

Background: ADAM15 confers apoptosis resistance to chondrocytes upon exposure to genotoxic stress.
Results: Apoptosis induction provokes direct ADAM15 binding to focal adhesion kinase (FAK) and an associated enhanced phosphorylation of the FAK-Src complex.
Conclusion: ADAM15 interacts with FAK-Src, thereby enhancing survival signals.
Significance: The anti-apoptotic ADAM15 signaling has potential relevance to tumorigenesis beyond its impact on chondrocyte survival.
Keywords: Apoptosis, Cartilage, Cartilage Biology, Chondrocytes, Focal Adhesion Kinase, Signaling, ADAM15
Abstract
ADAM15, a disintegrin and metalloproteinase, is capable of counteracting genotoxic stress-induced apoptosis by the suppression of caspase-3 activation. A cell line expressing the membrane-bound ADAM15 without its cytoplasmic tail, however, lost this anti-apoptotic property, suggesting a crucial role of the intracellular domain as a scaffold for recruitment of survival signal-transducing kinases. Accordingly, an enhanced phosphorylation of FAK at Tyr-397, Tyr-576, and Tyr-861 was detected upon genotoxic stress by camptothecin in ADAM15-transfected T/C28a4 cells, but not in transfectants expressing an ADAM15 mutant without the cytoplasmic tail. Accordingly, a specific binding of the cytoplasmic ADAM15 domain to the C terminus of FAK could be shown by mammalian two-hybrid, pulldown, and far Western studies. In cells expressing full-length ADAM15, a concomitant activation of Src at Tyr-416 was detected upon camptothecin exposure. Cells transfected with a chimeric construct consisting of the extracellular IL-2 receptor α-chain and the cytoplasmic ADAM15 domain were IL-2-stimulated to prove that the ADAM15 tail can transduce a percepted extracellular signal to enhance FAK and Src phosphorylation. Our studies further demonstrate Src binding to FAK but not a direct Src interaction with ADAM15, suggesting FAK as a critical intracellular adaptor for ADAM15-dependent enhancement of FAK/Src activation. Moreover, the apoptosis induction elicited by specific inhibitors (PP2, FAK 14 inhibitor) of FAK/Src signaling was significantly reduced by ADAM15 expression. The newly uncovered counter-regulatory response to genotoxic stress in a chondrocytic survival pathway is potentially also relevant to apoptosis resistance in neoplastic growth.
Introduction
ADAM15 is a transmembrane-anchored protein with a large extracellular part, consisting of a prodomain at its N terminus followed by a metalloproteinase, a disintegrin, a cysteine-rich domain, and a cytoplasmic tail of 100 amino acid residues (1). ADAM15 is implicated in neoplastic and inflammatory extracellular matrix (ECM)3 remodeling (2–5) and neovascularization (6), and its up-regulation is strongly associated with the progression of aggressive forms of certain types of neoplasms such as prostate or breast cancer (7, 8). However, the pathogenic effect of ADAM15 does not remain confined to tumorigenesis and metastasis but is also operative in non-neoplastic conditions of tissue remodeling such as degenerative and inflammatory joint disease. An up-regulated ADAM15 expression has been demonstrated in osteoarthritic (OA) cartilage (9) as well as in the inflamed synovial membrane of rheumatoid arthritis joints (4). However, the evidence for a catalytic function of ADAM15 is sparse and seems to be confined to a sheddase activity for a rather restricted set of cell surface proteins (10–12). Thus, a direct proteolytic action of ADAM15 in cartilage remodeling in vivo is missing.
An alternative mode of action in OA cartilage is suggested by an increasing body of literature that pertains to an emerging role for ADAM15 in cell-matrix interactions. Aging ADAM15-deficient mice develop an accelerated cartilage degeneration compared with wild-type mice (13), thereby suggesting a homeostatic rather than a destructive role of ADAM15 in cartilage remodeling. Accordingly, ADAM15 has been shown to reinforce integrin-dependent cell adhesion to ECM components critically involving its extracellular domain and to modulate outside-in signaling in OA chondrocytes (14). A role of ADAM15 in cell adhesion is further supported by studies demonstrating a specific interaction of α5 and αv integrins with its disintegrin domain (15). In addition, ADAM15 was shown to display a modulatory effect on the autophosphorylation site Tyr-397 of FAK upon chondrocyte-collagen adhesion, which was dependent on the presence of its cytoplamic domain (14). FAK functions as a critical scaffolding molecule that integrates signals transmitted by integrins and growth factor receptors into triggers of growth, differentiation, and survival pathways (16–18). Accordingly, a modulatory effect of ADAM15 on FAK might have a significant impact on chondrocyte vitality that is crucial for the constant biosynthetic replenishment of ECM molecules to maintain cartilage integrity (19, 20). We could recently demonstrate, that ADAM15 expression leads to a significantly increased cell viability and apoptosis resistance of primary human OA chondrocytes after inducing DNA damage by the topoisomerase inhibitor camptothecin (21). This anti-apoptotic effect of ADAM15 was accompanied by an up-regulation of the anti-apoptotic protein X-linked inhibitor of apoptosis with a concomitantly reduced expression of activated caspase-3 (21).
In the present study, we investigated whether the detected anti-apoptotic properties of ADAM15 are conferred by the intracellular domain. In addition, we asked the question of a direct molecular interaction of the cytoplasmic tail of ADAM15 (cytoADAM15) with FAK based on the earlier published modulatory impact of ADAM15 on FAK signaling (14). We provide unequivocal evidence for direct binding and identified the FAK domain that interacts with cytoADAM15. Moreover, we demonstrate that FAK phosphorylation at Tyr-397, Tyr-576, and Tyr-861 induced by camptothecin is considerably enhanced in ADAM15 expressing T/C28a4 chondrocytic cells. This ADAM15-dependent reinforcement of FAK phosphorylation in response to an apoptosis-inducing stimulus is critically dependent on the cytoplasmic domain of ADAM15. Moreover, the increased activation of FAK is accompanied by an enhanced phosphorylation of Src at Tyr-416 that is critical for the stabilization of a catalytically active conformation (22). Accordingly, ADAM15 also counteracts the apoptotis-inducing effect of interference with FAK/Src signal transduction by specific pharmacologic agents, FAK 14 inhibitor (23, 24) and the Src inhibitor PP2 (25). Thus, ADAM15 leads to an amplified FAK-Src complex activation in response to genotoxic stress, thereby reinforcing counter-regulatory survival pathways. This ADAM15-dependent mechanism is likely not confined to chondrocytic cell survival but also relevant to apoptosis resistance in neoplastic growth.
EXPERIMENTAL PROCEDURES
Materials
Mouse and goat anti-ADAM15 antibodies were from R&D Systems (recognizing the prodomain between amino acids 1–200 (14) catalog no. MAB935, AF935). Rabbit anti-ADAM15 antibodies (directed against the cytoplasmic domain, catalog no. ab39159) were from Abcam. Anti-phospho-FAK (Tyr-397) (catalog no. 44-624G) was from BIOSOURCE, anti phospho-FAK (Tyr-576/577) (catalog no. 2183-1) and anti-phospho-FAK (Tyr-861) (catalog no. 2153–1) were from Epitomics, and monoclonal anti-FAK (clone 4.47, catalog no. 05-537) was from Upstate. Rabbit anti-Src (Tyr-416) was from Cell Signaling (catalog no. 2101). Rabbit anti-tubulin (catalog no. 1799-1) was from Epitomics, mouse anti-human CD25 (clone 7G7B6, catalog no. 174-020) was from Ancell, and anti-phospho-antibody (Tyr-99) was from Santa Cruz Biotechnology (catalog no. sc-7020). Camptothecin, PP2, and PP3 were from Calbiochem. Human IL-2 (Proleukin®) was from Novartis. FAK 14 inhibitor was from Tocris Bioscience.
Isolation of Human OA Chondrocytes
OA chondrocytes were isolated as described previously (21) from the hip or knee joints of patients at the time of endoprosthetic joint replacement for severe OA. All patients had provided written informed consent and approval was obtained from the Ethics Committee of the Goethe University Hospital (Frankfurt am Main, Germany).
Cloning of ADAM15 and Chimeric IL-2Rα/ADAM15 Construct
Full-length ADAM15 (GenBankTM accession no. AAS72992.1) and a deletion mutant lacking the cytoplasmic domain (ADAM15Δcyto) were cloned as described previously (14). The ADAM15 clone is also referred to as ADAM15A (8) or isoform variant 2 (26)). A chimeric construct consisting of the extracellular part of IL-2 receptor-α (IL2Rα, CD25) and the transmembrane and cytoADAM15 was cloned using Fusion PCR: 1) CD25 was amplified with the primer I and II using a CD25 cDNA containing plasmid (27). 2) The transmembrane and cytoplasmic domains of ADAM15 were amplified with primers III and IV (primer sequences in supplemental Table S1) using the full-length ADAM15 plasmid as template. In a third step, the PCR products from 1) and 2) were combined and amplified by PCR using primer I and IV. The PCR product was cloned into the pExchange-1 vector (Stratagene).
Generation of Permanent Cell Lines Transfected with ADAM15 and Mutant Constructs
Full-length ADAM15, ADAM15Δcyto, and the chimeric IL2Rα/cytoADAM15 construct were stably transfected into the chondrocyte cell line T/C28a4 (28). The transfection and selection procedures were performed as described previously (14).
Cell Culture
Transfected cells were grown in DMEM with 10% FCS as described previously (14). For all subsequent tests, cells were grown to subconfluency (4 × 106 cells/75 cm2 tissue culture flask). Human OA chondrocytes were kept in DMEM/Hams F12 medium containing 10% FCS. For IL-2 experiments, cells (5 × 105) expressing the chimeric IL-2Rα/ADAM15cyto protein were seeded into six-well tissue culture plates, grown for 24 h, and stimulated with 200 IU/ml IL-2.
Determination of Caspase-3/7 Activity
Transfected cells and primary OA chondrocytes (1 × 104) were seeded into 96-well white tissue culture plates (Greiner) and grown in DMEM containing 10% FCS for 24 h. Cells were treated with 20 μm camptothecin in DMEM for 0–8 h at 37 °C. Caspase-3/7 activity was measured using the CaspaseGlo 3/7® assay (Promega) on a Mithras LB 940 luminometer plate reader (Berthold Technologies).
Cell Viability Assay
The transfected cells (1 × 104) were grown in 96-well plates for 24 h at 37 °C and treated with various concentrations of camptothecin (0–100 μm) for 18 h. The number of viable cells was determined by quantification of ATP using the CellTiterGlo® luminescent cell viability assay from Promega.
Mammalian Two-hybrid
The interaction of ADAM15 with FAK and Src was analyzed using a mammalian two-hybrid assay (Stratagene). All primers used are listed in supplemental Table S1. CytoADAM15 (amino acids 717–814) was cloned into the BamHI/NotI site of the pCMV-AD prey vector using a full-length ADAM15 plasmid as template for PCR amplification. The FAK domains 33–355, 422–676, 707–1052, 707–913, and 914–1052 were cloned accordingly into the pCMV-BD bait vector using reverse-transcribed cDNA from chondrocytes. Src (1–192) and Src (1–450) was cloned into the pCMV-AD vector. 1 × 104 HEK cells were seeded into a 96-well plate, grown for 24 h, and transfected with the bait and prey vector (10 ng each), the firefly luciferase reporter (250 ng) and a Renilla luciferase plasmid (5 ng; pRL-TK vector, Promega) to control for equal transfection rates, using JetPei according to the manufacturer's instructions (Biomol). 48 h after transfection, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). Quadruplicate wells were transfected, and the assay was performed at least five times. Measured firefly values were normalized to the Renilla luciferase values.
Introduction of Point Mutations
The Tyr-861 of FAK was mutated into Phe-861 using the QuikChange site-directed mutagenesis kit from Stratagene according to the manufacturer's instructions.
Preparation of Cell Lysates and Western Blotting
Cell lysates were prepared and immunoblotted as described previously (21). Signals were exposed to x-ray films and scanned, and signal densities were measured using ImageJ software.
Generation of Recombinant FAK and ADAM15
The FAK domains 33–355, 422–676, and 707–913 were subcloned into the BamHI/NotI site of pGEX-6P using the above mentioned primers and expressed as Gst fusion proteins in BL21 Escherichia coli (Amersham Biosciences). CytoADAM15 (amino acids 717–814) was also subcloned into pGEX-6P with a Myc tag attached to the 3′-end (5′-GGATCCCAGATCCTC TTCAGAGATGAGTTTCTGCTCGAGGTAGAGCGAGGACACTGT-3′). Protein expression and purification using glutathione-Sepharose affinity chromatography was performed as described (14). For the protein binding assays, the Gst tag of the cytoADAM15 was removed with PreScission protease according to the manufacturer's instructions (Amersham Biosciences).
Pulldown Assays
Vector-transfected cells were lysed in 10 mm HEPES, pH 7.4, 1% Triton X-100 plus proteinase inhibitor mixture. The respective Gst-tagged FAK fragments were co-incubated with the recombinant Myc-tagged cytoADAM15 protein at various concentrations with the cell lysate (150 μg/300 μl) and Gst-Sepharose for 2 h at 4 °C, thoroughly washed with PBS, 1% Triton X-100, and analyzed by Western blotting using anti-myc antibodies.
Silencing of ADAM15 by RNA Interference
The knockdown of ADAM15 in chondrocytes was performed as described (21). Briefly, chondrocytes were seeded into 96-well plates, and the protein expression of ADAM15 was silenced using two different siRNAs for ADAM15 and a control siRNA for 40 h prior to treatment with FAK 14 inhibitor or PP2.
Immunocytochemistry
Chamber slides (BD Falcon) were coated with bovine collagen type II (50 μg/ml; MDBioSciences) and blocked with PBS/1% BSA. 1 × 104 OA chondrocytes and ADAM15-transfected cells were seeded into the chamber slides and grown for 24 h. After fixation in 4% paraformaldehyde in PBS and blocking with PBS/1% BSA, cells were stained with goat anti-ADAM15 (1:100) and mouse anti-FAK (1:100) for 2 h and visualized with Alexa Fluor 488 anti-goat and Alexa Fluor 594 anti-mouse conjugated antibodies (1:200; Molecular Probes) using confocal laser scanning microscopy. Nuclei were counterstained with DAPI.
Statistics
Data presented are the means ± S.D. of quadruplicates of at least five independently performed assays. Statistical significance was determined using unpaired Student's t test. A p value of < 0.05 was considered statistically significant.
RESULTS
Cytoplasmic Domain of ADAM15 Confers Anti-apoptotic Properties to ADAM15
To elucidate underlying mechanisms of the recently uncovered anti-apoptotic effect of ADAM15 (21), T/C28a4 chondrocytes were stably transfected with either full-length ADAM15 or a mutant construct encoding a transmembrane-anchored variant that lacks the cytoplasmic domain (ADAM15Δcyto) (14). Both the wild-type and mutant ADAM15 are expressed to the same degree as visualized by Western blotting (14). Cell surface expression of both proteins at a roughly comparable level was confirmed by FACS analysis (supplemental Fig. S1). The expression of ADAM15 in T/C28a4 transfected cells remained below the level detected in the majority of distinct primary chondrocyte populations derived from 15 OA patients (supplemental Fig. S2).
Cells expressing full-length ADAM15 displayed a significantly reduced caspase-3/7 activation in response to apoptosis induction by camptothecin compared with vector-transfected control chondrocytes (Fig. 1A) (21). However, the caspase induction in ADAM15Δcyto-transfected cells did not differ from that determined in vector control transfectants. Accordingly, the viability of ADAM15-transfected cells remained high (∼80%) despite exposure to increasing concentrations of camptothecin that caused a significant drop of the survival rates in ADAM15Δcyto- and vector-transfected cells (Fig. 1B). These results indicate a critical role of the cytoplasmic domain for the anti-apoptotic properties of ADAM15.
FIGURE 1.
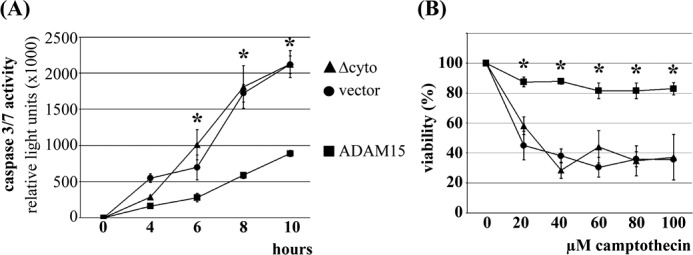
Anti-apoptotic property of full-length ADAM15. A, ADAM15-, ADAM15Δcyto-, and vector-transfected cells were treated with 20 μm camptothecin and caspase-3/7 activity as a marker for apoptosis was measured for 0–10 h. B, the ATP content, which correlates with cell viability, was determined after exposure of the cells for 18 h using increasing camptothecin concentrations. Both assays show that the removal of the cytoplasmic tail of ADAM15 resulted in a loss of the anti-apoptotic and cell protective capacity compared with chondrocytes transfected with full-length ADAM15. *, p < 0.0004, when comparing the caspase activity or viability of the ADAM15- versus vector-transfected cells.
Increased Phosphorylation of FAK in ADAM15-transfected Cells upon Genotoxic Apoptosis Induction
Based on our earlier studies suggesting ADAM15-dependent modulation of integrin-mediated FAK phosphorylation upon cell adhesion to collagen (14), we studied the FAK activation in ADAM15-expressing cells in response to camptothecin-induced apoptosis. Vector control, ADAM15, and ADAM15Δcyto-transfected cells were treated with camptothecin, and FAK phosphorylation at Tyr-397, Tyr-576, Tyr-861, and Tyr-925 was analyzed by immunoblotting. An enhanced, sustained phosphorylation of FAK at Tyr-861, Tyr-576, and Tyr-397 was determined in ADAM15-transfected cells throughout all time points measured (∼2.5-fold higher phosphorylation level for all three tyrosines when compared with that of vector-transfected cells). The vector control cells, however, did not activate FAK considerably above base-line level (Fig. 2A). Furthermore, no phosphorylation signal was detectable at Tyr-925 neither in vector- nor ADAM15-transfected cells upon camptothecin stimulation (data not shown). Cells transfected with ADAM15Δcyto did not exhibit any FAK phosphorylation signal at Tyr-397, Tyr-576, and Tyr-861 above the low background in vector-transfected cells (Fig. 2B). These data clearly suggest a crucial involvement of the cytoplasmic tail of ADAM15 in the enhancement of genotoxic stress-induced FAK phosphorylation.
FIGURE 2.
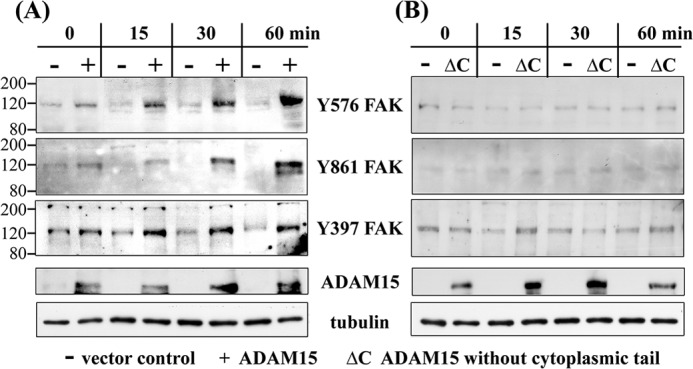
Enhanced phosphorylation of FAK in ADAM15-transfected chondrocytes after apoptosis induction by camptothecin. A, immunoblots of cell lysates of vector-transfected (−) and ADAM15-transfected cells (+) treated with 2 μm camptothecin for up to 60 min, showing a stronger phosphorylation at Tyr-576, Tyr-861, and Tyr-397 of FAK in the ADAM15-expressing cells in comparison with vector-transfected cells. B, T/C28a4 cells transfected with ADAM15 lacking the cytoplasmic tail (ΔC), however, did not display an activation of FAK. Blots were controlled for ADAM15 and/or ADAM15ΔC expression, and loading was monitored using anti tubulin antibodies. The immunoblots are representative of at least five repeated experiments.
ADAM15 Binds to C Terminus of FAK
Based on the modifying effect of ADAM15 on camptothecin-induced FAK phosphorylation, we further investigated whether a direct molecular interaction might underlie the functional link between both molecules. The FERM domain (amino acids 33–355), the catalytic domain (422–676), the C-terminal FAK region (707–913), and cytoADAM15 were expressed recombinantly and subsequently used in pulldown studies as well as Far Western. The Gst-tagged FAK domains and the cytoADAM15 harboring both a Gst and Myc tag were expressed in E. coli. Upon purification on Gst-Sepharose and subsequent analysis by SDS-PAGE, the proteins were verified by immunoblotting using anti-Gst antibodies (Fig. 3A, upper panels). For the subsequent binding studies, the Gst tag of cytoADAM15 was removed by preScission protease and the Myc tag used for detection of specific ADAM15 binding to the recombinant FAK fragments. The electrophoretic mobilities of the Gst-tagged as well as the cleaved cytoplasmic ADAM15 domains on SDS-PAGE and their identification by immunoreactivity with anti-Myc and anti-ADAM15 antibodies in respective Western blots are shown in Fig. 3A (lower panels). Binding of ADAM15 to the C terminus of FAK was revealed by pulldown studies applying anti-Gst antibodies for the precipitation of the Gst-tagged FAK domains and anti-Myc antibodies for detection of bound cytoADAM15 (Fig. 3B). The blots were stripped, and the precipitated Gst-tagged FAK proteins were visualized by anti-Gst antibodies for control (Fig. 3B). In a Far Western analysis, all three Gst-FAK domains were dotted onto nitrocellulose and incubated with the Myc-tagged cytoADAM15 protein. ADAM15 binding remained restricted exclusively to the C-terminal FAK fragment (707–913) (data not shown).
FIGURE 3.
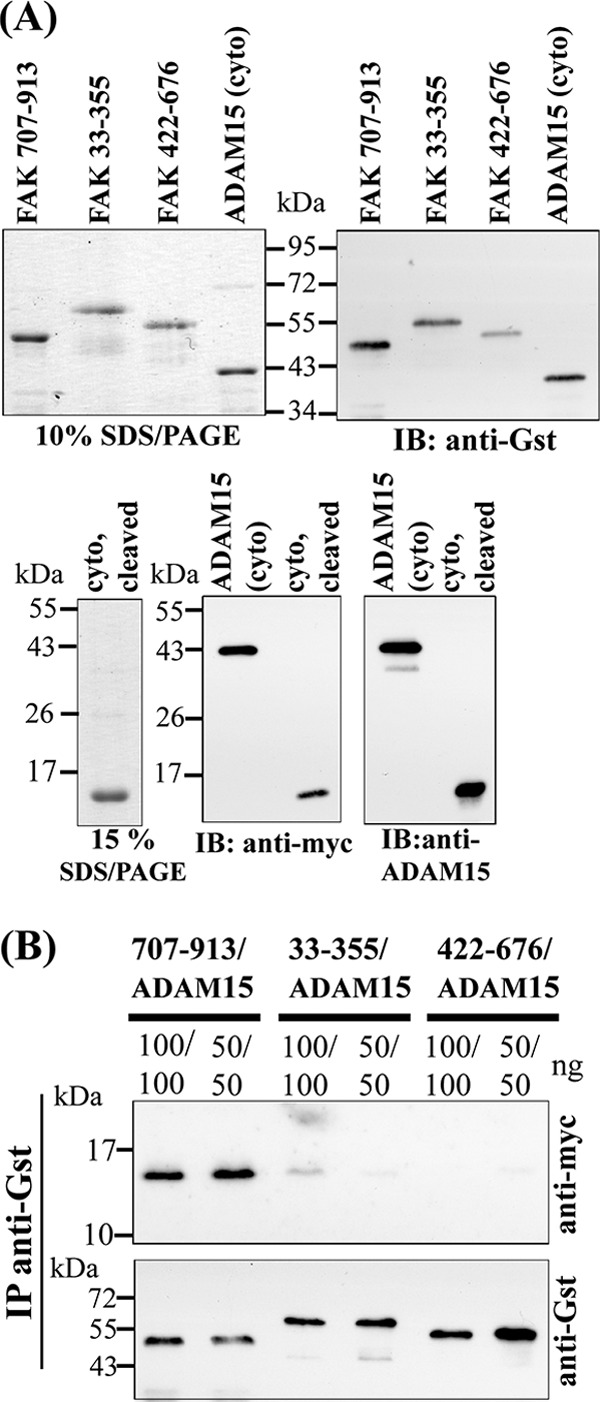
Generation of recombinant ADAM15 and FAK proteins for protein binding assays. A, upper left panel: Coomassie stained SDS-PAGE of the purified Gst-tagged FAK fragments and the Gst- and Myc-tagged cytoplasmic domain of ADAM15 (cyto) and upper right panel: the detection of these proteins with anti-Gst antibodies by Western blotting. A, lower panels: SDS-PAGE of the cytoplasmic domain of ADAM15 with the Gst tag cleaved off by PreScission Protease, followed by immunodetection using anti-Myc and anti-ADAM15 antibodies. B, pulldown assays: cell lysates spiked with Myc-tagged ADAM15 were co-incubated with either of the three different FAK proteins. Upon immunoprecipitation (IP) using anti-Gst-Sepharose and subsequent immunoblotting (IB), bound ADAM15 was detected by anti-Myc antibodies. Blots were stripped, and the Gst-tagged FAK proteins were verified by anti-Gst antibodies, thereby demonstrating the interaction of ADAM15 with the C-terminal FAK-region (amino acids 707–913).
An independent experimental approach served to confirm the interaction of FAK with ADAM15 and further fine-map the area of FAK. Mammalian two-hybrid technology was employed to elucidate whether cytoADAM15 exhibits a specific binding affinity to one of the functional FAK domains. For this purpose, the FERM (33–355), the catalytic (422–676), the entire C-terminal FAK region (707–1052), or the segregated focal adhesion-targeting domain (FAT domain, 914–1052) were cloned separately into the bait vector and cytoADAM15 into the prey vector (Fig. 4A). Bait, prey, and the firefly luciferase reporter were co-transfected with a Renilla control plasmid into HEK cells, and the monitoring of transcribed firefly luciferase activity served as a quantitative indicator for protein interaction. ADAM15 exhibited very strong binding to the C terminus of FAK (707–1052), and further fragmentation of this region clearly demonstrated specific interaction with a FAK fragment covering amino acids 707–913 but not with the FAT domain (Fig. 4B). Virtually no binding of ADAM15 to both the FERM and the catalytic domain of FAK was detectable. Because ADAM15 expression resulted in an enhanced phosphorylation of FAK at Tyr-861 upon apoptosis induction, we investigated whether this modification within the interaction domain of FAK might have an impact on ADAM15 binding. However, site-directed mutagenesis of FAK at position 861 replacing the Tyr by a Phe remained functionally silent (Fig. 4B). Further fine-mapping revealed the FAK fragment 730–790 as the smallest containing all requirements for cytoADAM15 binding within the FAK region 707–913 (Fig. 4C). Further shortening of either the N or C terminus by 10 amino acids already resulted in a ∼70% loss of cytoADAM15 binding.
FIGURE 4.

Interaction of ADAM15 with FAK. A, the domain structure of FAK: the FERM, the catalytic domain, and the C-terminal domain with the FAT domain. Black bars, two proline-rich regions (PXXP motifs) are binding sites for SH2/SH3 domain containing proteins. B and C, mammalian two-hybrid assay. The distinct FAK domains, cloned as segregated fragments into the pCMV-BD bait vector, were co-transfected with the cytoplasmic domain of ADAM15 in pCMV-AD prey vector and a firefly luciferase reporter into HEK293 cells. B, an exclusive interaction of ADAM15 with a C-terminal region of FAK (amino acids 707–913), but not with the FERM, the kinase, or the FAT domain is demonstrated. Mutation of Tyr-861 into Phe-861 in the interaction domain of FAK had no influence on ADAM15 binding. C, left panel, further fine-mapping of the Fak domain 707–913 revealed fragment 730–790 (underlined) as the smallest region with binding capacity equivalent to 707–913. Shortening of 10 amino acids either at the N or C terminus abrogates the binding to ADAM15 by ∼70%. Shown is a representative experiment. C, right panel, depicts the mean (± S.D.) of five repeated assays of ADAM15 binding with each distinct FAK fragment. Measured luciferase values were normalized to the values obtained from a co-transfected Renilla luciferase control plasmid.
ADAM15 Co-localizes with FAK
ADAM15-transfected cells were grown on collagen type II and double-stained for ADAM15 and FAK. The merged image clearly reveals a co-localization of both proteins in focal contacts (Fig. 5A). This finding was confirmed in primary human OA chondrocytes seeded on collagen type II (Fig. 5B).
FIGURE 5.

Co-localization of ADAM15 and FAK. ADAM15 transfected chondrocytic T/C28a4 cells (A) and human osteoarthritic chondrocytes (B) were grown on collagen type II and double-stained with goat anti-ADAM15 and mouse anti-FAK antibodies visualized with Alexa Fluor 488 (green) and 594 (red) conjugated secondary antibodies using confocal laser scanning microscopy (40× objective). C, shown is a magnification of the white boxed area in B with co-localized spots marked by white arrows. The merged images clearly reveal a co-localization of ADAM15 and FAK in focal contacts.
Src Kinase Phosphorylation Is Enhanced in ADAM15-transfected Cells upon Apoptosis Induction
ADAM15-, ADAM15Δcyto-, and vector-transfected cells were stimulated with camptothecin (20 μm) and analyzed for Tyr-416Src phosphorylation by immunoblotting. An enhanced Src phosphorylation was observed in whole cell lysates of ADAM15-transfected cells compared with those derived from vector-control cells (Fig. 6A). Only a very low phosphorylation level of Src at Tyr-416 could be detected in ADAM15Δcyto-transfected cells (data not shown). To identify the binding partner of Src, immunoprecipitations using either anti-FAK or anti-ADAM15 antibodies were performed (Fig. 6B), revealing the co-precipitation of Src both with FAK and ADAM15. Accordingly, our results demonstrate ADAM15, FAK, and Src in a precipitable complex containing all three proteins. In this complex, ADAM15 contributes to enhanced Src activation as shown by a consistently stronger Tyr-416 phosphorylation signal in precipitates of ADAM15-transfected cells compared with those derived from vector control (Fig. 6C).
FIGURE 6.

Stronger phosphorylation of Src kinase in ADAM15-transfected cells is dependent on direct binding to FAK. A, upon stimulation with camptothecin for 60 min, an enhanced phosphorylation of Tyr-416 Src was detected in whole cell lysates of ADAM15-transfected cells (+) as compared with those derived from respective vector control cells (−) by immunoblotting. B, to analyze whether the stronger phosphorylation of Src results from an interaction with ADAM15 or with FAK, co-immunoprecipitations (IP) were performed by using as the precipitating antibody either 1) anti-ADAM15 or 2) anti FAK. C, lysates of ADAM15 (+) and vector control cells (−) upon camptothecin exposure were immunoprecipitated with either anti-ADAM15 1) or anti-FAK antibodies 2) and immunoblotted using anti Tyr-416 Src antibodies. A stronger phosphorylation of Tyr-416 Src was observed in the ADAM15-transfected cells compared with the vector control cells, independent of the antibody used for the precipitation. Blots were stripped, and precipitated Src was visualized using anti-Src antibodies. Src was pulled down in ADAM15 precipitated lysates only (vector-transfected cells, negative). In all anti-FAK antibody-precipitated lysates, equal amounts of Src protein were demonstrated. Blots were restripped and controlled for precipitated ADAM15 1) or FAK 2) using respective antibodies. D, mammalian two-hybrid was used to analyze the binding partner of Src. The N-terminal SH2/SH3 domain as well as full-length Src were cloned into the prey vector and distinct FAK domains into bait vector. Bait and prey were co-transfected with the firefly luciferase reporter and a Renilla luciferase control plasmid into HEK cells. A strong binding of the SH2/SH3 domain as well as full-length Src to the C-terminal region of FAK (707–913) was measured, whereas the FERM, the catalytic, and the FAT domain did not bind to Src. Mutation of Tyr-861 into Phe-861 of FAK did not affect Src interaction. However, no direct binding of ADAM15 to SH2/SH3 Src or full-length Src could be detected by contrast to the already proven interaction with the C-terminal FAK domain (707–1052, Fig. 4) that served as a positive control in this experiment.
Src Directly Interacts with FAK, but Not with ADAM15
Because the above-described results left the question open whether Src can directly bind to cytoADAM15 as well as to FAK in the formed trimolecular complex mammalian two-hybrid studies were employed for investigation. To elucidate FAK/Src-interaction, the distinct FAK domains cloned into the bait vector were co-transfected with the prey vector harboring either the SH2/SH3 domain or full-length Src into HEK293 cells. The measured luciferase activity revealed binding of Src to the C-terminal FAK region (707–1052). An equally strong binding of both Src constructs to the truncated C-terminal FAK region (707–913) was also noted but no interaction with the FERM, the catalytic, or the FAT domain. The FAK-Src interaction was not influenced by a replacement mutation of Tyr-861 into Phe-861 (Fig. 6D). Neither the SH2/SH3 domain nor full-length Src could be demonstrated to bind to cytoADAM15 (Fig. 6D). Accordingly, our results provide strong evidence that the enhanced Src phosphorylation in ADAM15-transfected cells is dependent on the interaction of ADAM15 with FAK, which in turn, can bind to Src.
Signaling of Chimeric IL2Rα/cytoADAM15 Construct
The IL-2 receptor-α (CD25), capable of IL-2 binding but devoid of any intrinsic signaling competence, was fused to cytoADAM15 and stably transfected into the chondrocytic cells to study its potential of transducing signals that are unequivocally traceable to an ectodomain-specific stimulus. The expression of the chimeric construct was analyzed by immunoblotting with either anti-cytoADAM15 or anti-CD25 antibodies, thus verifying the integrity of the chimeric protein of ∼60 kDa (Fig. 7A, left panels). No expression of the T cell-specific CD25 was detected in vector-transfected cells. The surface expression of IL2Rα/cytoADAM15 was also checked by FACS analysis (data not shown). Co-precipitation studies using anti-CD25 antibodies revealed binding of FAK to the chimeric protein in IL2Rα/cytoADAM15 cell lysates only (Fig. 7A, right panel), thereby confirming the interaction of ADAM15 with FAK. To investigate whether this interaction can be triggered by a receptor-specific stimulus, the chimeric cells were treated with IL-2 (2 and 5 min), and cell lysates subsequently were immunoprecipitated with anti-FAK antibodies and analyzed for Src activation at Tyr-416 by Western blotting. Vector-transfected cells served as identically treated controls. An enhanced phosphorylation of Src upon IL-2 stimulation was detected in cells expressing the IL2Rα/cytoADAM15 chimera, whereas only very low Src background phosphorylation independent of IL-2 was recorded in the control cells (Fig. 7B). The amount of total precipitated Src was controled by reprobing of the stripped blots with a respective antibody. Furthermore, the IL-2-specific FAK activation at Tyr-397, Tyr-576, and Tyr-861 was analyzed by Western blotting of lysates from stimulated cells. An increased phosphorylation of FAK at all three tyrosines could be detected in response to IL-2 stimulation in the IL2Rα/cytoADAM15-expressing cells, whereas IL-2 did not influence the low FAK phosphorylation levels of any investigated tyrosine in the vector control cells (Fig. 7C).
FIGURE 7.
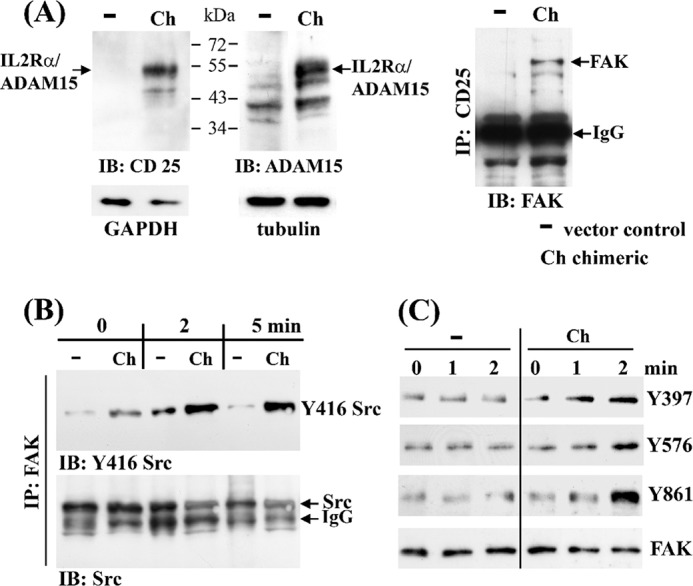
Signaling of ADAM15 expressed as a IL-2 receptor-α/cytoADAM15 chimera. The T/C28a4 chondrocyte cell line was stably transfected with a chimeric construct composed of the extracellular IL-2 receptor-α (CD25) fused to the cytoplasmic tail of ADAM15, and the chimeric protein was detected by immunoblotting using either anti-CD25 or anti-ADAM15 antibodies at ∼60 kDa (A, left panels). A, right panel, shows co-immunoprecipitation of FAK only in cell lysates of cells transfected with the chimeric IL2Rα/cytoADAM15 construct (Ch) as compared with vector transfected cells (−) using anti-CD25 antibodies. B, immunoprecipitation (IP) using anti-FAK antibodies show co-precipitated Src in both vector and chimera-transfected cells, but an enhanced phosphorylation of Tyr-416 Src is noted exclusively in the IL2Rα/cytoADAM15 cells following stimulation with IL-2. C, immunoblots (IB) of lysates derived from IL-2 stimulated cells exhibit a stronger phosphorylation of FAK at Tyr-397, Tyr-576, and Tyr-861 in IL2Rα/cytoADAM15 cells. Equal loading was controlled by reprobing with anti-FAK antibodies.
ADAM15 Expression Counteracts Proapoptotic Effect of FAK/Src Inhibition
To further study the functional role of ADAM15 interference with FAK/Src signaling for its anti-apoptotic properties, two compounds specifically interfering with either FAK or Src signaling were applied. Serum withdrawal in DMEM alone already results in a significantly enhanced apoptosis of vector-transfected cells compared with ADAM15-tranfected cells (Fig. 8, A and B) (21). Under these proapoptotic conditions, treatment with either FAK inhibitor 14, which specifically inhibits phosphorylation of FAK at Tyr-397 in the low μm range (23) or PP2 for up to 24 h caused a considerable increase in caspase activation. However, the caspase activity remained significantly lower in ADAM15 compared with control cells (by a factor of ∼2.0 in case of FAK 14 inhibitor and ∼3.5 of PP2, Fig. 8, A and B). PP3, the inactive analog of PP2, did not display an effect different from medium alone (data not shown). ADAM15 silencing by two specific siRNAs for 40 h in ADAM15-transfected cells and subsequent incubation with either 0.25 μm FAK 14 inhibitor or 5 nm PP2 for up to 30 h resulted in a significantly higher caspase activity in the ADAM15-silenced cells compared with cells that were silenced with a nonfunctional siRNA or with transfection agent alone (Fig. 8, C and D). ADAM15 protein expression was reduced by ∼90% after 48 h as evidenced by immunoblotting (21).
FIGURE 8.

Inhibition of FAK and Src signaling by FAK 14 inhibitior and PP2 in the presence of ADAM15. Vector- (black bars) and ADAM15-transfected cells (open bars) were treated with 0.25 μm and 0.5 μm FAK 14 inhibitor (A) and 5 nm and 10 nm PP2 (B) for the time points shown, and caspase-3/7 activity was determined. A significantly increased caspase activity was detected in the control cells in contrast to ADAM15-transfected cells that exhibited higher apoptosis resistance to both inhibitors. Incubation with DMEM alone also displayed a significantly higher caspase activity in vector control cells. C and D, ADAM15 transfected cells were silenced using siRNA I and II for 40 h and treatment with 0.25 μm FAK 14 inhibitor (C) or 5 nm PP2 (D) resulted in a significantly higher caspase activity as compared with cells silenced with a nonfunctional siRNA (N) or with transfection agent alone (0). Incubation with DMEM alone served as a control. Shown are representative results of least five repeated experiments. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Increased Phosphorylation of FAK and Src in OA Chondrocytes upon Genotoxic Stress
The interaction of ADAM15 with the FAK-Src complex is not confined to T/C28a4 cells. Also, primary OA chondrocytes showed a markedly increased phosphorylation of FAK at Tyr-576, Tyr-861, and Tyr-397 and Src at Tyr-416 upon camptothecin treatment (Fig. 9, lanes N and 0). Moreover, silencing of ADAM15 using specific siRNAs I and II and following camptothecin stimulation resulted in a significantly reduced phosphorylation signal of all three Tyr residues of FAK and Tyr-416 Src as compared with OA chondrocytes silenced with a nonfunctional siRNA or with transfection agent alone (∼3.0-fold, Fig. 9), corroborating the previous finding of an enhancement of FAK/Src phosphorylation in response to genotoxic stress in ADAM15 expressing primary OA chondrocytes. Silencing of ADAM15 in OA chondrocytes reduced protein expression by ∼85% (21).
FIGURE 9.
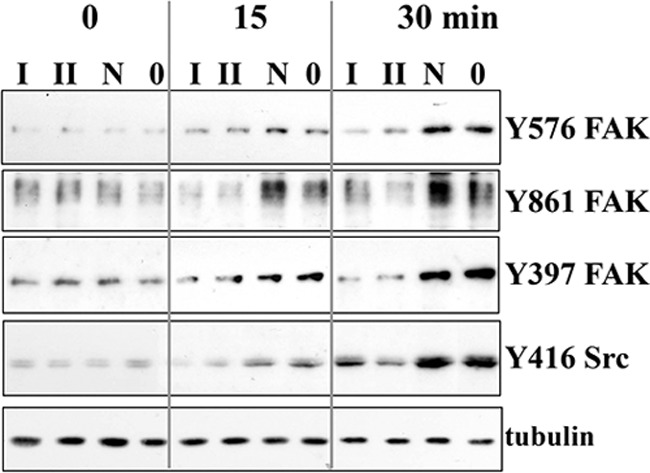
Enhanced phosphorylation of FAK and Src in osteoarthritic chondrocytes after apoptosis induction by camptothecin. Immunoblots of cell lysates of OA chondrocytes that were silenced using ADAM15-specific siRNAs I and II and a nonfunctional siRNA (N) or transfection agent alone (0) and treated with camptothecin, showing marked induction of phosphorylation of FAK at Tyr-576, Tyr-861, and Tyr-397 and Src at Tyr-416 by camptothecin after 15 and 30 min (N and 0), which is strongly reduced after silencing of ADAM15 (I and II). Shown are representative results of an analysis performed on primary chondrocytes derived from five OA cartilage samples.
DISCUSSION
This study was undertaken to investigate the anti-apoptotic function of ADAM15 conferred by its cytoplasmic domain via interaction with the FAK-Src complex. In this work, the ADAM15 isoform originally cloned from an osteoblast cell line (29), also referred to as ADAM15 A (8) or variant 2 (26), was analyzed. It represents the major form (∼90%) expressed in a variety of tissues (29). Other splice variants are of low abundance (0.2–10%) (30) and were linked to aggressive forms of breast cancer (8, 10). In our experimental setting, camptothecin, a DNA damage-promoting topoisomerase I inhibitor, was applied to induce apoptosis in the chondrocytic cell line T/C28a4, transfected either with full-length ADAM15 or the deletion mutant ADAM15Δcyto (21). In this model of genotoxic stress-induced apoptosis, the protective effect of ADAM15 turned out as being strictly dependent on the conserved expression of its cytoplasmic tail. These findings in conjunction with our earlier results on an involvement of ADAM15 in the amplification of X-linked inhibitor of apoptosis-dependent anti-apoptotic pathways (21) and on the modulation of integrin-mediated FAK signaling (14) in cell adhesion stimulated us to investigate the direct molecular interaction between cytoADAM15 with FAK and its potential consequences for FAK-Src complex activation. We hypothesized that such an interaction might provide a mechanism to explain the anti-apoptotic effects of ADAM15 because the activating phosphorylation of FAK at different tyrosine residues has already been shown to trigger distinct survival pathways, including one that involves NF-κB activation and X-linked inhibitor of apoptosis up-regulation (31). Our pulldown and mammalian two-hybrid studies provide unequivocal evidence for a direct binding of cytoADAM15 to a region (730–790) between the kinase and the FAT domain of FAK that is directly adjacent to the two docking sites for SH3 consensus motif-containing proteins (32). Accordingly, the expression of full-length ADAM15 but not of the deletion mutant ADAM15Δcyto also resulted in a stronger phosphorylation at tyrosines Tyr-397, Tyr-576, and Tyr-861 of FAK and concomitantly Src Tyr-416. Enhanced phosphorylation at the respective FAK tyrosine residues has already been incriminated in the blocking of apoptosis pathways induced by diverse stimuli, such as UV irradiation, disruption of cellular anchorage to the ECM, or hyperosmotic stress (33–35). These results provide experimental evidence for a critical role of the interaction between FAK and cytoADAM15 in the enhancement of survival pathways.
The extended studies in cells transfected with IL2Rα/cytoADAM15 fusion chimeras further demonstrate by IL-2 stimulation that signals derived from specific extracellular triggering can be transduced via the cytoplasmic domain into enhanced FAK and Src phosphorylation. Accordingly, the cytoplasmic domain of ADAM15 can function either as a transmission device for a well defined ectodomain signal demonstrated in the IL2Rα/cytoADAM15-transfected cells or as a complex signaling scaffold in wild-type ADAM15 integrating diverse stimuli derived from connected concomitantly activated pathways exemplified by its modulatory impact on a complex genotoxic stress response. Whereas our studies provide clear evidence for the signaling competence of cytoADAM15 in its interaction with the FAK-Src complex, the nature of the extracellular stimuli activating this ADAM15-dependent signaling pathway under (patho)physiologic conditions, however, remains to be elucidated. Ligation of ECM components with the extracellular ADAM15 prodomain (14) or interaction of αv and/or α5 integrins in cis or trans on the cell surface with the ADAM15 disintegrin domain (15) might constitute hypothetical scenarios. Our immunostainings of ADAM15 and FAK in the focal contacts comprising of transmembrane integrins and cytoplasmic proteins linking the ECM to the cytoskeleton provide evidence that the spatial requirements for such interactions are fulfilled. Likewise, colocalization with Src at the cell periphery and Src interaction has been shown for another ADAM family member (ADAM12 (36, 37)). It is tempting to speculate in the light of data presented here and the compiled work of others that up-regulation of ADAM15 in (patho)physiologic conditions might have a dual impact on integrin-associated signaling. As a transmembrane anchored protein, ADAM15 might precipitate local integrin clustering via its extracellular disintegrin domain, thereby eventually elicitating integrin-transmitted signals to the FERM domain of FAK independent of cell matrix contact. Conversely, ADAM15 might involve the interaction of its cytoplasmic tail with the FAK region (730–790) to convert signals perceived by its extracellular prodomain from the ECM into an activation of the FAK-Src complex, thereby bypassing the integrin-dependent outside-in signaling pathway.
Further unanswered questions relate to the involvement of the catalytic domain of ADAM15 that has remained beyond the scope of the present investigation. Although the available direct evidence for catalytic activity of ADAM15 is rather limited (9–11, 38), its contribution to the uncovered anti-apoptotic effects cannot be excluded entirely. Thus, ADAM15 has been incriminated in ectodomain shedding of EGF receptor ligands (39), rendering it possible that the observed impact of its cytoplasmic domain on the amplification of FAK-Src complex phosphorylation represents only a part of a bidirectional signaling circuit that might also involve an inside-out activation of the catalytic ADAM15 domain. However, direct experimental evidence for such an intriguing role of the catalytic domain is missing and remains to be investigated in future studies.
In conclusion, our investigation provides clear experimental evidence for a direct molecular interaction between ADAM15 and FAK leading to an enhanced phosphorylation of the FAK-Src complex and a concomitant amplification of anti-apoptotic survival pathways. The novel function of ADAM15 elucidated in chondrocytic cells under genotoxic stress as a model mimicking the proapoptotic milieu in OA cartilage might also be relevant beyond its homeostatic role in cartilage degeneration to other pathologic conditions such as neoplastic disease. This is in compliance with already published evidence in the latter conditions (2, 40, 41) incriminating ADAM15 expression and FAK-Src complex activation in aggressive malignant growth.
Acknowledgments
The chondrocyte cell line T/C28a4 (28) was kindly provided by Dr. M. B. Goldring (Hospital for Special Surgery, NY, NY). The cartilage samples were kindly provided by Dr. Werner Ewald (Orthopedic University Hospital Friedrichsheim at the University Frankfurt am Main, Germany). The IL-2 receptor-α cDNA was a kind gift of Dr. Kerstin Danker (Charité Berlin, Campus Benjamin Franklin, Germany).
This work was supported by Deutsche Forschungsgemeinschaft Grant BU584/4-1.

This article contains supplemental Table S1 and Figs. S1 and S2.
- ECM
- extracellular matrix
- FAK
- focal adhesion kinase
- FERM
- N-terminal domain of FAK ezrin/radixin/moesin (ERM)
- FAT
- focal adhesion targeting
- Src
- c-src kinase
- OA
- osteoarthritis
- IL2Rα or CD25
- IL-2 receptor-α chain
- ADAM15Δcyto
- deletion mutant of ADAM15 lacking the cytoplasmic domain
- cytoADAM15
- cytoplasmic domain/tail of ADAM15.
REFERENCES
- 1. Edwards D. R., Handsley M. M., Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffy M. J., McKiernan E., O'Donovan N., McGowan P. M. (2009) Role of ADAMs in cancer formation and progression. Clin. Cancer Res. 15, 1140–1144 [DOI] [PubMed] [Google Scholar]
- 3. Blobel C. P. (2005) ADAMs: Key components in EGFR signaling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 4. Böhm B. B., Aigner T., Blobel C. P., Kalden J. R., Burkhardt H. (2001) Highly enhanced expression of the disintegrin metalloproteinase MDC15 (metargidin) in rheumatoid synovial tissue. Arthritis Rheum 44, 2046–2054 [DOI] [PubMed] [Google Scholar]
- 5. Marzia M., Guaiquil V., Horne W. C., Blobel C. P., Baron R., Chiusaroli R. (2011) Lack of ADAM15 in mice is associated with increased osteoblast function and bone mass. Biol. Chem. 392, 877–885 [DOI] [PubMed] [Google Scholar]
- 6. Horiuchi K., Weskamp G., Lum L., Hammes H. P., Cai H., Brodie T. A. (2003) Potential role for ADAM15 in pathological neovascularization in mice. Mol. Cell Biol. 23, 5614–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lucas N., Day M. L. (2009) The role of the disintegrin metalloproteinase ADAM15 in prostate cancer progression. J. Cell Biochem. 106, 967–974 [DOI] [PubMed] [Google Scholar]
- 8. Zhong J. L., Poghosyan Z., Pennington C. J., Scott X., Handsley M. M., Warn A., Gavrilovic J., Honert K., Krüger A., Span P. N., Sweep F. C., Edwards D. R. (2008) Distinct functions of natural ADAM-15 cytoplasmic domain variants in human mammary carcinoma. Mol. Cancer Res. 6, 383–394 [DOI] [PubMed] [Google Scholar]
- 9. Böhm B. B., Aigner T., Gehrsitz A., Blobel C. P., Kalden J. R., Burkhardt H. (1999) Up-regulation of MDC15 (metargidin) messenger RNA in human osteoarthritic cartilage. Arthritis Rheum. 42, 1946–1950 [DOI] [PubMed] [Google Scholar]
- 10. Maretzky T., Le Gall S. M., Worpenberg-Pietruk S., Eder J., Overall C. M., Huang X. Y., Poghosyan Z., Edwards D. R., Blobel C. P. (2009) Src stimulates fibroblast growth factor receptor-2 shedding by an ADAM15 splice variant linked to breast cancer. Cancer Res. 69, 4573–4576 [DOI] [PubMed] [Google Scholar]
- 11. Tousseyn T., Thathiah A., Jorissen E., Raemaekers T., Konietzko U., Reiss K., Maes E., Snellinx A., Serneels L., Nyabi O., Annaert W., Saftig P., Hartmann D., De Strooper B. (2009) ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the γ-secretase. J. Biol. Chem. 284, 11738–11747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Najy A. J., Day K. C., Day M. L. (2008) The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J. Biol. Chem. 283, 18393–18401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Böhm B. B., Aigner T., Roy B., Brodie T. A., Blobel C. P., Burkhardt H. (2005) Homeostatic effects of the metalloproteinase disintegrin ADAM15 in degenerative cartilage remodeling. Arthritis Rheum. 52, 1100–1109 [DOI] [PubMed] [Google Scholar]
- 14. Böhm B. B., Schirner A., Burkhardt H. (2009) J. Cell Mol. Med. 13(8B), 2634–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nath D., Slocombe P. M., Stephens P. E., Warn A., Hutchinson G. R., Yamada K. M., Docherty A. J., Murphy G. (1999) Interaction of metargidin (ADAM-15) with αvβ3 and α5β1 integrins on different haemopoietic cells. J. Cell Sci. 112, 579–587 [DOI] [PubMed] [Google Scholar]
- 16. Parsons J. T. (2003) Focal adhesion kinase: The first ten years. J. Cell Sci. 116, 1409–1416 [DOI] [PubMed] [Google Scholar]
- 17. Sonoda Y., Matsumoto Y., Funakoshi M., Yamamoto D., Hanks S. K., Kasahara T. (2000) Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J. Biol. Chem. 275, 16309–16315 [DOI] [PubMed] [Google Scholar]
- 18. Kurenova E., Xu L. H., Yang X., Baldwin A. S., Jr., Craven R. J., Hanks S. K., Liu Z. G., Cance W. G. (2004) Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell Biol. 24, 4361–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aigner T., Kim H. A. (2002) Apoptosis and cellular vitality: Issues in osteoarthritic cartilage degeneration. Arthritis Rheum 46, 1986–1996 [DOI] [PubMed] [Google Scholar]
- 20. Aigner T., Söder S., Gebhard P. M., McAlinden A., Haag J. (2007) Mechanisms of disease: Role of chondrocytes in the pathogenesis of osteoarthritis–structure, chaos, and senescence. Nat. Clin. Pract. Rheumatol. 3, 391–399 [DOI] [PubMed] [Google Scholar]
- 21. Böhm B., Hess S., Krause K., Schirner A., Ewald W., Aigner T., Burkhardt H. (2010) ADAM15 exerts an antiapoptotic effect on osteoarthritic chondrocytes via up-regulation of the X-linked inhibitor of apoptosis. Arthritis Rheum 62, 1372–1382 [DOI] [PubMed] [Google Scholar]
- 22. Yang S., Banavali N. K., Roux B. (2009) Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. U.S.A. 106, 3776–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Golubovskaya V. M., Nyberg C., Zheng M., Kweh F., Magis A., Ostrov D., Cance W. G. (2008) A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 51, 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng D., Golubovskaya V., Kurenova E., Wood C., Massoll N. A., Ostrov D., Cance W. G., Hochwald S. N. (2010) A novel strategy to inhibit FAK and IGF-1R decreases growth of pancreatic cancer xenografts. Mol. Carcinog. 49, 200–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. (1996) Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271, 695–701 [DOI] [PubMed] [Google Scholar]
- 26. Kleino I., Ortiz R. M., Huovila A. P. (2007) ADAM15 gene structure and differential alternative exon use in human tissues. BMC Mol. Biol. 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mechai N., Wenzel M., Koch M., Lucka L., Horstkorte R., Reutter W., Danker K. (2005) The cytoplasmic tail of the α3 integrin subunit promotes neurite outgrowth in PC12 cells. J. Neurosci. Res. 82, 753–761 [DOI] [PubMed] [Google Scholar]
- 28. Goldring M. B., Birkhead J. R., Suen L. F., Yamin R., Mizuno S., Glowacki J., Arbiser J. L., Apperley J. F. (1994) Interleukin-1 β-modulated gene expression in immortalized human chondrocytes. J. Clin. Invest. 94, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krätzschmar J., Lum L., Blobel C. P. (1996) Metargidin, a membrane-anchored metalloprotease-disintegrin protein with an RGD integrin binding sequence. J. Biol. Chem. 271, 4593–4596 [DOI] [PubMed] [Google Scholar]
- 30. Kleino I., Ortiz R. M., Yritys M., Huovila A. P., Saksela K. (2009) Alternative splicing of ADAM15 regulates its interactions with cellular SH3 proteins. J. Cell Biochem. 108, 877–885 [DOI] [PubMed] [Google Scholar]
- 31. Huang D., Khoe M., Befekadu M., Chung S., Takata Y., Ilic D., Bryer-Ash M. (2007) Focal adhesion kinase mediates cell survival via NF-κB and ERK signaling pathways. Am. J. Physiol. Cell Physiol. 292, C1339–1352 [DOI] [PubMed] [Google Scholar]
- 32. Mitra S. K., Hanson D. A., Schlaepfer D. D. (2005) Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 33. Kasahara T., Koguchi E., Funakoshi M., Aizu-Yokota E., Sonoda Y. (2002) Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid Redox Signal 4, 491–499 [DOI] [PubMed] [Google Scholar]
- 34. Reddig P. J., Juliano R. L. (2005) Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 24, 425–439 [DOI] [PubMed] [Google Scholar]
- 35. Lunn J. A., Jacamo R., Rozengurt E. (2007) Preferential phosphorylation of focal adhesion kinase tyrosine 861 is critical for mediating an anti-apoptotic response to hyperosmotic stress. J. Biol. Chem. 282, 10370–10379 [DOI] [PubMed] [Google Scholar]
- 36. Stautz D., Sanjay A., Hansen M. T., Albrechtsen R., Wewer U. M., Kveiborg M. (2010) ADAM12 localizes with c-Src to actin-rich structures at the cell periphery and regulates Src kinase activity. Exp. Cell Res. 316, 55–67 [DOI] [PubMed] [Google Scholar]
- 37. Kang Q., Cao Y., Zolkiewska A. (2000) Metalloprotease-disintegrin ADAM 12 binds to the SH3 domain of Src and activates Src tyrosine kinase in C2C12 cells. Biochem. J. 352, 883–892 [PMC free article] [PubMed] [Google Scholar]
- 38. Maretzky T., Yang G., Ouerfelli O., Overall C. M., Worpenberg S., Hassiepen U., Eder J., Blobel C. P. (2009) Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem. J. 420, 105–113 [DOI] [PubMed] [Google Scholar]
- 39. Hart S., Fischer O. M., Prenzel N., Zwick-Wallasch E., Schneider M., Hennighausen L., Ullrich A. (2005) GPCR-induced migration of breast carcinoma cells depends on both EGFR signal transactivation and EGFR-independent pathways. Biol. Chem. 386, 845–855 [DOI] [PubMed] [Google Scholar]
- 40. Bolós V., Gasent J. M., López-Tarruella S., Grande E. (2010) The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco. Targets Ther. 3, 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim L. C., Song L., Haura E. B. (2009) Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 6, 587–595 [DOI] [PubMed] [Google Scholar]