subunits of the Na+/K+ ATPase, whose role is to adjust its kinetic properties to the needs of a specific tissue or physiological state without affecting it elsewhere. In addition to their effects on the pump kinetics, FXYD proteins were shown to largely stabilize the active conformation of the Na+/K+ ATPase against heat or detergent inactivation and, presumably, other physiological challenges (4, 5). Other Na+/K+ ATPase-independent functions of FXYD proteins have been suggested as well (3).
This work examines intracellular trafficking to the plasma membrane of two FXYD proteins: FXYD1 (phospholemman (PLM)2) and FXYD7. PLM is primarily expressed in cardiac myocytes and skeletal muscle (6, 7). In Xenopus oocytes, PLM interacts with the Na+/K+ ATPase and lowers its affinity toward cell Na+ (8). Such a decrease will increase intracellular Na+, leading to an increase of cytoplasmic Ca2+ (by inhibiting Na+/Ca2+ exchange) and increased contractility. In addition, PLM directly interacts with the cardiac Na+/Ca2+ exchanger (NCX1) and inhibits its function (9–11). The intracellular carboxyl tail of PLM is phosphorylated by protein kinase A and protein kinase C on at least three residues: Ser-63, Ser-68, and Ser/Thr-69. Phosphorylation of these residues mediates effects of β adrenergic agonists and plays a role in the structural and functional interactions with both Na+/K+ ATPase and NCX1 (12–17). Finally, it was recently demonstrated that PLM also regulates cardiac L-type Ca2+ channels, suggesting a more general role in heart contractility (11, 18).
FXYD7 is a brain-specific protein expressed in both neurons and glia (19). In Xenopus oocyte it doubles the Na+/K+ ATPase K1/2 to extracellular K+ with no apparent effect on the Na+ affinity of the pump (19–21). FXYD7 undergoes O-glycosylation on three extracellular N-terminal threonine residues. These were reported to play a role in protein stability but not on the functional interactions with the Na+/K+ ATPase (19, 20).
This study characterizes intracellular trafficking and cell surface expression of both PLM and FXYD7. Expression of PLM in the cell surface of Xenopus oocytes requires coexpression of the α and β subunits of the Na+/K+ ATPase but not NCX1. Cell surface expression of PLM can be augmented either by a phosphorylation-mimicking mutation of Thr-69 or by truncating the three terminal arginine residues Arg-70–72. FXYD7, on the other hand, can be expressed in the oocyte surface irrespective of coexpression of the Na+ pump subunits. This is due to O-glycosylation of the extracellular domain of FXYD7. Subsequent studies in cultured mammalian cells confirmed an essential role of O-glycosylation in the surface expression of FXYD7.
EXPERIMENTAL PROCEDURES
Total and Surface Expression of FXYD Proteins in Xenopus Oocytes
Stage V-VI oocytes were injected with cRNA mixtures corresponding to various HA-tagged FXYD constructs ± rat α1 and pig β1 subunits of rat Na+/K+ ATPase or the rat cardiac Na+/Ca2+ exchanger (NCX1.1). The injected oocytes were incubated at 19 °C in ND96 (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 5 mm HEPES (pH 7.6), 1.8 mm CaCl2, 2.5 mm sodium-pyruvate) for 3 days. The oocytes were then fixed with 4% formaldehyde for 20 min at 4 °C. They were washed three more times with cold ND96 and blocked with 1% BSA in ND96 for 6–7 h. Monoclonal anti-HA antibody was added to the blocking medium (1:5000), and the oocytes were incubated overnight at 4 °C. They were then washed extensively, and a secondary, HRP-conjugated goat anti-mouse IgG (1:5000) was added for 2 h at 4 °C. Oocytes were divided among wells of a 96-well plate (1 oocyte/well), ECL substrate (SuperSignal ELISA Femto maximum sensitivity substrate, Pierce) was added (100 μl/well), and luminescence was quantified using ImageQuant LAS 4000 mini (General Electric). All luminescence values measured were within the linear concentration-dependent range.
Total expression of various FXYD constructs and α1 Na+/K+ ATPase was determined by Western blotting of microsomal fractions prepared according to Ref. 22. Samples were resolved on Tris-Tricine SDS-PAGE and blotted onto PVDF membranes. Membranes were cut to low, medium, and high molecular weight regions and hybridized with different antibodies according to the experimental design. Generally, surface expressions of different FXYD constructs to be compared were normalized to small changes of the total amount of FXYD protein expressed (usually 10–15%). This is justified because under the experimental conditions used, surface expression of FXYD proteins is limited by the amount of FXYD cRNA and not the amount of Na+/K+ ATPase cRNA coexpressed (Fig. 1). Also, linear dependence between the amount of cRNA injected and surface expression indicates that the heterologous expression of FXYD and Na+/K+ ATPase is far from saturating the translation or trafficking capacity of the cell. In the case of FXYD7, such normalization was problematic, because this protein runs as multiple bands, reflecting different degrees of glycosylation and, possibly, different cellular locations. However, all data reported are valid at least semiqualitatively, even without normalization, to total expression levels. That is, inhibition of surface expression by a particular mutation is also apparent under conditions where the mutated protein is expressed at a higher level than the control sample and vice versa.
FIGURE 1.

Cell surface expression of FXYD proteins in Xenopus oocytes. Groups of oocytes were either not injected (NI) or injected with 0.5 ng or 2 ng HA-tagged FXYD cRNA ± 10 ng α1 and 7 ng β1 Na+/K+ ATPase (NaK) cRNA. Three days later, cell surface expression of the HA epitope was determined. A, representative experiment showing the ECL signal in oocytes injected with various cRNA mixtures. Bottom panel, Western blot analysis of oocyte microsomes with anti α1 Na+/K+ ATPase and anti HA antibodies. FXYD7 migrates as at least three species that correspond to different degrees of O-glycosylation (19). B, quantification of FXYD surface expression. Means ± S.E. of 16–32 oocytes from three different frogs are depicted. Data were normalized to the mean chemiluminescence measured in oocytes from the same frog injected with PLM + Na+/K+ ATPase.
Cell Culture and Transfection
An H1299 cell clone expressing endogenous YFP-tagged α1-Na+/K+ ATPase generated by exon tagging was obtained from the library of annotated reporter cell clones. Cells were cultured in RPMI 1640 medium (Biological Industries, Beit Haemek, Israel) supplemented with 10% FCS plus penicillin and streptomycin. M-1 cells were purchased from the ATCC and cultured in a 1:1 mixture of DMEM and F12 media (Biological Industries) supplemented with 5% fetal calf serum, 5 μm dexamethasone, penicillin, and streptomycin. Stable transfections of CFP-tagged and non-tagged FXYD constructs were carried out using ICAFectin®441 (Eurogentec) or JetPei (Polyplus Transfection, France) according to the instructions of the manufacturers. Stable clones were selected using hygromycin B (Invitrogen) at 500 μg/ml for H1299 cells or 150 μg/ml for M-1 cells.
Surface Biotinylation in Oocytes and Mammalian Cells
Surface-expressed FXYD proteins were labeled in the oocyte membrane using p-diazobenzoyl biocytin (DBB), reactive toward the phenolic group of tyrosines and the imidazole group of histidines. DBB was synthesized from p-aminobenzoyl biocytin shortly before the reaction, as described in Ref. 23. Oocytes were incubated with DBB for 1 h at room temperature on a rotator. Unbound biotin was quenched using cold quenching buffer (0.1% BSA in ND96) followed by 6 washes with ND96. Oocytes were homogenized, membranal fractions were prepared, and biotinylated proteins were isolated on streptavidin beads as described in Ref. 24. Samples were solubilized in SDS-PAGE sample buffer, resolved electrophoretically, and analyzed by Western blotting as above.
For determining the apical versus the basolateral location of FXYD7 in M-1 cells, the cells were seeded on 12-mm Transwell inserts (Costar, pore size 0.4 μm) at a density of 2 × 105 cells/plate. They were grown for 7 days with daily changes of medium until confluent monolayers characterized by a transepithelial electric resistance of more than 1 kΩ × cm2 were established. The cells were washed three times with ice-cold PBS, and DBB was added to either the basolateral (lower) or the apical (upper) compartment to a final concentration of 0.5 mg/ml. Cells were incubated for 30 min at 4 °C on a rocker, and free biotin was removed by four washings in an ice-cold quenching buffer (0.1% BSA, 100 mm glycine in PBS). After two additional washes in PBS, the permeable supports were excised; and cells were scraped, lysed, and treated as above. H1299 cells cultivated on 10-cm culture dishes were washed in PBS and biotinylated by incubation with 1.5 mg/ml sulfo-NHS-SS-biotin (Pierce) for 30 min at 4 °C. Free biotin was removed by four 2-min incubations in ice-cold quenching buffer (0.1% BSA, 100 mm glycine in PBS) followed by two washes in cold PBS. Cells were lysed, and biotinylated proteins were isolated and quantified as described in Ref. 25.
Confocal and Fluorescence Microscopy
H1299 cells expressing the YFP-tagged α1 subunit of the Na+/K+ ATPase and various CFP-tagged FXYD constructs were seeded on Lab-Tek Chamber coverglass (Nunc). Twenty-four hours later, cells were visualized using a scanning confocal microscope (Olympus FV1000) through a ×60 oil immersion objective. The imaging stage was prewarmed to 37 °C, and CO2 was supplied. To determine the basolateral versus the apical location of FXYD7, M-1 cells were cultivated on permeable supports as above. Confluent monolayers were fixed for 30 min at room temperature in 2% paraformaldehyde, 75 mm L-lysine, and 10 mm sodium-metaperiodate. Following four washings in PBS, cells were permeabilized by a 1-h incubation in PBS containing 5% BSA + 0.05% saponin. The permeable support was cut out and stained with the sequential application of rabbit anti-FXYD7 (2 h, 1:40), Cy3-coupled anti-rabbit secondary antibody (1 h, 1:400), mouse anti-α1 Na+/K+ ATPase (2 h, 1:50), and Cy5-coupled anti-mouse secondary antibody (1 h, 1:400). Samples were mounted using Immu-Mount (Thermo-Shandon, Pittsburgh, PA) and visualized for Cy3 and Cy5 fluorescence.
Antibodies
A rabbit polyclonal antibody directed at the C-terminal sequence of PLM was described previously (8). A monoclonal antibody recognizing the N terminus of the α1 subunit of Na+/K+ ATPase (6H) was kindly provided by Dr. M. J. Caplan, Yale University School of Medicine. A mouse monoclonal anti-HA antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit polyclonal antibody against the C-terminal region (Arg-52-Val-80) of FXYD7 was a gift from Prof. Kathi Geering (University of Lausanne, Switzerland). Mouse monoclonal antibody against NCX1 (R3F1) was purchased from SWANT (Bellinzona, Switzerland). A mouse monoclonal anti-β tubulin was purchased from Sigma-Aldrich, and rabbit polyclonal anti-GRASP65 was purchased from Abcam (Cambridge, MA). Cy3-coupled anti-rabbit and Cy5-coupled anti-mouse secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA).
cDNAs
N-terminally HA-tagged rat PLM (HA-PLM) was prepared by replacing the signal peptide of PLM with an HA epitope. This construct served as a template for point mutations of Ser-63, Ser-68, and Thr-69, created by a single PCR step of circled cDNA using PfuUltra high fidelity DNA polymerase (Stratagene, La Jolla, CA). HA-FXYD7 was constructed by inserting an HA epitope upstream of the mouse FXYD7 coding region. Point mutations of extracellular threonine residues were introduced as above. PLM/FXYD7 chimera were created by PCR using primers with overlapping sequences. For expression in Xenopus oocytes, cDNAs were subcloned between 5′ and 3′ sequences of Xenopus β-globin in pGEM or pBluescript-derived vectors (26). cRNAs were synthesized from the T7 promoter of linearized plasmids. CFP was inserted in the carboxyl tails of HA-PLM and HA-FXYD7 between the last amino acid and the stop codon. To compare the relative abundances of FXYD7 and Na+/K+ ATPase we have also expressed α1 Na+/K+ ATPase tagged N-terminally with an HA epitope. For expression in mammalian cells, the various open reading frames were subcloned into the mammalian expression vector pIRES-hyg. Rat NCX1.1 cDNA was kindly provided by Prof. J. Lytton, Calgary University, Canada. The ER marker plasmid pDSRed2-ER was from Clontech.
RESULTS
Surface expression of FXYD proteins was measured in Xenopus oocytes injected with cRNA coding for PLM or FXYD7 tagged in their extracellular domain by the HA epitope. Intact oocytes were incubated with a monoclonal anti-HA antibody, and the externally bound antibody was quantified by ECL. A large difference in the amount of surface-expressed PLM was apparent between oocytes injected with PLM alone and those injected with PLM plus Na+/K+ ATPase (Fig. 1A). This difference is not likely to be due to rapid degradation of the FXYD protein expressed in the absence of the Na+/K+ ATPase. Western blotting of oocyte lysates with the anti-HA antibody demonstrates that the total expression of PLM is even higher in oocytes that do not express exogenous Na+/K+ ATPase (Fig. 1A, bottom panel). In contrast to the large effect of the Na+/K+ ATPase on PLM surface expression, FXYD7 was expressed on the oocyte surface at similar levels irrespective of the coexpression of exogenous Na+/K+ ATPase. The above observation was further established by averaging 16–32 oocytes from three different frogs in each group (Fig. 1B). Because the same antibody is being used to detect surface expression of PLM and FXYD7, the expression levels of the two proteins should be comparable. Thus, the data indicate that the amount of FXYD7 that reaches the plasma membrane without exogenous Na+/K+ ATPase is similar to the amount of surface PLM expressed with the pump. The experiment also demonstrates that increasing the amount of injected cRNA from 0.5 ng to 2.0 ng doubles surface expression of PLM (Fig. 1B). Thus, under the experimental conditions, surface expression of PLM is not limited by the amount of Na+/K+ ATPase coexpressed or the translation and trafficking capabilities of the cell.
In the above experiment, surface expression of FXYD proteins was measured as the binding of antibody to the extracellular HA epitope. In the case of PLM, this involved eliminating the signal peptide, a manipulation that may affect intracellular trafficking. Therefore, we wanted to confirm the data of Fig. 1 by a second, independent approach using non-modified PLM. This was done by surface biotinylation of intact oocytes and detecting the amount of PLM that could be precipitated by streptavidin attached to agarose beads. The commonly used N-hydroxysuccinimide ester derivatives of biotin are reactive toward primary amino groups and react with the ϵ-amino group of lysine residues and N-terminal α amines. Because the extracellular domain of PLM is relatively short and lacks lysine residues, we were unable to achieve significant surface biotinylation of this protein in oocytes. Better labeling of non-modified PLM could be obtained using the tyrosine- and histidine-specific biotinylating reagent DBB (23) (Fig. 2). The labeling was relatively weak because the ectodomain of rat PLM has only two tyrosine residues and a single histidine, all located very close to the membrane surface. Nevertheless, a marked difference in the surface expression of PLM-injected ± Na+/K+ ATPase was apparent. DBB did not label ERK, indicating that it does not permeate the cell membrane. Thus, the large effect of Na+/K+ ATPase on surface expression of PLM is confirmed by biotinylation of unmodified PLM and is not secondary to modification of the protein by HA tagging.
FIGURE 2.
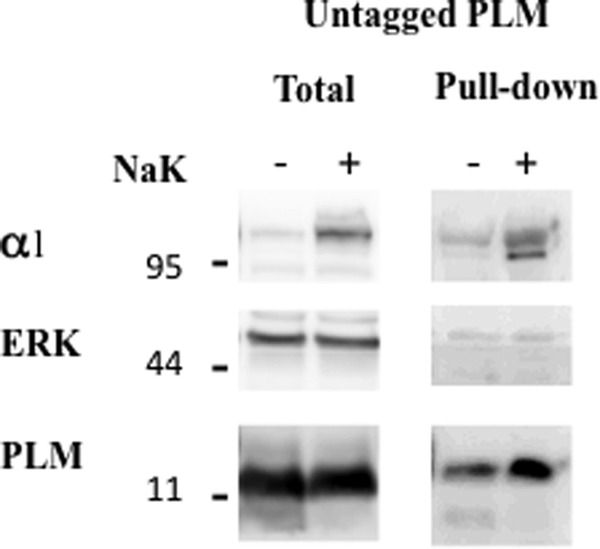
Cell surface biotinylation of PLM. Oocytes were injected with 2 ng of unmodified PLM ± 10 ng α1 and 7 ng β1 Na+/K+ ATPase (NaK). Three days later, oocytes were biotinylated by a 60-min incubation with 0.5 mg/ml DBB at room temperature. Oocytes were lysed, and biotinylated proteins were precipitated on streptavidin beads as described under “Experimental Procedures.” Total and pulled-down biotinylated proteins were resolved electrophoretically, and the blot was cut into low, medium, and high MW regions that were blotted for PLM, ERK, and α1 Na+/K+ ATPase.
Several studies have demonstrated inhibition of the cardiac Na+/Ca2+ exchanger (NCX1) by a direct interaction with PLM (9, 10). We therefore tested whether NCX1.1 can also drive PLM to the oocyte surface. However, unlike the Na+/K+ ATPase, coexpressing NCX1 with PLM was without effect on the surface expression of this FXYD protein (Fig. 3A). Experiments in transfected cells and PLM knockout mice have demonstrated that inhibition of NCX1 requires phosphorylation of Ser-68 and possibly Ser-63 in the carboxyl tail of PLM (16, 27). Because PLM expressed in oocytes is mostly unphosphorylated at these residues (28, 29), we have also tested expression of the PLM mutant in which both serine residues were replaced by glutamic acid. These mutations were shown to mimic effects of protein kinase A and protein kinase C on the interaction of PLM with NCX1 (16). However, they were without effect on surface expression of PLM in oocytes with or without NCX1 or Na+/K+ ATPase (Fig. 3A). Fig. 3, B and C, demonstrates that NCX1 is expressed in the oocyte and reaches the plasma membrane at a comparable efficiency to the Na+/K+ ATPase. Thus, insufficient surface expression of NCX1.1 cannot account for the negative result above.
FIGURE 3.

Effects of NCX1.1 and phosphorylation-mimicking mutations on surface expression of PLM. Oocytes were either non-injected (NI) or injected with 2 ng of either HA-PLM or HA-PLM construct in which Ser-63 and Ser-68 were mutated into glutamic acid (S63/68E). Some of the oocytes were injected, in addition, with cRNA for the α1 (10 ng) and β1 (7 ng) subunits of the Na+/K+ ATPase (NaK) or the 1.1 isoform of the cardiac Na+/Ca2+exchanger (NCX, 5 ng). Total and surface expression of the various proteins was measured 3 days later. A, surface expression of the HA epitope in the various groups of oocytes. Means ± S.E. of data from 12 oocytes are depicted and expressed as fraction of the mean value in oocytes injected with PLM + NaK. B, Western blot analysis of total proteins extracted from the same groups of oocytes with antibodies to HA, β tubulin, and either the α1 of Na+/K+ ATPase or NCX1. C, surface expression of NCX1.1 and α1Na+/K+ ATPase. Oocytes that were injected with either 7 ng of NCX1.1 or 10 ng α1 plus 7 ng β1 or non injected (NI) were biotinylated by a 30-min incubation at 4 °C with 1.5 mg/ml sulfo-NHS-SS-biotin. Five percent of the total lysate (total) and the whole streptavidin pull-down material (PD) were blotted with anti α1 and NCX1 antibody. An aliquot of rat heart microsomes served as a positive control for the specificity of the antibody.
Lansbery et al. (30) reported that in transfected Madin-Darby canine kidney cells, PLM resides primarily in the ER, and a shift in its localization toward the plasma membrane is achieved either by activating protein kinase C or by mutating Ser-63, Ser-68, and Ser-69 into aspartic acid. Such a translocation was also achieved by deleting three terminal arginine residues that may act as an ER retention signal. In our system, the double mutant S63/68E was without effect on the surface expression of PLM (Figs. 3A and 4). However, either the mutation of Thr-69 into aspartic acid or truncating three C-tail arginine residues (Δ3R) resulted in double the surface expression (Fig. 4). Significantly, the above increase was apparent only when the mutated PLM was expressed with but not without α1β1 Na+/K+ ATPase. It is also possible that mutating Ser-63/68 into aspartic acid (30) versus glutamic acid (this study) produces different effects. Yet, the second manipulation (Δ3R) was identical in both studies.
FIGURE 4.

Mutating Thr-69 and truncating C tail arginine residues elevate surface expression of PLM. Oocytes were injected with cRNA mixtures coding for the indicated PLM constructs (2 ng) ± α1 (10 ng) and β1 (7 ng) Na+/K+ ATPase (NaK). Left panel, surface expression of the HA epitope under the different conditions. The chemiluminescence signal was normalized to the total unmutated PLM protein expressed, and data are expressed as fractions of the mean value in oocytes injected with PLM+NaK (means ± S.E. of 16 oocytes). Probabilities for differences in surface expression between mutated and unmutated PLM were calculated using unpaired Student's t test. *, p < 0.0005; **, p < 0.0002. Right panel, Western blot analyses of the same oocytes with antibodies to HA, β tubulin, and α1Na+/K+ ATPase. The different lanes represent oocytes injected with the following cRNA mixtures: Lane 1, non-injected; lane 2, PLM; lane 3, S63/68E, lane 4, S63/68E+T69D; lane 5, Δ3R; lanes 6–9, as lanes 2–5 plus αβ Na+/K+ ATPase.
The finding that coexpression of the α1 and β1 subunits of the Na+/K+ ATPase elevates surface expression of PLM provides evidence that the interaction of the two proteins in the ER or Golgi apparatus is required to achieve exit of PLM from the intracellular compartment and trafficking to the membrane surface. The amount of PLM that is expressed on the oocyte surface in the absence of exogenous Na+/K+ ATPase may be transported by a “bulk flow” mechanism (31) or assembled with endogenous oocyte Na+/K+ ATPase. As shown in Fig. 1, high-level surface expression of FXYD7 does not require exogenous Na+/K+ ATPase, suggesting the presence of an “exit signal” that allows it to be transported to the oocyte surface without α1 and β1. To identify the domain(s) and residues involved, we compared surface expression of the HA epitope in oocytes injected with two PLM/FXYD7 chimeras. Previously, it was reported that a C-terminal valine residue in FXYD7 (Val-80) functions as an ER exit signal and that its deletion delays cell surface expression of this protein (20). This effect was, however, a transient one, and no significant difference between wild-type and mutated FXYD7 was apparent following a 48-h incubation. In agreement, we found that replacing the carboxy-terminal sequence of PLM by the corresponding FXYD7 region (i.e. chimera PP7) resulted in only a small increase in surface expression relative to PLM (Fig. 5). However, exchanging the extracellular N-terminal sequences had a larger effect, and surface expression of chimera 7PP injected without α1β1 was virtually as high as PLM coinjected with Na+/K+ ATPase. Thus, the N-terminal extracellular domain of FXYD7 appears to be the protein segment that enables it to be efficiently transported to the cell surface without α1β1 Na+/K+ ATPase.
FIGURE 5.
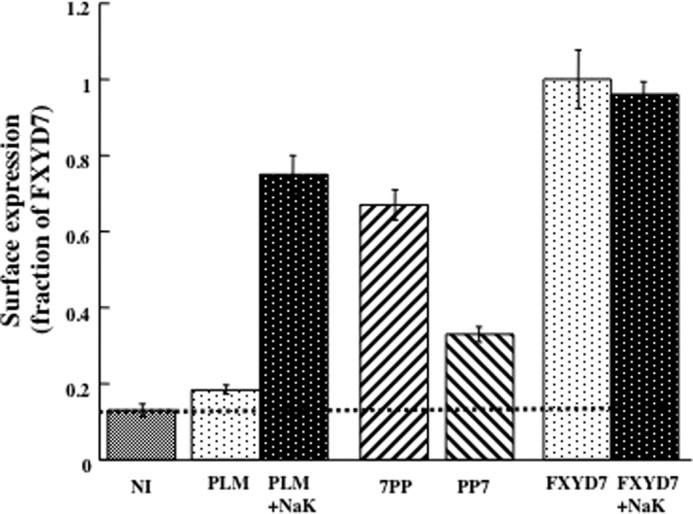
FXYD domains involved in surface expression. Oocytes were either not injected (NI) or injected with cRNA mixtures coding for HA-tagged PLM (2 ng) or FXYD7 (2 ng) ± α1(10 ng) and β1 (7 ng) Na+/K+ ATPase (NaK). Additional groups of oocytes were injected with 2 ng of two PLM/FXYD7 chimeras. One had the extracellular domain of HA-FXYD7 but the transmembrane and intracellular segments of PLM (7PP). The other had the extracellular and transmembrane domains of HA-PLM and the intracellular segment of FXYD7 (PP7). Surface expression of the HA tag was measured 3 days later, and the data are means ± S.E. of 24 oocytes. Data are expressed as a fraction of the mean chemiluminescence signal in oocytes expressing HA-FXYD7.
The extracellular N-terminal of FXYD7 contains three well conserved threonine residues that are predicted to be mucin-type N-acetylgalactosamine acceptor sites. Their mutation affects the electrophoretic mobility of multiple FXYD7 bands. Changes are also achieved by extensive treatment with neuraminidase plus O-glycosidase, suggesting that at least some of the residues are indeed O-glycosylated (19). We therefore examined whether mutating these residues affects surface expression. Double or triple mutations of these threonines into alanine (i.e. T3/5A and T3/5/9A, respectively) largely inhibited surface expression of FXYD7, and the impaired expression could not be negated by coexpression with the Na+/K+ ATPase (Fig. 6). A similar observation has been made by introducing these mutations in the chimera 7PP (data not shown). As demonstrated in Fig. 1A and as reported previously (19), FXYD7 runs as multiple bands likely to represent different degrees of O-glycosylation. These are shifted to lower molecular weights in the Thr/Ala mutants (Fig. 6, bottom panel). Large inhibition of surface expression was also apparent by mutating the conserved tyrosine, phenylalanine, and aspartic acid in the FXYD motif into alanines (AFAA). This effect was also independent of coexpression with the Na+/K+ ATPase (Fig. 6).
FIGURE 6.
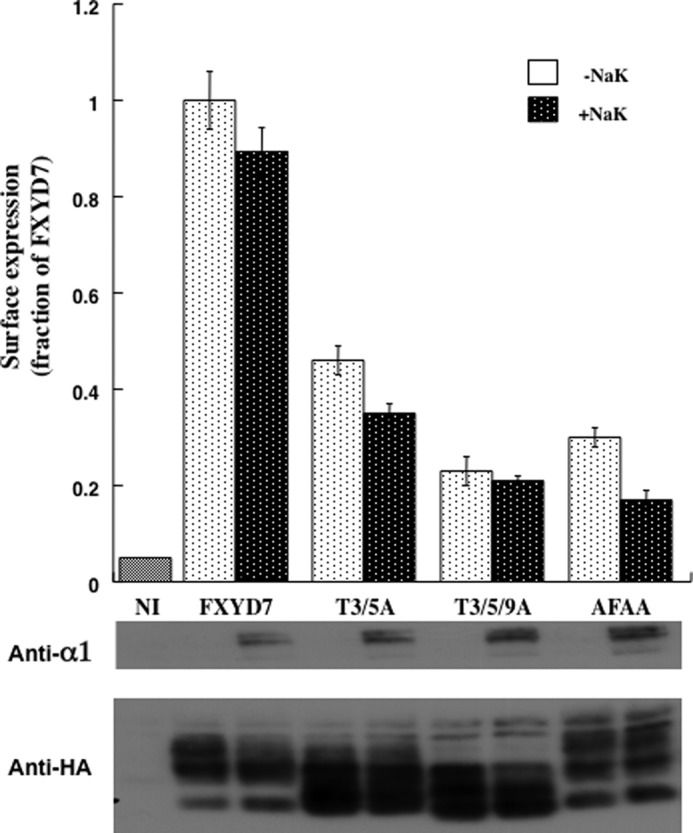
Mutating O-glycosylation sites or the FXYD motif inhibits surface expression of FXYD7. Oocyte were injected with 2 ng of cRNA coding for wild-type and mutated HA-tagged FXYD7 constructs ± the α1 (10 ng) and β1 (7 ng) subunits of Na+/K+ ATPase. Surface expression was measured 3 days later, and means ± S.E. of eight oocytes are depicted and expressed as a fraction of the mean value of oocytes expressing FXYD7. Bottom panel, Western blot analyses with anti-HA and anti-α1.
The relative roles of each of the three threonine residues were evaluated by comparing surface expression of FXYD7 carrying a single or double mutation (Fig. 7). The single mutant T3A had a large inhibitory effect, whereas T5A had a smaller but still significant effect. The double mutant T3/5A evoked an additive response. Thr-9 has less effect with higher variability, which appeared to be statistically insignificant.
FIGURE 7.
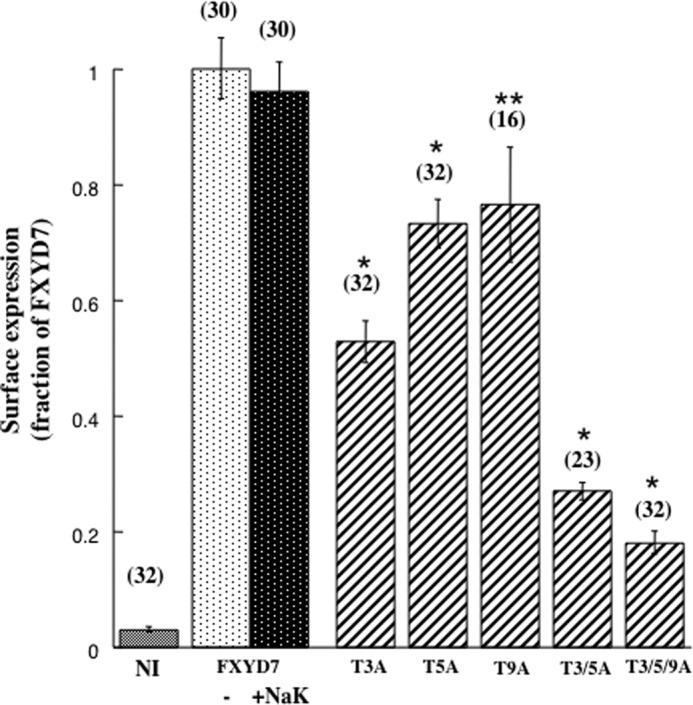
Effect of Mutating individual O-glycosylation sites on surface expression of FXYD7. Oocytes were injected with the above amounts of cRNA of either FXYD7 ± Na+/K+ ATPase or with the indicated FXYD7 mutants (without Na+/K+ ATPase). The number of oocytes averaged is indicated in brackets. *, p < 0.01; **, p > 0.05.
Next, we aimed to determine whether mutating O-glycosylation sites prevents surface expression of FXYD7 in mammalian cells too, and identify the intracellular location of the mutated protein. These studies have been carried out in the human lung carcinoma cell line H1299. These cells have been used before for the generation of a fluorescently labeled endogenous protein library by random integration of YFP into the H1299 genome (32). One of the identified cell clones expresses an α1 Na+/K+ ATPase protein tagged in its N-terminal by YFP inserted between the 5th and 6th amino acids. This provides a fluorescently labeled Na+/K+ ATPase expressed from the normal locus in the genome. The YFP-α1 H1299 cells were stably transfected with various FXYD constructs tagged by CFP in their C-tails. Fig. 8 depicts confocal images of such cells. The YFP tagged Na+/K+ ATPase was expressed primarily in the plasma membrane, whereas CFP-tagged PLM and FXYD7 were detected both in the plasma membrane and intracellular compartments (Fig. 8, a–c). The triple FXYD7 mutant T3/5/9A however, localized intracellularly and could not be detected in the plasma membrane (Fig. 8d). To better quantify plasma membrane expression, cells were surface-biotinylated, and the amount of streptavidin-pulled down FXYD protein was compared. Unlike Xenopus oocytes, FXYD7 and PLM could be labeled in intact H1299 cells by sulfo-NHS-SS biotin, presumably because of the better accessibility of the N-terminal α amine. Western blot analysis indicates that α1, PLM, and FXYD7 are effectively pulled down by streptavidin beads, indicating that they are mostly expressed in the cell surface (Fig. 9). T3/5/9A, on the other hand, could not be biotinylated at all, indicating a nearly complete intracellular localization. Another relevant observation is that in transfected mammalian cells, FXYD7 migrates as a doublet, and only the upper band is biotinylated. T3/5/9A FXYD7, on the other hand, migrates as a single band, corresponding to the lower MW species of FXYD7. We suggest that the lower band corresponds to the intracellular immature FXYD7, whereas the upper band is the fully glycosylated form of FXYD7 in the plasma membrane.
FIGURE 8.

Expression of FXYD proteins in H1299 cells. H1299 cells that stably express YFP-α1 Na+/K+ ATPase and CFP-tagged PLM, FXYD7, or T3/5/9A FXYD7 were seeded on LabTek chamber coverslides. Confocal images were taken 24 h later using an ×60 oil immersion objective. Scale bar = 20 μm.
FIGURE 9.
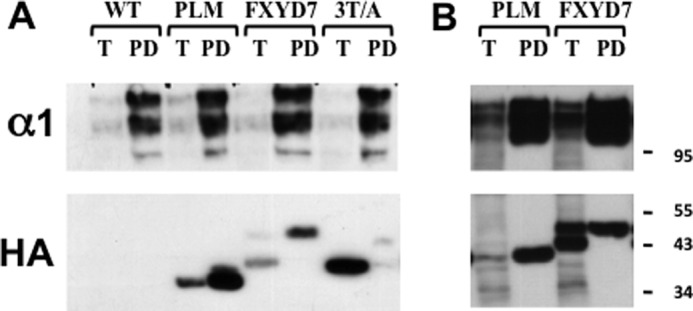
Surface biotinylation of FXYD proteins in H1299 cells. A, H1299 cells stably expressing YFP-α1 Na+/K+ ATPase and the above CFP-tagged FXYD proteins were surface-biotinylated with sulfo-NHS-SS biotin. Cells were lysed and subjected to streptavidin pull-down as described under “Experimental Procedures.” 5% of the total cell lysate (T) and all of the streptavidin bound proteins (PD) were resolved electrophoretically and blotted with anti-HA and anti-α1 antibodies. α1 Na+/K+ ATPase migrates as a doublet corresponding to normal and YFP-tagged polypeptides. B, a second experiment using the same protocol that better demonstrates the FXYD7 doublet.
Next, we determined the cellular localization of the unglycosylated FXYD7 by labeling transfected cells for both ER and Golgi markers (Fig. 10). In the first case, cells were transiently transfected with pDSRed2-ER, a plasmid that encodes DSRed2 flanked by the ER targeting sequences of calreticulin and the ER retention sequence KDEL (Fig. 10A). For Golgi labeling, cells were stained with antibody against the Golgi re-assembly stacking protein (GRASP65, Fig. 10B). Both internal organelles appear to contain T3/5/9A FXYD7. This is evident from the yellow and pink fluorescence in the merged images of Fig. 10, A and B, respectively. Localization in ER and Golgi is expected for proteins “en route” to the plasma membrane, especially in overexpression conditions. However, because the plasma membrane is stained for FXYD7 but not the triple mutant, we conclude that T3/5/9A cannot traffic beyond the Golgi apparatus. This is in agreement with the known confinement of O-glycosyltransferases to the cis and trans compartments of the Golgi apparatus (33).
FIGURE 10.

Cellular location of FXYD7. H1299 cells expressing CFP-tagged FXYD7 and T3/5/9A FXYD7 were imaged. A, cells were also transiently transfected with the ER marker pDSRed2-ER. B, cells expressing CFP tagged T3/5/9A FXYD7 were stained for GRASP65 plus secondary Cy3-conjugated antibody and imaged for CFP and Cy3. Scale bar = 10 μm.
Another interesting question is whether in mammalian cells, too, FXYD7 traffics to the plasma membrane when it is not associated with the Na+/K+ ATPase. Unlike in oocytes, one cannot significantly manipulate the Na+/K+ ATPase abundance in mammalian cells without impairing their viability. Thus, an alternative approach examined was expression of FXYD7 in polarized epithelial cells. It is well established that in epithelial cells the Na+/K+ ATPase is confined to the basolateral membrane. FXYD7 is natively expressed only in neurons and glia and, therefore, is not expected to have an endogenous basolateral targeting signal (19). Hence, Na+/K+ ATPase independent intracellular trafficking of FXYD7 should direct it to both the apical and basolateral surface. On the other hand, intracellular association of FXYD7 with the Na+/K+ ATPase will direct it to the basolateral membrane only. To assess these possibilities, we have stably transfected the epithelial cell line M-1 with FXYD7 and examined its cellular location in confluent monolayers using either immunofluorescence or surface biotinylation.
M-1 cells and FXYD7-transfected M-1 cells were cultivated on permeable supports for about 1 week until monolayers of tight epithelia characterized by a transepithelial resistance of >1 kΩ × cm2 had been formed. The cells were examined for apical versus basolateral expression of FXYD7 either by surface biotinylation with DBB or by immunofluorescence using anti-FXYD7 antibody. Fig. 11 summarizes data obtained by both techniques. In general, labeling by DBB was relatively inefficient. This was particularly the case for basolateral labeling, where the support on which cells are cultivated limits the accessibility to the biotinylating reagent (compare pull-down efficiency in Figs. 9 and 11). Nevertheless, the data clearly demonstrate that FXYD7 can be labeled from the basolateral but not apical side of the cell (Fig. 11A, arrow). The validity of the approach was established by demonstrating that α1 Na+/K+ ATPase, too, was labeled only from the basolateral side. Also, the fact that ERK was not biotinylated demonstrates that DBB does not permeate the cell membrane and labels only externally exposed protein domains. Independently, we have stained confluent cell monolayers for FXYD7 and α1 Na+/K+ ATPase. The two appear to colocalize and in images of the xz plane demonstrate only vertical labeling corresponding to the lateral location of the protein (Fig. 11, B and C).
FIGURE 11.

Plasma membrane localization of FXYD7 in polarized epithelia. M-1 cells were cultivated for 7 days on permeable supports as described under “Experimental Procedures.” A, DBB (0.5 mg/ml) was added to either the basolateral (bl.) or the apical (Ap.) side of confluent monolayers of wild-type or FXYD7-transfected cells. After biotinylation, quenching, and lysis, biotinylated proteins were pulled down using streptavidin beads. Five percent of the total lysate (T) and all of the streptavidin pulled-down (PD) material were run on SDS-PAGE and transferred to a membrane. The membrane was cut to high, medium, and low molecular weight regions and probed with the indicated antibodies. B, cells cultivated on permeable supports were fixed, permeabilized, and stained for FXYD7 (red) and α1 Na+/K+ ATPase (green). Shown are xy and xz planes. C, higher magnification of the yz section in a confluent monolayer stained for FXYD7.
DISCUSSION
Many studies have establish that FXYD proteins function as auxiliary subunits or regulators of the Na+/K+ ATPase and alter its kinetic properties in tissue-specific and physiological state-specific manners (for a review, see Refs. 2, 3). However, additional functions have been postulated as well. These include interactions with the cardiac Na+/Ca2+ exchanger (9, 16, 34–36), formation or regulation of ionic channels (11, 18, 37–40), and effects on cell adhesion and motility (25, 41–43). This paper characterizes surface expression in Xenopus oocytes and mammalian cells of two FXYD proteins, PLM and FXYD7. Stage VI oocytes have a very low level and slow turnover rate of endogenous Na+/K+ ATPase, which allows the convenient manipulation of Na+/K+ ATPase abundance to study its effect on FXYD surface expression. On the other hand, oocytes are inadequate for intracellular localization studies because of high autofluorescence.
A key observation is that translocation of PLM to the plasma membrane strongly depends on coexpression of the Na+/K+ ATPase, whereas FXYD7 reaches the oocyte cell surface equally well with and without Na+/K+ ATPase. The results obtained for PLM were as expected. It is well established that multimeric proteins assemble intracellularly and that such assembly is required for trafficking to the plasma membrane (31). The usually small but significant plasma membrane expression of PLM obtained in the absence of exogenously expressed α and β Na+/K+ ATPase may reflect passive bulk flow that does not involve receptor-mediated concentration of cargo proteins in transport vesicles nor the association with endogenous Na+/K+ ATPase subunits. The ability of FXYD7 to be transported to the cell surface of oocytes equally well with and without Na+/K+ ATPase is somewhat surprising and may suggest additional functions of this protein.
Unlike the Na+/K+ ATPase, NCX1 had no effect on the surface expression of PLM. This was the case both for the wild type and a PLM construct in which serine residues 63 and 68 were mutated into glutamic acid, phosphorylation-mimicking mutations shown before to enhance PLM inhibition of NCX1-mediated current (16). Because previous studies demonstrating PLM-NCX1 interactions were carried out using cardiac myocytes and transfected mammalian cells but not Xenopus oocytes, it is possible that the oocyte system lacks a component needed for the NCX1-PLM interaction. It is known that the cardiac T-tubule/SR microdomain contains an ankyrin-B-based macromolecular complex that includes Na+/K+ ATPase, NCX1, and the InsP3 receptor (44). It is possible that the negative result above reflects the lack of such a complex in oocytes that were not injected with Na+/K+ ATPase and/or ankyrin-B.
Lansbery et al. (30) reported that in Madin-Darby canine kidney cells, PLM resides primarily in the ER but that enhanced translocation to the plasma membrane is achieved either by the phosphorylation-mimicking mutations of Ser-63, Ser-68, and Thr-69 or by truncating the three terminal arginine residues Arg-70–72. In agreement, we found that both mutations significantly elevated plasma membrane expression of PLM in oocytes that coexpress Na+/K+ ATPase and that the single mutant T69D is sufficient to produce the full augmentation. Thr-69 was identified recently as another protein kinase C site whose phosphorylation augments the pump-mediated current in cardiac myocytes beyond the level achieved by phosphorylating Ser-63 and Ser-68 (12). Also, phosphorylation of this residue contributes to sildenafil-induced cardio protection against reperfusion injury (45). Although mutating the C-tail of PLM nearly doubled its surface expression in oocytes that coexpress α1β1 Na+/K+ ATPase, it did not augment PLM surface expression in oocytes not coinjected with Na+/K+ ATPase cRNA (Fig. 4). One interpretation is that phosphorylation and arginine truncation enhance interaction of PLM with the Na+/K+ ATPase rather than disrupt an ER retention signal to enable PLM to exit the ER. An intracellular interaction between Na+/K+ ATPase and FXYD proteins could not be resolved from the crystal structure of shark Na+/K+ ATPase (46). It was, however, inferred from chemical cross-linking of FXYD2 in pig kidney membrane and in transfected cells (47). Irrespective of the mechanism, the fact that a similar effect is achieved by truncating arginine residues and phosphorylation-mimicking mutations suggests a role for the electric charge in the carboxyl tail of PLM.
Unlike PLM, FXYD7 surface expression was high, irrespective of the coexpression of Na+/K+ ATPase. Because functional and structural interactions between FXYD7 and the Na+/K+ ATPase are well established (19–21), one may expect that the coexpression of Na+/K+ ATPase will further elevate FXYD7 surface expression. The fact that this was not the case may be accounted for by one of two ways: 1) FXYD7 is expressed at a large excess over Na+/K+ ATPase and, therefore, the coexpression of the pump does not significantly elevate its surface abundance; and 2) FXYD7 may reach the surface equally well with and without Na+/K+ ATPase. When both proteins are coexpressed, they will interact in the ER and reach the plasma membrane together. If, however, FXYD7 is expressed without exogenous Na+/K+ ATPase, it traffics equally fast without associating with pump subunits. In an attempt to discriminate between these possibilities, oocytes were injected with cRNAs having the HA epitope on both FXYD7 and α1, allowing comparison of the relative abundance of the two proteins by Western blotting of the microsomal membrane. These experiments established an ∼4-fold excess of FXYD7 over α1 (data not shown). This supports the first option above but does not rule out the second one.
The Na+/K+ ATPase-independent trafficking of FXYD7 critically depends on at least two of the three conserved N-terminal threonine residues shown to undergo O-glycosylation (19). Subsequent experiments in transfected mammalian cells demonstrated that the triple Thr/Ala mutant accumulates in the ER and Golgi apparatus, suggesting that O-glycosylation of FXYD7 is required for the final stages of translocation to the plasma membrane. This is in full agreement with the known confinement of O-glycosyltransferases to the Golgi apparatus (33). O-glycans are known to act as sorting signals that direct proteins to the apical surface in epithelia (48). In other cases, cytoplasmic O-glycosylation either inhibited or enhanced surface expression of membrane proteins. Such inhibition was reported for E-cadherin and synaptotagmin I (49, 50). On the other hand, inhibiting O-glycosylation impaired mucin secretion in HT-29 cells (51) and promoted proteolytic cleavage of the Cu2+ transporter hCTR1 (52).
In principle, inhibition of surface expression of Thr/Ala mutants could be secondary to an enhanced degradation of this protein, and such mechanism may in principle also account for the limited surface expression of PLM in oocytes that do not coexpress Na+/K+ ATPase. However, Western blot analyses shown in Figs. 1–4 and 6 demonstrate that lack of surface expression of either PLM or mutated FXYD7 do not correlate with a decrease in the steady-state amount of these proteins. On the contrary; their abundance appears to increase in the absence of exogenous Na+/K+ ATPase. In addition, an enhanced degradation of the triple Thr/Ala mutant reported in (53) was prevented by the coexpression of α1 and β1 Na+/K+ ATPase. In our experiments, the coexpression of Na+/K+ ATPase did not reverse inhibition of surface expression by the Thr/Ala mutation (Fig. 6).
Interestingly, marked inhibition of FXYD7 surface expression was also apparent by mutating the FXYD motif common to all members of the family. Mutating the conserved Phe, Tyr, and Asp residues into Ala had no effect on O-glycosylation of FXYD7, as evident by the multiple bands seen on a Western blot analysis (Fig. 6 and Ref. 20). The crystal structure of shark rectal gland Na+/K+ ATPase highlights interactions of this motif with both α1 and β1, suggesting a role in association of the FXYD-Na+/K+ ATPase complex (46). The fact that mutating this domain markedly affects translocation of an FXYD protein that is capable of cell surface expression without α1β1 Na+/K+ ATPase suggests an additional, yet unknown role of this motif in the intracellular trafficking of FXYD proteins.
Independent transport to the cell surface and assembly in the plasma membrane is an unusual mechanism for the construction of a multimeric protein complex. Therefore, the data may indicate an additional, Na+/K+ ATPase-independent role of FXYD7. As discussed above, such functions have been postulated for other FXYD proteins. Alternatively, it is possible that FXYD7 is not involved in the constitutive regulation of Na+/K+ ATPase and that an equilibrium exists in the plasma membrane between FXYD7 that is and is not in complex with Na+/K+ ATPase. Evidence for such an equilibrium involving PLM oligomerization has been suggested by demonstrating fluorescence energy transfer between YFP- and CFP-tagged PLM (54).
Intracellular trafficking of FXYD7 not associated with the Na+/K+ ATPase has been demonstrated in Xenopus oocytes but not in mammalian cells. Similar experiments in mammalian cells require the comparison of FXYD7 surface expression at different stoichiometric ratios with α1β1 Na+/K+ ATPase and, in particular, under conditions where it is overexpressed relative to the pump. We were unable to establish such conditions and, hence, attempted an indirect approach of FXYD7 targeting to the apical versus basolateral membrane in M-1 cells. M-1 is an established cell line derived from mouse kidney collecting duct (55). When cultivated on porous supports, it forms polarized tight epithelium-expressing Na+/K+ ATPase in the basolateral membrane and epithelial Na+ channels in the apical membrane. This is evident from the development of transepithelial electrical potential difference and short circuit current that can be blocked by the apical application of amiloride (55). We found that FXYD7, too, is directed in the polarized epithelium to the basolateral membrane, suggesting an intracellular association with the Na+/K+ ATPase. The Validity of this conclusion depends on the assumption that FXYD7 does not have an endogenous basolateral targeting motif. This is likely because this protein is expressed in vivo only in non-epithelial cells. Also, O-glycosylation that appears to take place in FXYD7 is known to be an apical and not basolateral targeting signal (48). We also tested for the difference in the lateral diffusion of FXYD7 and α1Na+/K+ ATPase by measuring fluorescence recovery after photobleaching of CFP- and YFP-tagged proteins (data not shown). These experiments, however, appear inconclusive because the lateral diffusion of proteins in the membrane depends only weakly on their size (56).
The difference between Xenopus oocytes and mammalian cells may be explained by the fact that in oocytes injected with FXYD7 without Na+/K+ ATPase, this protein is far more abundant than the endogenous Na+/K+ ATPase. Such overexpression is not achieved in mammalian cells, which express a high level of the pump. Thus, FXYD7 may in principle traffic to and reside in the plasma membrane when it is not associated with the Na+/K+ ATPase. However, this option is manifested only if its abundance is much higher than that of the pump, a situation not achieved in the transfected mammalian cells. Another possibility is that dependence on the Na+/K+ ATPase is altered with temperature (25 °C in oocytes versus 37 °C in mammalian cells).
In summary, the above study demonstrates the role of N-terminal O-glycosylation in the surface expression of FXYD7 and the role of C-terminal positive charges in the surface expression of PLM. It also raises the possibility that FXYD7 has a cellular role other than modulating the Na+/K+ ATPase.
Acknowledgments
We thank Prof. Meir Wilchek for advice in surface biotinylation and synthesis of DBB.
This study was supported by Israel Science Foundation Research Grant 688/11.
- PLM
- phospholemman
- DBB
- p-diazobenzoyl biocytin
- ER
- endoplasmic reticulum
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein.
REFERENCES
- 1. Sweadner K. J., Rael E. (2000) The FXYD gene family of small ion transport regulators or channels. cDNA sequence, protein signature sequence, and expression. Genomics 68, 41–56 [DOI] [PubMed] [Google Scholar]
- 2. Geering K. (2006) FXYD proteins: new regulators of Na-K-ATPase. Am. J. Physiol. Renal Physiol. 290, F241–F250 [DOI] [PubMed] [Google Scholar]
- 3. Garty H., Karlish S. J. (2006) Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 68, 431–459 [DOI] [PubMed] [Google Scholar]
- 4. Lifshitz Y., Petrovich E., Haviv H., Goldshleger R., Tal D. M., Garty H., Karlish S. J. (2007) Purification of the human α2 isoform of Na,K-ATPase expressed in Pichia pastoris. Stabilization by lipids and FXYD1. Biochemistry 46, 14937–14950 [DOI] [PubMed] [Google Scholar]
- 5. Mishra N. K., Peleg Y., Cirri E., Belogus T., Lifshitz Y., Voelker D. R., Apell H. J., Garty H., Karlish S. J. (2011) FXYD proteins stabilize Na,K-ATPase. Amplification of specific phosphatidylserine-protein interactions. J. Biol. Chem. 286, 9699–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palmer C. J., Scott B. T., Jones L. R. (1991) Purification and complete sequence determination of the major plasma membrane substrate for cAMP-dependent protein kinase and protein kinase C in myocardium. J. Biol. Chem. 266, 11126–11130 [PubMed] [Google Scholar]
- 7. Chen L. S., Lo C. F., Numann R., Cuddy M. (1997) Characterization of the human and rat phospholemman (PLM) cDNAs and localization of the human PLM gene to chromosome 19q13.1. Genomics 41, 435–443 [DOI] [PubMed] [Google Scholar]
- 8. Crambert G., Fuzesi M., Garty H., Karlish S., Geering K. (2002) Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc. Natl. Acad. Sci. U.S.A. 99, 11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahlers B. A., Zhang X. Q., Moorman J. R., Rothblum L. I., Carl L. L., Song J., Wang J., Geddis L. M., Tucker A. L., Mounsey J. P., Cheung J. Y. (2005) Identification of an endogenous inhibitor of the cardiac Na+/Ca2+ exchanger, phospholemman. J. Biol. Chem. 280, 19875–19882 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X. Q., Wang J., Carl L. L., Song J., Ahlers B. A., Cheung J. Y. (2009) Phospholemman regulates cardiac Na+/Ca2+ exchanger by interacting with the exchanger's proximal linker domain. Am. J. Physiol. Cell Physiol. 296, C911-C921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J., Gao E., Rabinowitz J., Song J., Zhang X. Q., Koch W. J., Tucker A. L., Chan T. O., Feldman A. M., Cheung J. Y. (2011) Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am. J. Physiol. Heart Circ. Physiol. 300, H859–H868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fuller W., Howie J., McLatchie L. M., Weber R. J., Hastie C. J., Burness K., Pavlovic D., Shattock M. J. (2009) FXYD1 phosphorylation in vitro and in adult rat cardiac myocytes: threonine 69 is a novel substrate for protein kinase C. Am. J. Physiol. Cell Physiol. 296, C1346–C1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Despa S., Bossuyt J., Han F., Ginsburg K. S., Jia L. G., Kutchai H., Tucker A. L., Bers D. M. (2005) Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 97, 252–259 [DOI] [PubMed] [Google Scholar]
- 14. Silverman B. Z., Fuller W., Eaton P., Deng J., Moorman J. R., Cheung J. Y., James A. F., Shattock M. J. (2005) Serine 68 phosphorylation of phospholemman: acute isoform specific activation of cardiac Na/K ATPase. Cardiovasc. Res. 65, 93–103 [DOI] [PubMed] [Google Scholar]
- 15. Han F., Bossuyt J., Despa S., Tucker A. L., Bers D. M. (2006) Phospholemman phosphorylation mediates the protein kinase C-dependent effects on Na+/K+ pump function in cardiac myocytes. Circ. Res. 99, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 16. Zhang X. Q., Ahlers B. A., Tucker A. L., Song J., Wang J., Moorman J. R., Mounsey J. P., Carl L. L., Rothblum L. I., Cheung J. Y. (2006) Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger. Role of phosphorylation. J. Biol. Chem. 281, 7784–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song J., Gao E., Wang J., Zhang X. Q., Chan T. O., Koch W. J., Shang X., Joseph J. I., Peterson B. Z., Feldman A. M., Cheung J. Y. (2012) Constitutive overexpression of phosphomimetic phospholemman S68E mutant results in arrhythmias, early mortality, and heart failure. Potential involvement of Na+/Ca2+ exchanger. Am. J. Physiol. Heart Circ. Physiol. 302, H770-H781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo K., Wang X., Gao G., Huang C., Elmslie K. S., Peterson B. Z. (2010) Amino acid substitutions in the FXYD motif enhance phospholemman-induced modulation of cardiac l-type calcium channels. Am. J. Physiol. Cell Physiol. 299, C1203-C1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Béguin P., Crambert G., Monnet-Tschudi F., Uldry M., Horisberger J. D., Garty H., Geering K. (2002) FXYD7 is a brain-specific regulator of Na,K-ATPase α 1-β isozymes. EMBO J. 21, 3264–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crambert G., Li C., Swee L. K., Geering K. (2004) FXYD7, mapping of functional sites involved in endoplasmic reticulum export, association with and regulation of Na,K-ATPase. J. Biol. Chem. 279, 30888–30895 [DOI] [PubMed] [Google Scholar]
- 21. Li C., Crambert G., Thuillard D., Roy S., Schaer D., Geering K. (2005) Role of the transmembrane domain of FXYD7 in structural and functional interactions with Na,K-ATPase. J. Biol. Chem. 280, 42738–42743 [DOI] [PubMed] [Google Scholar]
- 22. Geering K., Beggah A., Good P., Girardet S., Roy S., Schaer D., Jaunin P. (1996) Oligomerization and maturation of Na,K-ATPase. Functional interaction of the cytoplasmic NH2 terminus of the β subunit with the α subunit. J. Cell Biol. 133, 1193–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilchek M., Ben-Hur H., Bayer E. A. (1986) p-Diazobenzoyl biocytin. A new biotinylating reagent for the labeling of tyrosines and histidines in proteins. Biochem. Biophys. Res. Commun. 138, 872–879 [DOI] [PubMed] [Google Scholar]
- 24. Lubarski I., Karlish S. J., Garty H. (2007) Structural and functional interactions between FXYD5 and the Na+-K+-ATPase. Am. J. Physiol. Renal Physiol. 293, F1818-F1826 [DOI] [PubMed] [Google Scholar]
- 25. Lubarski I., Asher C., Garty H. (2011) FXYD5 (dysadherin) regulates the paracellular permeability in cultured kidney collecting duct cells. Am. J. Physiol. Renal Physiol. 301, F1270-F1280 [DOI] [PubMed] [Google Scholar]
- 26. Liman E. R., Tytgat J., Hess P. (1992) Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871 [DOI] [PubMed] [Google Scholar]
- 27. Song J., Zhang X. Q., Ahlers B. A., Carl L. L., Wang J., Rothblum L. I., Stahl R. C., Mounsey J. P., Tucker A. L., Moorman J. R., Cheung J. Y. (2005) Serine 68 of phospholemman is critical in modulation of contractility, [Ca2+]i transients, and Na+/Ca2+ exchange in adult rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 288, H2342-H2354 [DOI] [PubMed] [Google Scholar]
- 28. Bibert S., Roy S., Schaer D., Horisberger J. D., Geering K. (2008) Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na,K-ATPase isozymes. J. Biol. Chem. 283, 476–486 [DOI] [PubMed] [Google Scholar]
- 29. Mounsey J. P., Lu K. P., Patel M. K., Chen Z. H., Horne L. T., John J. E., 3rd, Means A. R., Jones L. R., Moorman J. R. (1999) Modulation of Xenopus oocyte-expressed phospholemman-induced ion currents by coexpression of protein kinases. Biochim. Biophys. Acta 1451, 305–318 [DOI] [PubMed] [Google Scholar]
- 30. Lansbery K. L., Burcea L. C., Mendenhall M. L., Mercer R. W. (2006) Cytoplasmic targeting signals mediate the delivery of phospholemman to the plasma membrane. Am. J. Physiol. Cell Physiol. 290, C1275–C1286 [DOI] [PubMed] [Google Scholar]
- 31. Lee M. C., Miller E. A., Goldberg J., Orci L., Schekman R. (2004) Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20, 87–123 [DOI] [PubMed] [Google Scholar]
- 32. Sigal A., Danon T., Cohen A., Milo R., Geva-Zatorsky N., Lustig G., Liron Y., Alon U., Perzov N. (2007) Generation of a fluorescently labeled endogenous protein library in living human cells. Nat. Protoc. 2, 1515–1527 [DOI] [PubMed] [Google Scholar]
- 33. Hanisch F. G. (2001) O-glycosylation of the mucin type. Biol. Chem. 382, 143–149 [DOI] [PubMed] [Google Scholar]
- 34. Cheung J. Y. (2008) Regulation of cardiac contractility: high time for FXYD. Am. J. Physiol. Heart Circ. Physiol. 294, H584-H585 [DOI] [PubMed] [Google Scholar]
- 35. Wang J., Zhang X. Q., Ahlers B. A., Carl L. L., Song J., Rothblum L. I., Stahl R. C., Carey D. J., Cheung J. Y. (2006) Cytoplasmic tail of phospholemman interacts with the intracellular loop of the cardiac Na+/Ca2+ exchanger. J. Biol. Chem. 281, 32004–32015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang X. Q., Qureshi A., Song J., Carl L. L., Tian Q., Stahl R. C., Carey D. J., Rothblum L. I., Cheung J. Y. (2003) Phospholemman modulates Na+/Ca2+ exchange in adult rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 284, H225-H233 [DOI] [PubMed] [Google Scholar]
- 37. Moorman J. R., Palmer C. J., John J. E., 3rd, Durieux M. E., Jones L. R. (1992) Phospholemman expression induces a hyperpolarization-activated chloride current in Xenopus oocytes. J. Biol. Chem. 267, 14551–14554 [PubMed] [Google Scholar]
- 38. Morrison B. W., Moorman J. R., Kowdley G. C., Kobayashi Y. M., Jones L. R., Leder P. (1995) Mat-8, a novel phospholemman-like protein expressed in human breast tumors, induces a chloride conductance in Xenopus oocytes. J. Biol. Chem. 270, 2176–2182 [DOI] [PubMed] [Google Scholar]
- 39. Attali B., Latter H., Rachamim N., Garty H. (1995) A corticosteroid-induced gene expressing an “IsK-like” K+ channel activity in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 92, 6092–6096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Minor N. T., Sha Q., Nichols C. G., Mercer R. W. (1998) The γ subunit of the Na,K-ATPase induces cation channel activity. Proc. Natl. Acad. Sci. U.S.A. 95, 6521–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ino Y., Gotoh M., Sakamoto M., Tsukagoshi K., Hirohashi S. (2002) Dysadherin, a cancer-associated cell membrane glycoprotein, down-regulates E-cadherin and promotes metastasis. Proc. Natl. Acad. Sci. U.S.A. 99, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nam J. S., Hirohashi S., Wakefield L. M. (2007) Dysadherin: a new player in cancer progression. Cancer Lett. 255, 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller T. J., Davis P. B. (2008) FXYD5 modulates Na+ absorption and is increased in cystic fibrosis airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L654-L664 [DOI] [PubMed] [Google Scholar]
- 44. Mohler P. J., Davis J. Q., Bennett V. (2005) Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 3, 2158–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Madhani M., Hall A. R., Cuello F., Charles R. L., Burgoyne J. R., Fuller W., Hobbs A. J., Shattock M. J., Eaton P. (2010) Phospholemman Ser-69 phosphorylation contributes to sildenafil-induced cardioprotection against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 299, H827-H836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 47. Füzesi M., Gottschalk K. E., Lindzen M., Shainskaya A., Küster B., Garty H., Karlish S. J. (2005) Covalent cross-links between the γ subunit (FXYD2) and α and β subunits of Na,K-ATPase. Modeling the α-γ interaction. J. Biol. Chem. 280, 18291–18301 [DOI] [PubMed] [Google Scholar]
- 48. Potter B. A., Hughey R. P., Weisz O. A. (2006) Role of N- and O-glycans in polarized biosynthetic sorting. Am. J. Physiol. Cell Physiol. 290, C1–C10 [DOI] [PubMed] [Google Scholar]
- 49. Zhu W., Leber B., Andrews D. W. (2001) Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J. 20, 5999–6007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kanno E., Fukuda M. (2008) Increased plasma membrane localization of O-glycosylation-deficient mutant of synaptotagmin I in PC12 cells. J. Neurosci. Res. 86, 1036–1043 [DOI] [PubMed] [Google Scholar]
- 51. Hennebicq-Reig S., Lesuffleur T., Capon C., De Bolos C., Kim I., Moreau O., Richet C., Hémon B., Recchi M. A., Maës E., Aubert J. P., Real F. X., Zweibaum A., Delannoy P., Degand P., Huet G. (1998) Permanent exposure of mucin-secreting HT-29 cells to benzyl-N-acetyl-α-d-galactosaminide induces abnormal O-glycosylation of mucins and inhibits constitutive and stimulated MUC5AC secretion. Biochem. J. 334, 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maryon E. B., Molloy S. A., Kaplan J. H. (2007) O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J. Biol. Chem. 282, 20376–20387 [DOI] [PubMed] [Google Scholar]
- 53. Crambert G., Béguin P., Uldry M., Monnet-Tschudi F., Horisberger J. D., Garty H., Geering K. (2003) FXYD7, the first brain- and isoform-specific regulator of Na,K-ATPase. Biosynthesis and function of its posttranslational modifications. Ann. N.Y. Acad. Sci. 986, 444–448 [DOI] [PubMed] [Google Scholar]
- 54. Bossuyt J., Despa S., Martin J. L., Bers D. M. (2006) Phospholemman phosphorylation alters its fluorescence resonance energy transfer with the Na/K-ATPase pump. J. Biol. Chem. 281, 32765–32773 [DOI] [PubMed] [Google Scholar]
- 55. Stoos B. A., Naray-Fejes-Toth A., Carretero O. A., Ito S., Fejes-Toth G. (1991) Characterization of a mouse cortical collecting duct cell line. Kidney Int. 39, 1168–1175 [DOI] [PubMed] [Google Scholar]
- 56. Kucik D. F., Elson E. L., Sheetz M. P. (1999) Weak dependence of mobility of membrane protein aggregates on aggregate size supports a viscous model of retardation of diffusion. Biophys. J. 76, 314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]