

Background: Oligomers of amyloid-β peptides are implicated in the etiology of Alzheimer disease.
Results: Specific “off-pathway” oligomers of Aβ42 show unique replication properties upon interacting with monomers.
Conclusion: The results indicate that oligomers that are formed along pathways outside the fibril formation pathway may undergo replication.
Significance: Mechanistic details of Aβ soluble oligomers will enable better understanding of Alzheimer disease pathology.
Keywords: Aggregation, Alzheimer Disease, Amyloid, Neurodegeneration, Protein Aggregation, Protein Chemistry
Abstract
Aggregates of amyloid-β (Aβ) peptides have been implicated in the etiology of Alzheimer disease. Among the different forms of Aβ aggregates, low molecular weight species ranging between ∼2- and 50-mers, also called “soluble oligomers,” have emerged as the species responsible for early synaptic dysfunction and neuronal loss. Emerging evidence suggests that the neurotoxic oligomers need not be formed along the obligatory nucleation-dependant fibril formation pathway. In our earlier work, we reported the isolation of one such “off-pathway” 12–18-mer species of Aβ42 generated from fatty acids called large fatty acid-derived oligomers (LFAOs) (Kumar, A., Bullard, R. L., Patel, P., Paslay, L. C., Singh, D., Bienkiewicz, E. A., Morgan, S. E., and Rangachari, V. (2011) PLoS One 6, e18759). Here, we report the physiochemical aspects of LFAO-monomer interactions as well as LFAO-LFAO associations in the presence of interfaces. We discovered that LFAOs are a replicating strain of oligomers that recruit Aβ42 monomers and quantitatively convert them into LFAO assemblies at the expense of fibrils, a mechanism similar to prion propagation. We also found that in the presence of hexane-buffer or chloroform-buffer interfaces LFAOs are able to associate with themselves to form larger but non-fibrillar aggregates. These results further support the hypothesis that low molecular weight oligomers can be generated via non-fibril formation pathways. Furthermore, the unique replicating property of off-pathway oligomers may hold profound significance for Alzheimer disease pathology.
Introduction
Formation of assemblies by a protein called amyloid-β (Aβ)2 peptide in the cortical and hippocampal regions of the brain is mainly responsible for the cognitive decline and memory loss that occur in Alzheimer disease (AD). The brains of AD patients contain a large number of insoluble proteinaceous aggregates that are deposited as senile plaques. These aggregates are mainly composed of 40- and 42-amino acid-long peptides called Aβ40 and Aβ42, respectively (2). An overwhelming body of evidence has emerged in recent years that disputes the classic “amyloid cascade theory” and implicates the smaller, soluble aggregates as the primary neurotoxins responsible for neuronal loss as opposed to the fibrils (3–8). Studies have indicated that soluble oligomers, not the plaque load, in both cell culture and transgenic animal models are not only toxic but also better correlate with the level of cognitive disability (8–10). Several such observations suggest that low molecular weight (LMW) “soluble oligomers” ranging between ∼2- and 50-mers are involved in the early synaptic dysfunction in AD (3, 5, 7, 11). Needless to say, this has led to an unprecedented focus on identifying and characterizing smaller LMW aggregates of Aβ both in vitro and in vivo.
The process of Aβ aggregation is a nucleation-dependent phenomenon in which the formation of a critical mass of a self-assembled aggregates called the “nucleus” along with a concomitant conformational change is a prerequisite for the emergence of mature fibrils. Such a mechanism generates a classic sigmoidal growth curve containing a lag phase prior to the growth phase. In this process, it is clear that smaller oligomers, which are the intermediates of fibril formation, are formed transiently along the pathway. However, it is becoming evident from many recent reports that there are alternative pathways of aggregation (12–14) and that neurotoxic oligomers can also be populated via alternate pathways (15–17). Recently, a 12–16-mer oligomeric species of Aβ42 called “globulomers” was reported to be formed independently of the fibril formation pathway (18), and a similar observation was reported for a 9–15-mer species generated in the presence of SDS (19). Moreover, annular protofibrils (APFs) formed from prefibrillar oligomers (PFOs) failed to convert to fibrils even after an extended period of time, suggesting that these pathogenic oligomers are formed outside the nucleation-dependant fibril formation pathway (20). Reports such as these have shown that there are multiple pathways of aggregation that compete with fibril formation, especially during the prenucleation stage, giving rise to several kinds of oligomers. Therefore, it is important to understand the physiological consequence of such a process. One property of “off-pathway” oligomers is that they convert to fibrils, if at all, at a much slower rate than the fibril formation “on-pathway” counterparts and hence possess longer half-lives. This in turn can result in prolonged toxicity to the neuronal cells and therefore has profound significance in AD pathology. Unfortunately, there is a paucity of structural, mechanistic, and pathological understanding of LMW oligomers in general and more so with the oligomers that are not formed along the fibril formation pathway. In our previous work, we reported that non-esterified fatty acid (NEFA) interfaces can induce multiple pathways of Aβ aggregation in carefully controlled conditions of NEFA concentrations and Aβ-to-NEFA molar ratios (1). More importantly, we reported that two distinct off-pathway oligomers of Aβ42 can be generated: 4–5-mers (small fatty acid-derived oligomers) and 12–18-mers (large fatty acid-derived oligomers (LFAOs)). Here, we report the isolation and characterization of LFAOs along with two important physiochemical properties. (a) LFAOs seem to be a replicating species that quantitatively convert Aβ42 monomers into LFAOs at the expense of fibrils, a mechanism similar to prion protein propagation. (b) In the presence of interfaces, LFAOs are able to form larger but non-fibrillar diffuse aggregates, further supporting our hypothesis on alternate aggregation pathways.
EXPERIMENTAL PROCEDURES
Materials
Aβ42 was synthesized by the Peptide Synthesis Facility at the Mayo Clinic (Rochester, MN) using routine Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry. MALDI-TOF mass spectrometry revealed >90% purity of the peptide. Sodium dodecylsulfate (SDS) and thioflavin-T (ThT) were procured from Sigma. Lauric acid (C12:0) fatty acid was purchased as a sodium salt from NuCheck Prep Inc. (Elysian, MN). All other chemicals were obtained from VWR Inc.
Preparation of Aβ42 Monomers
Lyophilized stocks of synthetic Aβ42 were stored at −20 °C desiccated. Briefly, 1.5–2 mg of peptide was dissolved in 0.5 ml of 30 mm NaOH and stored for 15 min at room temperature prior to size exclusion chromatography (SEC) onto a 1 × 30-cm Superdex-75 HR 10/30 column (GE Healthcare) attached to an ÄKTA FPLC system (GE Healthcare) to remove any preformed aggregates as reported previously (1). The column was pre-equilibrated in 20 mm Tris-HCl (pH 8.0) at 25 °C and run at a flow rate of 0.5 ml/min. One-minute fractions were collected. Concentrations of Aβ were determined by UV-visible spectrometry on a Cary 50 spectrophotometer (Varian Inc.) using a molar extinction coefficient of 1450 cm−1 m−1 at 276 nm (ExPASy) corresponding to the single tyrosine residue in Aβ42. Peptide integrity after SEC was again confirmed by MALDI-TOF mass spectrometry, which showed a monoisotopic molecular mass of 4516.31 Da in good agreement with a calculated mass of 4513.13 Da. Monomeric Aβ42 fractions were stored at 4 °C and used within 2–5 days of SEC purification in all experiments to avoid any preformed aggregates in our reactions.
Aβ Aggregation Reactions
Preparation and Isolation of LFAOs
LFAOs were prepared and isolated as described previously (1). Briefly, the freshly purified Aβ42 (50 μm) was incubated with 50 mm NaCl and 5 mm C12:0 fatty acid at 37 °C for 48 h. After 48 h, the sample was subjected to SEC, and the fractions corresponding to the peak near the void volume (V0) were collected. Concentrations of collected fractions were determined by UV absorbance. All isolated LFAO fractions were stored at 4 °C and used within 2 days after SEC isolation in all experiments.
Agitation Experiments with Hexane-Buffer and Chloroform-Buffer Interfaces
To freshly purified 2 μm LFAO or Aβ42 monomer (control) in 20 mm Tris (pH 8.0), 5% (v/v) hexane (ρ = 0.6548 g/ml) and chloroform (ρ = 1.483 g/ml) were added independently and mixed vigorously using a vortex mixer for 1 min of agitation followed by 5 min of rest. After eight cycles of agitation (∼1 h), the samples were then dialyzed against 20 mm Tris-HCl (pH 8.0) at 25 °C using a 2-kDa-molecular mass-cutoff Slide-A-Lyzer G2 dialysis cassette (Thermo Scientific) for 23 h. Afterward, the dialyzed samples were subjected to immunoblotting, dynamic light scattering (DLS), and atomic force microscopy (AFM) analyses.
Suspension Experiment with Chloroform-Buffer Interface
The suspension method reported previously (21) was followed in our experiments. Freshly purified 2 μm LFAO or Aβ42 monomer (control) (0.3 ml) was suspended on top of a 100% chloroform solution (0.3 ml) in a 1.5-ml siliconized Eppendorf tube without mixing. The samples were kept at 25 °C for 24 h without any disturbance. After 24 h, the samples were removed from just above the interface without disturbing the interface. The samples from both reactions were subjected to immunoblotting, DLS, and AFM analyses.
LFAO Replication Experiment
Monomeric Aβ42 (20 μm) was incubated alone or with 2% (0.4 μm) LFAO seed in 20 mm Tris (pH 8.0) at 25 °C for 72 h. Aliquots of samples were removed at 0, 24, 48, and 72 h and subjected to immunoblotting and AFM after spinning at 19,000 × g for 20 min. The 0.4 μm LFAO seeds alone were also used as a control. Similarly, for SEC isolation of replicated LFAO, 50 μm Aβ42 monomer was incubated with 1 μm LFAO under the same conditions, keeping the Aβ42:LFAO ratio the same. Aliquots of samples were removed at 0, 24, 48, and 72 h and subjected to SEC on a Superdex-75 HR 10/30 column after spinning at 19,000 × g for 20 min to remove fibrils. Fraction 17 from each SEC fractionation at 0, 24, 48, and 72 h was subjected to immunoblotting and circular dichroism (CD).
LFAO Replication Experiment with Dansyl-Aβ42 Monomer
The above experiment was repeated with dansyl-Aβ42 (Dan-Aβ42) monomer, which was purified similarly to the wild-type Aβ42 mentioned above. A freshly purified 50 μm Dan-Aβ42 monomer sample was incubated with 1 μm LFAO under the same conditions mentioned above. Aliquots of samples were removed after 72 h and subjected to SEC on a Superdex-75 HR 10/30 column after spinning at 19,000 × g for 20 min to remove fibrils. Fractions 17 and 18 from the SEC fractionation were subjected to immunoblotting and emission fluorescence spectroscopy using a Cary Eclipse spectrometer (Varian Inc.) in scan mode. The dansyl emission was monitored at 450 nm after exciting at 350 nm using 10-nm slits.
Fluorescence Spectroscopy
ThT fluorescence was monitored in a microcuvette with a Cary Eclipse spectrometer (Varian Inc.) after 15-fold dilution of Aβ42 samples into 5 mm Tris-HCl (pH 8.0) containing 10 μm ThT. Continuous measurements of fluorescence were taken for 1 min with the excitation and emission wavelengths fixed at 450 and 482 nm, respectively, and the excitation and emission slits set at 10 nm.
Dynamic Light Scattering
The DLS was performed on a Zetasizer Nano S DLS instrument (Malvern Inc., Worcestershire, UK). Each sample measurement consisted of six runs of 10 s each with a pre-equilibration time of 40 s. After the measurement, the number (%) was exported and plotted against size using Origin 7.0 software.
Polyacrylamide Gel Electrophoreses (PAGE) and Immunoblotting
Samples were dissolved in loading buffer (1× Laemmli buffer) containing 1% SDS, applied without heating to 4–12% NuPAGE gels (Invitrogen) containing bis-Tris, and resolved in MES running buffer with 0.1% SDS. Dye-linked molecular mass markers (Blue Plus2 Prestained Standards, Invitrogen) were run in parallel for calibration. Gels were electroblotted onto 0.45-μm nitrocellulose membranes (BioTraceTM NT, Pall Life Sciences). Blots were boiled in a microwave oven in PBS for 2 min and blocked overnight with 1× PBS containing 5% nonfat dry milk and 0.1% Tween 20 before probing (1–2 h) with 1:1000–1:2500 dilutions of Ab9 monoclonal antibody, which detects amino acid residues of Aβ (1–16). Blots were then incubated with anti-mouse horseradish peroxide (HRP) conjugate and developed with ECL reagent (Thermo Scientific).
Atomic Force Microscopy
Mica was cleaved using a razor blade and taped to a magnetic sample holder. The mica stub was then covered with a 3-aminopropyltriethoxysilane solution (500 μl of 3-aminopropyltriethoxysilane in 50 ml of 1 mm acetic acid) for 20 min. The 3-aminopropyltriethoxysilane solution was then decanted, and the mica was rinsed four times with 150 μl of deionized water. After rinsing, the mica stub was dried with compressed N2 gas and stored in a desiccator for 1 h. Next, 150 μl of 0.25–1 μm Aβ sample was added to the mica and allowed to adsorb for 30 min. The sample was then decanted, and the mica stub was rinsed four times with 150 μl of deionized water. Finally, the mica stub was dried with compressed N2 gas and stored in a desiccator until imaging. The surface topography of each sample was explored by imaging the peptide after it had been adsorbed onto 3-aminopropyltriethoxysilane-treated, freshly cleaved mica. These images were obtained via a Dimension 3000 atomic force microscope (Digital Instruments) in tapping mode using RTESP etched silicon probes (length, 125 mm; nominal force constant, 40 newtons/m; and resonance frequency, 350 kHz) (Veeco Instruments). While under ambient environmental conditions, the scan rate was held constant at 1 Hz. All standard image-processing techniques were performed on Nanoscope version 5.30 r2 image analysis software. Nanoscope software and Gwyddion version 2.7 software were used to calculate feature heights by two methods: 1) section analysis to extract height profiles and 2) particle analysis to determine the statistical distribution of pixel heights for individual aggregates. Representative height profiles can be found in Fig. 3 and supplemental Figs. S4 and S5. Multiple areas were imaged for each sample, and whereas height, phase, and amplitude data were collected simultaneously, amplitude images are used most often in the text to discuss morphology variation between samples.
FIGURE 3.

Morphological changes during LFAO replication. Aliquots of the samples from Fig. 2A (before SEC) were probed at different time points to see the replication of oligomers after incubating 20 μm Aβ42 with 0.4 μm LFAO (2% seed) for 72 h at room temperature. A, B, and C represent the amplitude AFM images of seeded sample after 0, 48, and 72 h, respectively (z scale, 0–0.8 V). D, E, and F represent the corresponding height data from A, B, and C, respectively (z scale, 0–40 nm). The white scale bar represents 2.5 μm, and the inset shows a field of dimensions 1 × 1 μm surrounding the particles indicated with arrows. The white boxes in D–F indicate the particles for which height analyses were conducted. Height profiles were extracted from the flattened height data and can be seen from the z direction in the height images and from the x,y direction below each image. These height profiles demonstrate how approximate feature heights were determined for each sample. The determined feature heights were confirmed via particle analysis (Nanoscope version 5.30 r2 image analysis software) shown at the bottom as a secondary technique to increase confidence in reported values.
Analytical Ultracentrifugation (AUC)
Sample Preparation
FITC-labeled Aβ42 (FITC-Aβ42) was purchased in a lyophilized form (Bachem Inc.) and stored at −20 °C prior to use. A stock containing 5 mm FITC-Aβ42 in DMSO was prepared as described previously (18). The stock was then diluted to 100 μm in 20 mm Tris (pH 8.0) and used for generating FITC-labeled LFAO (FITC-LFAO). Briefly, for the generation and isolation of FITC-LFAO, 40 μm wild-type Aβ42 and 10 μm FITC-Aβ42 were incubated with 5 mm C12:0 fatty acid under the same conditions as described above for LFAO preparation, and LFAOs were isolated using a similar protocol. For the replication experiments, monomeric Aβ42 (20 μm) was incubated with 2% (0.4 μm) FITC-LFAO seed in 20 mm Tris (pH 8.0) at 25 °C for 72 h. After 72 h, the sample was subjected to sedimentation velocity analysis. The 0.4 μm FITC-LFAO seeds alone were used as a control. Similarly, FITC-fibrils were prepared by mixing 45 μm WT Aβ42 with 5 μm FITC-Aβ42 and incubating with 150 mm NaCl in 20 mm Tris (pH 8.0) at 37 °C for 2–3 days. The ThT fluorescence was monitored daily until it reached a plateau. The sample was then centrifuged at 19,000 × g for 20 min. The pellet was resuspended in 20 mm Tris (pH 8.0) and used for AUC analysis.
Sedimentation Velocity
Samples were mixed by brief vortexing and then spun in a tabletop centrifuge for approximately 5 s to ensure that no sample was lost on the walls of the tube. Samples were then loaded into 1.2-cm-path length sedimentation velocity cells (Sedvel60) and placed in a Beckman XLA analytical ultracentrifuge equipped with a fluorescence detection system (Aviv FDS). The temperature on the centrifuge was equilibrated until it remained constant at 20 °C for at least 5 min. The centrifuge was then accelerated to 5000 rpm at which point the focus depth and gain for the fluorescence detection system were adjusted to maximize the signal collected. The centrifuge was then accelerated to 60,000 rpm, and data collection began immediately after final velocity was reached. Each scan was averaged over five consecutive scans to increase the signal-to-noise ratio. The run was stopped when the fluorescence intensity versus radial distance profiles remained constant between scans, indicating that the boundary had pelleted. The samples were then rerun for ∼30 min at 60,000 rpm using absorbance optics to collect pseudo-absorbance data. All data were transferred to a separate computer for analysis; the pseudo-absorbance data were used to calculate the meniscus position for each sample using the meniscus wizard in the software program DCDT2+ (version 2.3.2) (22). The software program Sedfit (Sedfit89) (23) was used to generate c(s) distributions for the fluorescence detection system data with 0.1-S resolution. The c(s) distribution for each sample was integrated and divided by the area, and all data are presented as normalized c(s) distributions. The software program Sedfit was also used to generate c(M) distributions after generating the c(s) distribution by assuming a constant diffusion coefficient for all samples and an f/f0 value of 1.2. The c(M) distributions were normalized using the same method as described for the c(s) distributions.
Circular Dichroism
CD spectra were obtained in the far-UV region with a Jasco J-815 spectropolarimeter. Samples were placed in a 0.1-cm-path length quartz cuvette (Hellma) and monitored in continuous scan mode (260–190 nm). The acquisition parameters were 50 nm/min with 8-s response time, 1-nm bandwidth, and 0.1-nm data pitch, and data sets were the average of two scans. Spectra of appropriate blanks were subtracted from each data set as indicated. The corrected, averaged spectra were smoothed using a “mean movement” algorithm with a convolution width of 25 using the Jasco spectrum analysis program.
RESULTS
LFAOs Are Not Discrete but Disperse Mixture of Oligomeric Species
In our previous work, we showed that Aβ42 upon incubation with varying concentrations of NEFAs generates two distinct types of oligomers, LFAOs (12–18-mers) and small fatty acid-derived oligomers (4–5-mers), at near and above critical micelle concentrations of NEFA, respectively (1). In the same report, we also indicated that LFAOs can be isolated using Superdex-75 SEC. In the current report, first we quantified the amount of NEFA (lauric acid), if any, associated with the isolated LFAOs and estimated the molecular size distribution of LFAOs by SEC and AUC. An Aβ42 sample incubated with buffered 5 mm lauric acid at 37 °C for 48 h was subjected to fractionation on a Superdex-75 SEC column (Fig. 1A, smooth line). The sample eluted in two major peaks, one between fractions 17 and 20 and the other between fractions 22 and 25. In addition, a small peak near the void volume of the column (fraction 16) was also observed. Immunoblots of the inclusive volume fractions 22–25 suggested they were Aβ42 monomers consistent with our previous observation (1) (data not shown). LFAOs eluted in the partially included volume (fractions 17 and 18) as observed previously (Fig. 1A). The presence of NEFA was quantified using a free fatty acid assay kit (BioVision Inc., Milpitas, CA) as shown previously (24). Such examination indicated only a negligible amount (<0.1%) of NEFA present in these fractions containing isolated LFAOs (supplemental Fig. S1).
FIGURE 1.
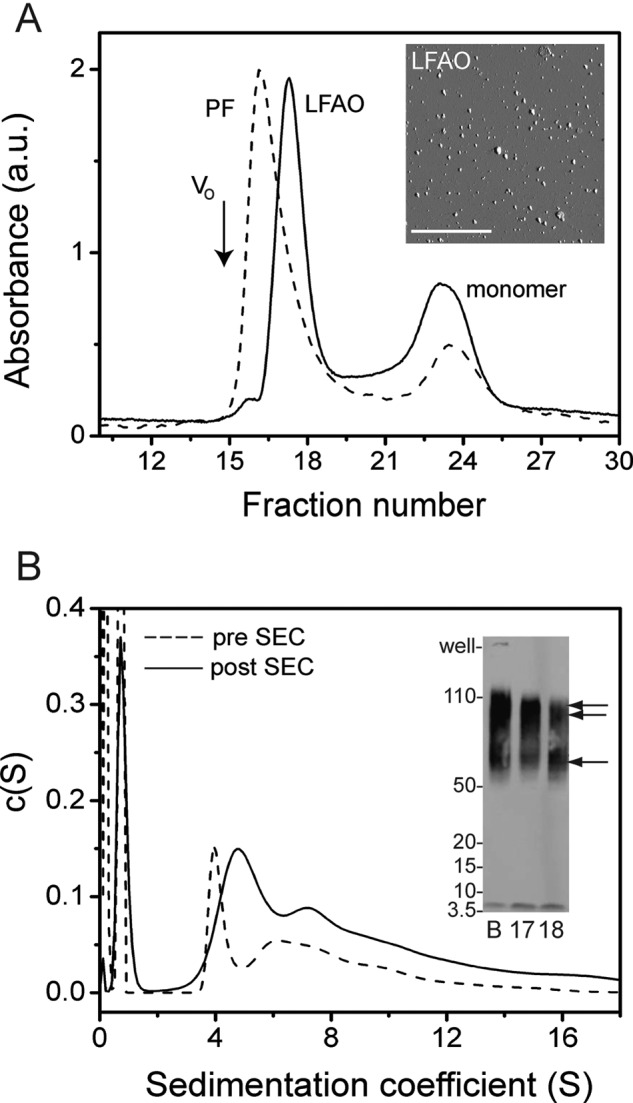
Isolation and characterization of LFAOs. A, SEC fractionation of LFAOs using a Superdex-75 column in 20 mm Tris (pH 8.0) at room temperature with a flow rate of 0.5 ml/min and a fraction size of 1 ml. As a comparison, a sample of PFs of Aβ42 was also fractionated. V0 indicates the void volume based on globular protein standards. Inset, AFM image of fractionated LFAOs reproduced from Kumar et al. (1). Scale bar, 2.5 μm. B, normalized c(s) distribution profile generated from a sedimentation velocity experiment performed at 50,000 rpm. Inset, immunoblot of LFAOs before (B) and after fractionation (fractions 17 and 18). The single arrow indicates the 50–70-kDa band, whereas the double arrow indicates the 80–110-kDa band. The results shown are a single data set of three independent acquisitions. a.u., absorbance units.
To ascertain the molecular mass of LFAOs based on retention volumes, the SEC profile was compared with those of globular protein standards (gel filtration standard; Bio-Rad catalogue number 151-1901) (data not shown). Based on this analysis, the molecular mass of LFAOs was estimated to be between 60 and 200 kDa. In addition, protofibrils (PFs) of Aβ42, which are high molecular mass species (>200,000 Da; >500-mers), were also generated following a previously established protocol (25, 26) and subjected to fractionation under similar conditions. The PF sample exhibited two elution peaks, the PFs at the void volume (V0; Fig. 1A, black arrow) at fraction 15 and monomers at fractions 22–25 (Fig. 1A, dashed line). The PF fractionation also supported our conclusion with globular protein standards that the molecular mass of LFAOs is between 60 and 200 kDa. Subjecting the LFAO fractions (fractions 17 and 18) to another cycle of fractionation through SEC yielded 7.3% monomers, which is comparable with 10.6% obtained in the first SEC fractionation. This suggests that LFAOs undergo partial dissociation upon dilution (supplemental Fig. S2). The AFM image of the isolated LFAOs showed largely a bimodal distribution of non-fibrillar punctate dots ranging between 7–10 and 16–19 nm in height (Fig. 1A, inset).
The molecular size distribution of LFAOs was also estimated by sedimentation velocity experiments in an analytical ultracentrifuge. As described under “Experimental Procedures,” FITC-labeled Aβ42 was introduced into the oligomeric assembly as a reporter to allow monitoring via an analytical ultracentrifuge-fluorescence detection system. More importantly, labeling also reduces the amount of sample needed while increasing the sensitivity at low concentrations. The c(s) sedimentation coefficient distribution profiles obtained from these experiments are shown in Fig. 1B. The sizes of the LFAOs were analyzed both before and after fractionation to observe size differences if any. The Aβ42 sample in the presence of 5 mm lauric acid prior to SEC showed a distribution of peaks with a sharp peak centered around 4 S and a more disperse peak between 6 and 12 S along with a peak at ∼1 S (Fig. 1B, dotted lines). The 1 S peak corresponds to monomeric Aβ as determined by the control sample (supplemental Fig. S3A). An immunoblot of the Aβ42 sample incubated in 5 mm lauric acid prior to SEC indicated a disperse band between 56 and 110 kDa (Fig. 1B, inset, lane B). The AUC analyses of the SEC-fractionated LFAOs (fraction 17) showed an overall distribution similar to that prior to SEC fractionation containing predominantly two species, a main peak centered at 5 S and a second disperse peak between 7 and 12 S along with some monomers (1 S) (Fig. 1B, smooth line). As a comparison, we analyzed Aβ42 fibrils using AUC, which showed a heterogeneous mixture of species between 40 and 200 S (supplemental Fig. S3B). It is noteworthy that a considerable decrease in the relative amount of monomers was also observed after fractionation based on the signal: 52.3 and 14.8% monomers before and after fractionation, respectively. The LFAOs showed a similar distribution of oligomers even after a 10 to 50-fold dilution in AUC (data not shown). It is also noteworthy that recently a similar sedimentation distribution profile was obtained for amyloid-derived diffusible ligands of Aβ42 (27). In contrast to the LFAOs, the Aβ42 fibril control showed widely disperse sedimentation coefficient values between 50 and 200 S (supplemental Fig. S3B). The immunoblots of both the SEC-fractionated and unfractionated LFAO samples showed largely a disperse band between 56 and 110 kDa with two distinct band distributions centered around 56–70 and 80–110 kDa (Fig. 1B, inset). The band distributions in the immunoblots complement the bimodal distributions observed both in AFM and AUC data. Together, the data suggest that the LFAOs are not discrete oligomeric species but are a disperse distribution of oligomers ranging between 56 and 110 kDa (12–24-mers).
LFAOs Are Replicating Strains
Typically, “seeding” is a process in which a small amount of preformed aggregate when added to monomeric Aβ enhances the aggregation rate towards fibril formation. Because the aggregate morphology is dependent on the physiochemical property of the seed itself, we wanted to explore whether LFAOs can “replicate,” or in other words seed their own formation, by the addition of monomeric Aβ, a mechanism analogous to prion protein propagation. This property has been recently demonstrated by Kayed et al. (28), who reported that specific soluble oligomers called PFOs were able to seed their own replication upon interacting with monomeric Aβ. The property of replication could be a manifestation of the unique structure of the oligomeric assembly and more importantly may complement our previously reported hypothesis that such oligomers are formed along a pathway different from fibril formation (1, 19). To explore this, a 20 μm sample of freshly purified, “seed-free” Aβ42 was incubated with 0.4 μm isolated LFAOs as seeds (2% molar ratio) at room temperature. Aliquots from this sample were subjected to immunoblotting after 24, 48, and 72 h of incubation to monitor the reaction progress (Fig. 2A). The amount of LFAOs loaded in each lane of the SDS-polyacrylamide gel was kept constant at 28 ng based on the initial seed concentration to make quantitative estimates of the blots. The immunoblot in Fig. 2A showed an LFAO band within 24 h of incubation with monomers that was clearly more intense compared with the 0-h incubation sample (lane 3). Importantly, these bands are centered at 100 kDa, which is the higher band among the two observed for the isolated LFAOs (lane C), whereas the lower molecular mass (∼56–70-kDa) band is absent (Fig. 2A, 24 h). These bands became increasingly more intense after 48 and 72 h, whereas the intensity of the monomer bands diminished, suggesting the conversion of monomers to LFAOs. Also, after 72 h, a high molecular mass species that failed to enter the gel was observed (lane T; 72 h) that was not present in the supernatant after the sample was centrifuged at 19,000 × g for 20 min. This suggests that with time fibrils started to emerge, possibly due to monomers progressing toward on-pathway fibrils independently. This observation is not surprising considering that the on-pathway fibril formation reaction is expected to proceed in parallel with monomer-LFAO interactions. Concomitantly, the replication process was monitored by ThT fluorescence (Fig. 2C). Aβ42 seeded with 2% LFAO (Fig. 2C, ○) showed only a negligible amount of seeding with a lag time of ∼72 versus ∼84 h for the control (Fig. 2C, ■). It is important to point out that upon seeding with 20% seeds a significant reduction in lag times was observed in our previous report (1). The immunoblots recorded during the lag phase (24, 48, and 72 h) showed the presence of a substantial amount of LFAOs in the seeded samples that was clearly absent in the control, suggesting that LFAOs had undergone replication during this time period. More importantly, the immunoblots also seem to indicate that the LFAO seeds are able to induce the formation of the 80–110-kDa oligomers at the expense of 56–70-kDa species observed within the unseeded LFAOs.
FIGURE 2.
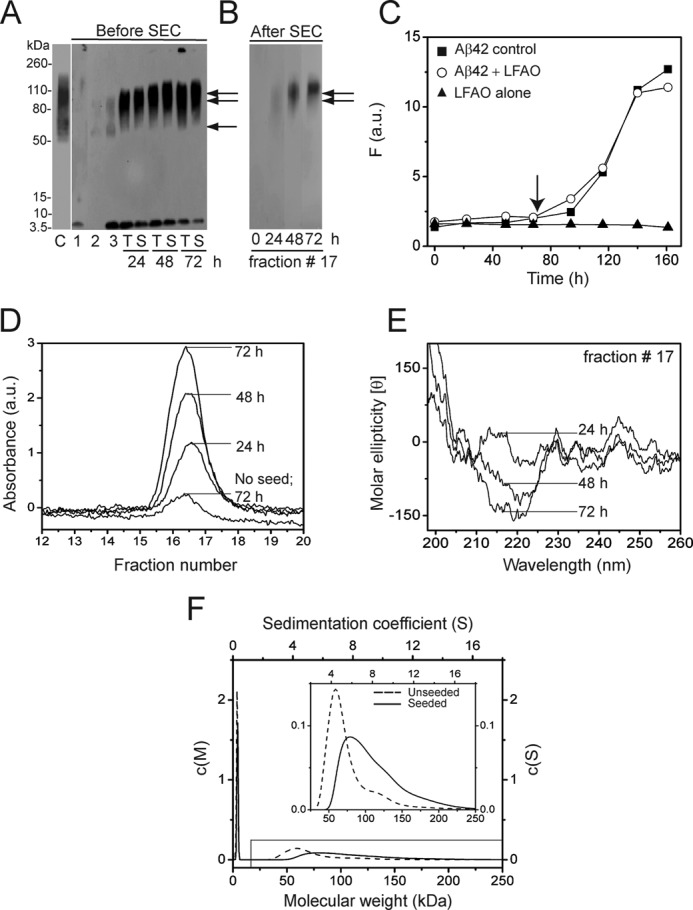
LFAOs replicate upon interacting with monomers. A, immunoblot of 20 μm Aβ42 incubated with 0.4 μm LFAO (2% seed) for 72 h at room temperature before and after SEC. Lane C represents LFAO control (338 ng). Lanes 1 and 2 are Aβ42 control after 72 h in the absence of LFAO seed and LFAO alone (28 ng), respectively. Lane 3 is an aliquot of the 20 μm Aβ42 seeded with 0.4 μm LFAO immediately after incubation (0 h). Lanes T and S represent the total sample and supernatant of the sample after centrifugation at 19,000 × g for 20 min, respectively, at the indicated times of incubation. The volume of sample loaded was kept constant to ensure that 28 ng of the parent LFAO was maintained. B, immunoblot of fraction 17, fractionated by SEC after 24, 48, and 72 h of incubation of the samples in A (lanes S). C, ThT fluorescence data of the seeded reaction. The arrow indicates the 72-h point to which the seeding reaction was monitored. D, fraction 17 of the seeded samples that were fractioned on a Superdex-75 column after 24, 48, and 72 h along with a control sample (no seed) after 72 h. E, corresponding far-UV CD spectra of fraction 17. F, normalized relative molecular mass distributions (c(M) profiles) obtained from sedimentation velocity data for control LFAOs (dotted line) and seeded oligomers (smooth line). Inset, expanded region of the c(M) plot indicated in the figure. The results shown are representative of at least two reproducible results. F, fluorescence; a.u., absorbance units.
We then reasoned that because replication leads to an increase in the amount of LFAOs formed a quantitative analysis would provide further proof of this process. To do so, a higher concentration of Aβ42 sample, 50 μm, was seeded with 1 μm LFAOs (2% molar seed ratio) to facilitate fractionation by SEC while maintaining a constant ratio of Aβ42:LFAO seed. Immunoblots of aliquots at 24, 48, and 72 h time showed a banding pattern similar to that seen in Fig. 2A (data not shown). The same aliquots were also subjected to fractionation by SEC. Prior to loading onto the Superdex-75 column, the samples were centrifuged to remove any fibrils present in the sample, and the supernatant was subjected to fractionation. As observed before in Fig. 1A, the samples eluted near the void volume (Fig. 2D, fractions 16–18), and immunoblotting confirmed that these species were predominantly LFAOs (Fig. 2B, lane 24 h). The amount of LFAOs eluted in these fractions increased progressively with time (as inferred by the increase in absorbance), suggesting a quantitative increase in the amount of LFAOs generated. Overall, the sample showed a 4-fold increase in the amount of LFAOs as compared with a non-seeded control after 72 h (Fig. 2D). Importantly, immunoblots for fraction 17 from SEC also indicated a progressive increase in the amount of 80–100-kDa LFAOs (Fig. 2B). Far-UV CD of the fractionated sample (fraction 17) also showed a progressive increase in β-sheet conformation, further supporting the increase in LFAO content upon seeding (Fig. 2E). Finally, to verify the formation of 80–110-kDa species at the expense of 56–70-kDa species, the LFAO sample was monitored both before and after replication by sedimentation velocity analysis after 72 h of incubation (Fig. 2F). Prior to seeding, a major species was observed that was centered at ∼60 kDa along with some monomers as observed previously (Fig. 2F, dotted lines). After replication, the data clearly showed a shift toward higher molecular masses (80–110 kDa), complementing the shift in banding pattern toward higher molecular mass observed in immunoblots (Fig. 2F, smooth line). A small decrease in the relative amount of monomer (∼6%) present after seeding was also observed in AUC data (Fig. 2F), further supporting the immunoblot data that indicated that quantitative amounts of monomers are converted to LFAOs upon initiating the seeding reaction.
AFM images of the supernatants of the seeded samples shown in Fig. 2A are presented in Fig. 3. Immediately after incubation (Fig. 3, A and D), samples exhibited only a few punctate spherical particles of ∼8 nm in height that are attributed to LFAOs. After 48 h of incubation (Fig. 3, B and E), samples exhibited a significant increase in the number of spherical LFAO particles with a slight increase in the height (∼12 nm) that seem to align along a longitudinal axis (inset). Samples imaged after 72 h of incubation (Fig. 3, C and F) showed a further increase in the number of particles with similar height (∼14 nm) with simultaneous presentation of individual punctate spherical particles aligned linearly (inset) and a small number of smooth fibrillar structures. Importantly, many of these linearly aligned globular particles presented an overall appearance of “nascent” PFs. It is noteworthy that a similar “ordering” of spherical particles was observed previously when Aβ40 was treated in the presence of an aqueous-organic interface (21). These images support the AUC and immunoblotting data and indicate that the seeded LFAOs replicate.
Finally, to ensure that monomers are incorporated into the replicated oligomers, Dan-Aβ42 was used as a fluorescent probe. We expected that seeding of Dan-Aβ42 with LFAOs would lead to oligomers containing the labeled peptide, which can be detected by intrinsic fluorescence as shown in the schematic (Fig. 4A). A seeding experiment was performed as described above using 50 μm Dan-Aβ42 with 1 μm LFAOs at room temperature and subjecting the seeded sample to SEC fractionation and immunoblotting after 72 h (Fig. 4B). The immunoblot clearly showed an increase in LFAO formation after 72 h compared with 0 h (Fig. 4C, lanes 4, 5, and 6). The immunoblot of the fractionated sample (fractions 17 and 18) also showed the presence of replicated LFAOs (Fig. 4C, lanes 8 and 9). Clearly, the unreacted monomers fractionated in the inclusion volume at fraction 24 (Fig. 4C, lane 10). The fluorescence spectra of fractions 17 and 18 obtained by exciting the sample at 350 nm while monitoring dansyl emission at 450 nm are shown in Fig. 4D. Both fractions clearly exhibited dansyl emission at 450 nm, indicating that Dan-Aβ42 monomers have been incorporated into the LFAO assembly during the seeding reaction, providing unambiguous evidence for such a mechanism. Together, the collective data suggest that LFAOs are a replicating strain that can induce the formation of similar oligomeric species upon interacting with Aβ42 monomers.
FIGURE 4.
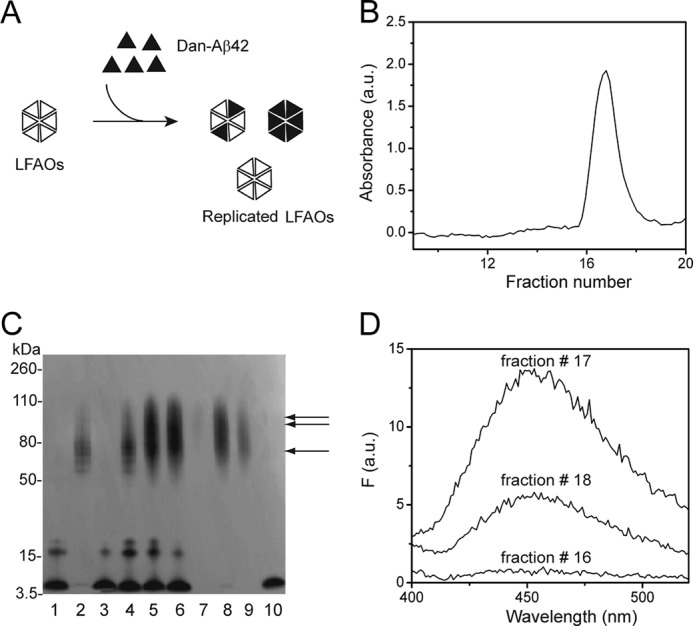
Incorporation of Dan-Aβ42 monomer into replicated oligomers after seeding. A, schematic for incorporation of Dan-Aβ42 into replicated LFAOs after seeding for 72 h. B, SEC fractionation of 50 μm Dan-Aβ42 seeded with 1 μm LFAO (2% seed) for 72 h at room temperature. C, immunoblot showing the comparison of seeded sample before and after SEC. Lane 1, 50 μm Dan-Aβ42 monomer (1.8 μg); lane 2, LFAO seed (36 ng); lane 3, Dan-Aβ42 control in the absence of seeds after 72 h; lane 4, aliquot of the seeding reaction immediately after incubation at 0 h; lanes 5 and 6, the total reaction and supernatant of the reaction, respectively; lanes 7–9, SEC fractions 16, 17, and 18 show replicated oligomers (arrows); lane 10, monomer fractionated in the included volume (fraction 24). The LFAO seed amount of 36 ng was kept constant in the immunoblot. D, fractionated sample. Fractions 16, 17, and 18 show the Dan-Aβ42 emission at 450 nm upon exciting at 350 nm with both excitation and emission slits set 10 nm each. The results are one of three reproducible data sets. F, fluorescence; a.u., absorbance units.
LFAOs Form Non-fibrillar, Diffuse Aggregates in Aqueous-Organic Phase Interfaces
Buffer-Hexane and Buffer-Chloroform Agitation Experiments
Aqueous-organic phase interfaces have been known to influence Aβ aggregation tremendously (20). Specifically, prefibrillar oligomers formed distinct ring-/porelike structures called APFs promoted by water-hexane interfaces and failed to form on-pathway fibrils upon long term incubation (20). We wanted to explore whether LFAOs were able to form similar non-fibrillar aggregates upon exposure to aqueous-organic phase interfaces. For these experiments, two interfacial systems were used: hexane-water (as reported previously (20)) and chloroform-water. First, the freshly isolated LFAOs (2 μm) were mixed and agitated with 5% (v/v) hexane in water following a protocol described by Kayed and et al. (20) (see “Experimental Procedures”). The agitated sample was dialyzed for 23 h at room temperature after which the samples were analyzed by DLS. LFAOs in buffer alone showed a monodisperse peak with a diameter of ∼10 nm (Fig. 5A, black peak) in agreement with the 7–10-nm range of heights observed in AFM images (Fig. 1A, inset). In contrast, LFAOs treated in hexane-water showed the formation of a mixture of large species with a hydrodynamic diameter of ∼700 nm (Fig. 5A, gray peak). The corresponding immunoblot of the sample did not show a high molecular mass species; however, a sharp decrease in the intensity of the LFAO band was observed (Fig. 5B, lane 6). It is interesting to note that the control Aβ42 monomers (in the absence of LFAOs) showed a high molecular mass band that failed to enter the gel upon similar hexane treatment (Fig. 5B, lane 3).
FIGURE 5.
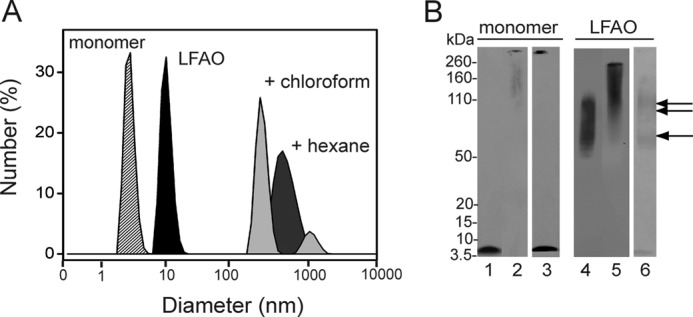
LFAO agitation in interfacial environments. A, the increase in size of LFAO after agitation with 5% (v/v) hexane or chloroform in buffer followed by dialysis for 23 h was monitored by DLS. B, immunoblot of the agitation experiments along with monomer controls. Lane 1 shows control monomer Aβ42. Lanes 2 and 3 show the 23-h dialyzed Aβ42 monomer sample treated with chloroform and hexane, respectively. Lane 4 shows control LFAO. Lanes 5 and 6 show 23-h dialyzed LFAO treated with chloroform and hexane, respectively. The single and double arrows indicate 50–70- and 80–110-kDa bands, respectively.
To see whether the formation of large amorphous aggregates in hexane can be replicated in other organic solvents, the effect of chloroform was investigated. Chloroform was used rather than hexane because it is denser than water, and buffer can be readily suspended on top of the organic layer. The LFAO sample (2 μm) was incubated with 5% chloroform, agitated, and dialyzed in a similar fashion as the hexane sample. A polydisperse distribution of species containing two major peaks (∼700 and 1000 nm), similar to that observed in the hexane-treated sample, was obtained by DLS (Fig. 5A, light gray peaks). The sample treated with chloroform showed a disperse band between 200 and 260 kDa in the immunoblot (Fig. 5B, lane 5). The immunoblot of the control monomer treated similarly also showed a high molecular mass band that failed to enter the gel (Fig. 5B, lane 2).
AFM images of the LFAO and control Aβ42 monomers subjected to aqueous-organic solvent mixtures are shown in Fig. 6. Large spheroidal aggregates with heights of ∼33 nm were observed in LFAO samples treated with hexane (Fig. 6A and supplemental Fig. S4A). These aggregates were at least 3–4 times larger in apparent diameter than the individual LFAO molecules and appear to result from LFAO association (Fig. 6A, inset). Fibrillar structures were observed in Aβ42 control monomer samples treated with hexane (Fig. 6B). It is noteworthy that LFAOs failed to form the ringlike structures seen with APFs (20), suggesting that different molecular organization processes occur for different soluble oligomers. LFAO samples treated with chloroform showed large clusters of aggregates ∼21 nm in height (Fig. 6C and supplemental Fig. S4C). However, no fibrillar material was observed in Aβ42 monomers treated with chloroform (Fig. 6D) despite the fact that a high molecular mass band that failed to enter the gel was observed for the same sample in the immunoblot (Fig. 5B, lane 2). It is likely that dilution of the sample during the preparation procedure for AFM analysis dissolved the unstable aggregates formed in chloroform interfaces. This observation is similar to the one reported by Nichols et al. (29) in which Aβ40 generated unstable aggregates in a chloroform-buffer interface that were completely disaggregated upon dilution. The collective data show that when LFAOs are exposed to aqueous-organic interfaces large, non-fibrillar, disperse aggregates are formed. This suggests that LFAOs assemble into aggregates that may not lie along the fibril formation pathway.
FIGURE 6.

Morphology of aggregates formed after treatment with chloroform and hexane. A and B, AFM images of LFAO and monomer Aβ42 control, respectively, agitated with hexane. A, LFAOs show the presence of small globular aggregates and large clumps of aggregates. The inset shows a 1-μm expansion of the aggregate indicated by the white arrow. B, control monomers treated with hexane show the presence of fibrils. The inset shows a 1-μm expansion of the aggregate indicated by the white arrow. C and D, AFM images of LFAO and monomer Aβ42 control, respectively, agitated with chloroform. C, LFAOs show the presence of several large clumps of aggregates. The inset shows a 1-μm expansion of the aggregates indicated by the white arrows. D, control monomers treated with chloroform failed to show the presence of any aggregates. The white scale bar represents 1 μm, and the square represents a 5 × 5-μm field. The amplitude images are presented here and the z scale is 0–0.8 V.
Chloroform-Buffer Suspension Experiment
The agitation experiments mentioned above cause rapid exchange of phases, leading to increased surface area of interfaces, which in turn affects association of LFAOs. Because agitation is in some respects a harsh method of aggregation, we wanted to see whether similar changes occurred in a milder form of interfacial treatment by simply suspending the aqueous layer containing Aβ on top of a more dense chloroform layer. One such aqueous-organic phase suspension experiment has been reported in which the buffer containing Aβ40 was gently suspended on top of a chloroform layer and aggregation was monitored (21). These conditions led to a rapid aggregation of Aβ40 peptide to form both globular and short, protofibril-like structures. A similar procedure was followed for LFAOs, and within 2 h of incubation, a monodisperse peak centered at ∼80 nm in diameter emerged in DLS that increased to ∼150-nm peak in the next 24 h (Fig. 7A). A similar trend was observed in the immunoblot of the sample in which a band at ∼150 kDa was seen within 2 h of incubation, and after 24 h, a much larger molecular mass band was observed that failed to enter the gel (Fig. 7B, lanes 4 and 5).
FIGURE 7.
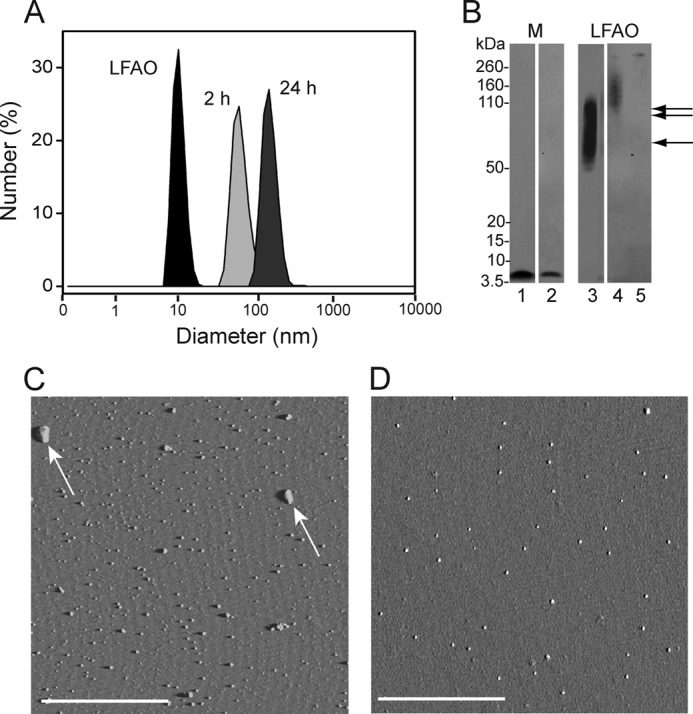
Chloroform suspension experiment. A, LFAO was suspended over 100% chloroform solution, and the sample was removed from above the interface after 2 and 24 h to monitor the increase in size by DLS. DLS data indicate that the size of LFAO (black peak) was shifted from 11.03 to ∼80 nm in 2 h (light gray peak) and to ∼160 nm in 24 h (dark gray peak), indicating the formation of large aggregates. B, immunoblot showing Aβ42 monomer (M) and LFAO sample removed from above the interface after 24 h. Lanes 1 and 2 show control Aβ42 monomer and monomer sample removed from above the interface (in the aqueous buffer side), respectively, after 24 h. Lanes 3 shows control LFAO. Lanes 4 and 5 show the LFAO sample removed from above the interface after 2 and 24 h, respectively. The single and double arrows indicate 50–70- and 80–110-kDa bands, respectively. C, AFM image of chloroform interface sample after 24 h showing the presence of large clumps of aggregates (white arrows). D, AFM image of control monomer treated with chloroform interface shows no appreciable aggregate structure. The white scale bar represents 2.5 μm. The amplitude images are presented here and the z scale is 0–0.8 V.
Numerous punctate dots of globular species with widely varying heights (7–30 nm) were seen in the AFM image of the same sample after 24 h of incubation (Fig. 7C and supplemental Fig. S5A). The species resembled the LFAO aggregates observed in the agitation experiments, but they were much larger in size. In sharp contrast, control Aβ42 monomers neither showed any significant increase in size as witnessed by immunoblots (Fig. 7B, lane 2) nor showed morphology similar to that of the LFAOs (Fig. 7D). In fact, the height of the particle seen in the AFM image was smaller (∼6 nm) than that of pure LFAOs (supplemental Fig. S5B). Interestingly, the globular particles observed with the monomer control were not seen on a Western blot, and DLS did not indicate the presence of such a species. Again, one possible explanation for this observation is that the unstable aggregates formed in chloroform-buffer interfaces disaggregate upon dilution, generating monomeric Aβ42 as observed previously (29). Nevertheless, the data clearly show the absence of fibrillar material with LFAOs, complementing our previous observations as well as our hypothesis that LFAOs are formed along an alternative pathway of aggregation similar to APFs and fibrillar oligomers observed by Kayed et al. (28).
DISCUSSION
In this report, we have shown that LFAOs are not discrete but diffuse oligomeric species with a broad molecular mass range of 56–110 kDa. Within this molecular mass range, two populations of LFAO species exist at 56–70 and 80–110 kDa, which approximately translate to 12–15-mers and 18–24-mers. It is fundamentally important to understand whether there are any residual NEFAs associated with LFAOs as they may affect the physiochemical properties of LFAOs. Our analysis indicated that NEFAs are essentially absent in the isolated LFAO samples (supplemental Fig. S1). It is also important to note that any residual amount of NEFA, if present, may not influence Aβ aggregation properties as their concentration would be well below its critical micelle concentration as reported previously (1). AUC-sedimentation velocity data revealed that LFAOs are mixtures of oligomers with sedimentation coefficients of 5 and 7–12 S and some monomers. This observation is similar to that reported for amyloid-derived diffusible ligands, which were also found to be a heterogeneous mixture containing species larger than 90 kDa that displayed a disperse c(s) distribution between 10 and 25 S in sedimentation velocity experiments (27). A sequential back-to-back SEC fractionation of LFAO samples showed no significant reduction in the concentration of monomers present in the sample, suggesting that LFAOs undergo dissociation upon dilution (supplemental Fig. S2). This further supports other findings that LFAOs are disperse rather than discrete aggregates.
A significant feature of LFAOs is their replicating property, which has potentially far reaching implications for AD pathology. First, the observation of LFAOs acting as “seeds” to produce more of the non-fibrillar aggregates implies the conversion of non-pathogenic species to potentially more toxic strains, similar to a mechanism observed with prion diseases (Fig. 8A) (30–34). The direct implication of this property in AD is the possibility that the LFAOs can act as a “molecular sink” to recruit more Aβ monomers away from fibrils. Although qualitative analysis has revealed this important property of LFAOs, a rigorous quantitative increase in the amount of LFAOs converted could also be of profound practical application. Briefly, by following a cyclic amplification method similar to the one introduced by Soto and co-workers (35, 36) for amplifying prion aggregates with a low concentration seed from a physiological source, one will be able to “scale up” the amount of the seed using exogenous monomers to the levels amenable for in vitro biophysical analyses while preserving the structural integrity of the source. We are currently exploring this aspect and will report it later.
FIGURE 8.
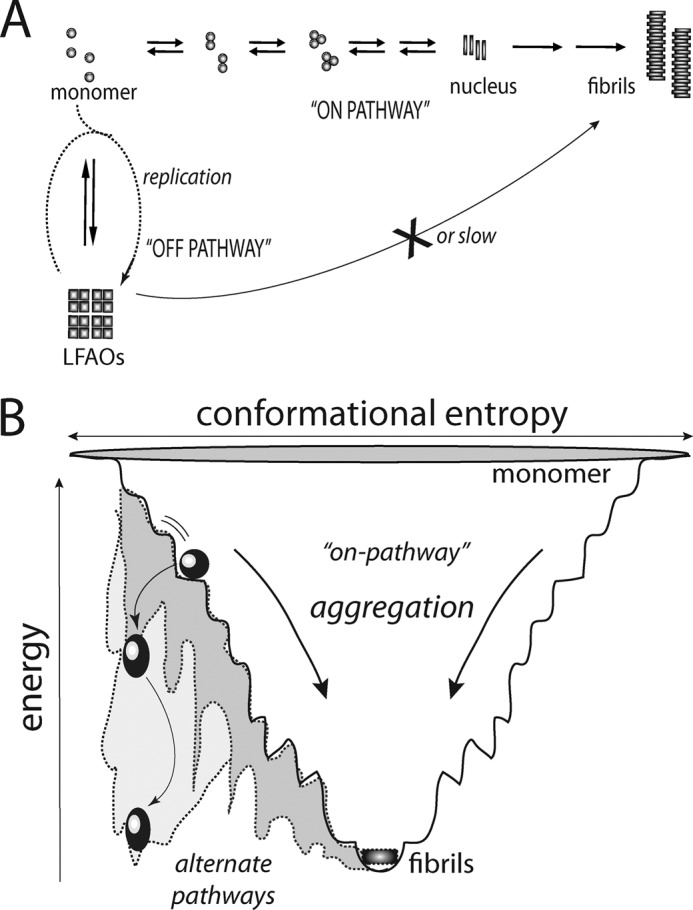
Schematic diagram depicting generation of off-pathway oligomers and replication. A, the classic on-pathway fibril formation process is depicted along with an alternative off-pathway. The process of replication undergone by off-pathway oligomers is shown in dotted lines. B, the aggregation process is shown as a thermodynamic funnel for intrinsically disordered proteins such as Aβ toward fibril formation. This path is considered to be the nucleation-dependant on-pathway. The spherical and oval balls represent LMW oligomers. However, the oligomers may adopt a different pathway such that they are trapped in local energy minima as off-pathway species (oval balls; gray area of the funnel). Such off-pathway species would have a significant energy barrier to overcome to proceed toward fibrils. Also, the off-pathway oligomers can recruit more monomers, which dictates them to adopt such alternate pathways (replication process shown in A). This may further increase the energy barrier for replicated off-pathway oligomers to overcome while significantly increasing their half-lives.
Comparison of LFAOs with Other Oligomers Indicates Similarities and Differences
There are only a handful of reports on the replication property of Aβ oligomers, and of those, the reports by Glabe and co-workers (20, 28, 37) are the most recent. They identified three specific soluble oligomer subtypes classified mainly on the basis of their immunoreactivity toward conformation-specific antibodies. These subtypes are PFOs, fibrillar oligomers (FOs), and APFs, and they are recognized by the conformation-specific antibodies A11, OC, and αPF, respectively (28). Among these, both PFOs and FOs showed a self-replication property by converting monomers into oligomers of the same type (28, 38). The main distinction between PFOs and FOs is that the PFOs are recognized by the polyclonal antibody A11, whereas the FOs are detected by the fibril-specific antibody OC (39). Our attempts to detect LFAOs with the oligomer-specific antibody A11 did not yield conclusive results. Although we could not get a positive signal with A11 for LFAOs, it would be inconclusive to say LFAOs may be structurally different from the epitope recognized by A11 as we could not generate appropriate positive controls for A11-specific oligomers even after several attempts. Nevertheless, a biochemical difference between the two oligomers is that FOs resemble fibril structure and seed fibril formation, whereas PFOs do not. The LFAOs reported here seem to resemble FOs rather than PFOs based on morphological heights, which range between 5 and 15 nm for FOs (38) as compared with 8–13 nm reported here for LFAOs. However, further comparative analyses of LFAOs with other self-replicating oligomers such as FOs could not be made largely due to the paucity of stringent biophysical investigations of the replication process. On the other hand, the APFs that are generated from hexane-water interfaces are characterized by a unique porelike structure that leads to membrane permeabilization and disruption of ion homeostasis, causing cell death (40). As shown in Fig. 6, similar treatment of LFAOs with hexane-water and chloroform-water interfaces failed to generate ringlike structures similar to APFs. Perhaps this difference is due to the fact that LFAOs are more similar to FOs than PFOs, and APFs are generated from PFOs.
Physiochemical Properties of LFAOs Showcase Reactions along Off-fibril Formation Pathway
This report also reiterates the fact previously reported by us and many others that oligomers need not be formed along the obligatory fibril formation pathway. The one important trait of all the oligomers discussed above such as FOs and PFOs is that they could nucleate the formation of oligomers from monomers but not fibrils (28, 38). Our current report also suggests that both interaction of LFAOs with monomers and association with similar oligomers lead to the formation of more non-fibrillar products. Not only are LFAOs formed along an alternative pathway, but they are also able to compete and divert monomers from the nucleation-dependant fibril formation pathway (Fig. 8B). Furthermore, association of LFAOs with each other in the presence of interfaces led to the formation of larger but non-fibrillar aggregates. This association property of LFAOs provides valuable insight into the potentially similar behavior of endogenous LMW oligomers in the presence of lipid interfaces in physiological environments. These data indicate that there may be many energy minimum states along the aggregation landscape (as depicted in Fig. 8B), and therefore, there is a likelihood of oligomers getting trapped in a “kinetic minimum” along the aggregation landscape and thus eliciting prolonged toxicity. Therefore, it is not unrealistic to hypothesize that the nucleation-dependant pathway could potentially be a “less pathogenic route” compared with the alternate pathway(s) that populates LMW oligomers. Future experiments will test this hypothesis.
Acknowledgment
We thank Dr. Gordon Cannon from the Department of Chemistry and Biochemistry for generously sharing the DLS instrument.
This work was supported, in whole or in part, by National Institutes of Health Grants 5P20RR016476-11 from the National Center for Research Resources and 8 P20 GM103476-11 from the NIGMS through the Mississippi IDeA Network of Biomedical Research Excellence. This work was also supported by American Heart Association Grant 10GRNT4190124 (to V. R.), National Science Foundation Major Research Instrumentation Grant 1040372 (to J. C. and V. R.), and United States Department of Education Graduate Assistance in Areas of National Need Fellowship Program Award P200A090066.

This article contains supplemental Figs. S1–S5.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- LFAO
- large fatty acid-derived oligomer
- LMW
- low molecular weight
- APF
- annular protofibril
- PFO
- prefibrillar oligomer
- NEFA
- non-esterified fatty acid
- ThT
- thioflavin-T
- SEC
- size exclusion chromatography
- DLS
- dynamic light scattering
- AFM
- atomic force microscopy
- Dan
- dansyl (5-dimethylaminonaphthalene-1-sulfonyl)
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AUC
- analytical ultracentrifugation
- PF
- protofibril
- FO
- fibrillar oligomer.
REFERENCES
- 1. Kumar A., Bullard R. L., Patel P., Paslay L. C., Singh D., Bienkiewicz E. A., Morgan S. E., Rangachari V. (2011) Non-esterified fatty acids generate distinct low-molecular weight amyloid-β (Aβ42) oligomers along pathway different from fibril formation. PLoS One 6, e18759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 3. Hartley D. M., Walsh D. M., Ye C. P., Diehl T., Vasquez S., Vassilev P. M., Teplow D. B., Selkoe D. J. (1999) Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 5. Kawarabayashi T., Shoji M., Younkin L. H., Wen-Lang L., Dickson D. W., Murakami T., Matsubara E., Abe K., Ashe K. H., Younkin S. G. (2004) Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 24, 3801–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., Klein W. L. (2004) Synaptic targeting by Alzheimer's-related amyloid β oligomers. J. Neurosci. 24, 10191–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 8. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 9. Dickson D. W., Crystal H. A., Bevona C., Honer W., Vincent I., Davies P. (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298; discussion 298–304 [DOI] [PubMed] [Google Scholar]
- 10. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shankar G. M., Li S., Mehta T. H., Garcia-Munoz A., Shepardson N. E., Smith I., Brett F. M., Farrell M. A., Rowan M. J., Lemere C. A., Regan C. M., Walsh D. M., Sabatini B. L., Selkoe D. J. (2008) Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jahn T. R., Radford S. E. (2008) Folding versus aggregation: polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 469, 100–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldsbury C., Frey P., Olivieri V., Aebi U., Müller S. A. (2005) Multiple assembly pathways underlie amyloid-β fibril polymorphisms. J. Mol. Biol. 352, 282–298 [DOI] [PubMed] [Google Scholar]
- 14. Gorman P. M., Yip C. M., Fraser P. E., Chakrabartty A. (2003) Alternate aggregation pathways of the Alzheimer β-amyloid peptide: Aβ association kinetics at endosomal pH. J. Mol. Biol. 325, 743–757 [DOI] [PubMed] [Google Scholar]
- 15. Bitan G., Lomakin A., Teplow D. B. (2001) Amyloid β-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J. Biol. Chem. 276, 35176–35184 [DOI] [PubMed] [Google Scholar]
- 16. Huang T. H., Yang D. S., Fraser P. E., Chakrabartty A. (2000) Alternate aggregation pathways of the Alzheimer β-amyloid peptide. An in vitro model of preamyloid. J. Biol. Chem. 275, 36436–36440 [DOI] [PubMed] [Google Scholar]
- 17. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L. (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gellermann G. P., Byrnes H., Striebinger A., Ullrich K., Mueller R., Hillen H., Barghorn S. (2008) Aβ-globulomers are formed independently of the fibril pathway. Neurobiol. Dis. 30, 212–220 [DOI] [PubMed] [Google Scholar]
- 19. Rangachari V., Moore B. D., Reed D. K., Sonoda L. K., Bridges A. W., Conboy E., Hartigan D., Rosenberry T. L. (2007) Amyloid-β(1–42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 46, 12451–12462 [DOI] [PubMed] [Google Scholar]
- 20. Kayed R., Pensalfini A., Margol L., Sokolov Y., Sarsoza F., Head E., Hall J., Glabe C. (2009) Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 284, 4230–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nichols M. R., Moss M. A., Reed D. K., Hoh J. H., Rosenberry T. L. (2005) Rapid assembly of amyloid-β peptide at a liquid/liquid interface produces unstable β-sheet fibers. Biochemistry 44, 165–173 [DOI] [PubMed] [Google Scholar]
- 22. Philo J. S. (2006) Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 354, 238–246 [DOI] [PubMed] [Google Scholar]
- 23. Schuck P. (1998) Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75, 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shao W., Yu Z., Chiang Y., Yang Y., Chai T., Foltz W., Lu H., Fantus I. G., Jin T. (2012) Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS One 7, e28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghosh P., Kumar A., Datta B., Rangachari V. (2010) Estimating the dynamics of protofibril elongation involved in Aβ42 peptide aggregation in Alzheimer's disease, in Proceedings of the First ACM International Conference on Bioinformatics and Computational Biology, Niagara Falls, NY, August 2–4, 2010, pp. 451–453, Association for Computing Machinery, New York [Google Scholar]
- 26. Nichols M. R., Moss M. A., Reed D. K., Lin W. L., Mukhopadhyay R., Hoh J. H., Rosenberry T. L. (2002) Growth of β-amyloid(1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 41, 6115–6127 [DOI] [PubMed] [Google Scholar]
- 27. Freir D. B., Nicoll A. J., Klyubin I., Panico S., Mc Donald J. M., Risse E., Asante E. A., Farrow M. A., Sessions R. B., Saibil H. R., Clarke A. R., Rowan M. J., Walsh D. M., Collinge J. (2011) Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R., 3rd, Pensalfini A., Yeung S., Head E., Marsh J. L., Glabe C. (2010) Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 5, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nichols M. R., Moss M. A., Reed D. K., Hoh J. H., Rosenberry T. L. (2005) Amyloid-β aggregates formed at polar-nonpolar interfaces differ from amyloid-β protofibrils produced in aqueous buffers. Microsc. Res. Tech. 67, 164–174 [DOI] [PubMed] [Google Scholar]
- 30. Collins S. R., Douglass A., Vale R. D., Weissman J. S. (2004) Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2, e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cordeiro Y., Lima L. M., Gomes M. P., Foguel D., Silva J. L. (2004) Modulation of prion protein oligomerization, aggregation, and β-sheet conversion by 4,4′-dianilino-1,1′-binaphthyl-5,5′-sulfonate (bis-ANS). J. Biol. Chem. 279, 5346–5352 [DOI] [PubMed] [Google Scholar]
- 32. Jones E. M., Surewicz W. K. (2005) Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 121, 63–72 [DOI] [PubMed] [Google Scholar]
- 33. King C. Y., Diaz-Avalos R. (2004) Protein-only transmission of three yeast prion strains. Nature 428, 319–323 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka M., Chien P., Yonekura K., Weissman J. S. (2005) Mechanism of cross-species prion transmission: an infectious conformation compatible with two highly divergent yeast prion proteins. Cell 121, 49–62 [DOI] [PubMed] [Google Scholar]
- 35. Saá P., Castilla J., Soto C. (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 [DOI] [PubMed] [Google Scholar]
- 36. Saborio G. P., Permanne B., Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 37. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu J. W., Breydo L., Isas J. M., Lee J., Kuznetsov Y. G., Langen R., Glabe C. (2010) Fibrillar oligomers nucleate the oligomerization of monomeric amyloid β but do not seed fibril formation. J. Biol. Chem. 285, 6071–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kayed R., Head E., Sarsoza F., Saing T., Cotman C. W., Necula M., Margol L., Wu J., Breydo L., Thompson J. L., Rasool S., Gurlo T., Butler P., Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lasagna-Reeves C. A., Glabe C. G., Kayed R. (2011) Amyloid-β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J. Biol. Chem. 286, 22122–22130 [DOI] [PMC free article] [PubMed] [Google Scholar]