

Background: Recent GWA studies have demonstrated an association between the endocytic clathrin adaptor PICALM and Alzheimer disease.
Results: Manipulation of PICALM expression selectively alters APP endocytosis and Aβ production in neural cells and plaque deposition in APP transgenic mice.
Conclusion: PICALM is involved in intracellular APP processing and plaque pathogenesis.
Significance: Understanding cellular mechanisms involved in APP processing may reveal novel targets for intervention.
Keywords: Alzheimer Disease, Amyloid, Amyloid Precursor Protein, Endocytosis, Pathology, PICALM
Abstract
One of the pathological hallmarks of Alzheimer disease is the accumulation of amyloid plaques in the extracellular space in the brain. Amyloid plaques are primarily composed of aggregated amyloid β peptide (Aβ), a proteolytic fragment of the transmembrane amyloid precursor protein (APP). For APP to be proteolytically cleaved into Aβ, it must be internalized into the cell and trafficked to endosomes where specific protease complexes can cleave APP. Several recent genome-wide association studies have reported that several single nucleotide polymorphisms (SNPs) in the phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) gene were significantly associated with Alzheimer disease, suggesting a role in APP endocytosis and Aβ generation. Here, we show that PICALM co-localizes with APP in intracellular vesicles of N2a-APP cells after endocytosis is initiated. PICALM knockdown resulted in reduced APP internalization and Aβ generation. Conversely, PICALM overexpression increased APP internalization and Aβ production. In vivo, PICALM was found to be expressed in neurons and co-localized with APP throughout the cortex and hippocampus in APP/PS1 mice. PICALM expression was altered using AAV8 gene transfer of PICALM shRNA or PICALM cDNA into the hippocampus of 6-month-old APP/PS1 mice. PICALM knockdown decreased soluble and insoluble Aβ levels and amyloid plaque load in the hippocampus. Conversely, PICALM overexpression increased Aβ levels and amyloid plaque load. These data indicate that PICALM, an adaptor protein involved in clathrin-mediated endocytosis, regulates APP internalization and subsequent Aβ generation. PICALM contributes to amyloid plaque load in brain likely via its effect on Aβ metabolism.
Introduction
A hallmark pathological feature of AD2 is the amyloid plaque, extracellular deposits of aggregated amyloid β peptide (Aβ). Aβ is a proteolytic fragment of the amyloid precursor protein (APP), a transmembrane protein found primarily in neurons (1), which can be cleaved by several enzyme complexes. The major fraction of APP is proteolyzed by α-secretase, which cleaves the protein within the Aβ region, rendering the resulting fragments, soluble APPα and α C-terminal fragment (α-CTF), nonpathogenic (2). This is known as the non-amyloidogenic pathway. Alternatively, Aβ can be generated by the serial proteolytic cleavage of APP with β- and then γ-secretases, and this is known as the amyloidogenic pathway (3). Both enzyme complexes cleave APP intracellularly, requiring internalization of APP (2, 4). After Aβ is generated in the endocytic pathway, some of it is secreted into the extracellular space where it is found even in normal individuals.
The requirement for APP endocytosis to generate Aβ was found after mutation of an internalization motif in the cytoplasmic tail of APP inhibited APP internalization and prevented Aβ generation (5). Subsequent studies demonstrated that APP internalization and Aβ production were clathrin-mediated (2, 6). Clathrin interacts with a variety of assembly proteins and adaptor proteins, including AP1, AP2, and AP3 (7), which regulate and aid in the formation of clathrin-coated pits (8). Despite strong evidence that clathrin-mediated endocytosis is required for Aβ generation, the precise molecular mechanisms of APP internalization have not been fully defined.
Recent genome wide association studies have identified several novel loci that were significantly associated with AD risk (9–12). One of the identified genes, phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM), which had no previously known association with AD, appears to play a critical role in clathrin-mediate endocytosis.
PICALM, also known as CALM, is a cytoplasmic adaptor protein that plays a critical role in clathrin-mediated endocytosis. PICALM binds to clathrin, phosphatidylinositol, and AP2 to aid in the formation of clathrin-coated pits (13–15). Deletion of the PICALM homolog AP180 in Drosophila and yeast resulted in impairment in clathrin-mediated endocytosis (16–18). Deletion of the PICALM homolog UNC-11 in Caenorhabditis elegans resulted in the mislocalization of VAMP2, a protein involved in docking and fusion of synaptic vesicles at presynaptic terminals (19), an effect replicated in human HEK293 cells (20), suggesting a role in synaptic VAMP2 endocytosis. A more recent study demonstrated direct interactions between PICALM and the R-SNAREs VAMP8, VAMP3, and VAMP2, indicating a role in the fusion of endocytic clathrin-coated vesicles with endosomes and subsequent trafficking (21).
Based on the role of PICALM in clathrin-mediated endocytosis, we hypothesize that PICALM may play a role in APP endocytosis and thus regulate Aβ generation. In this study, we tested this hypothesis in a cell culture model of APP processing and an animal model of AD.
EXPERIMENTAL PROCEDURES
Cell Culture
N2a-APP695 cells (neuroblastoma N2a cells overexpressing APP695) were grown in DMEM/Opti-MEM (50:50) supplemented with 5% FBS and 200 μg/ml G418.
Plasmid Construction and Cell Transfection
PICALM cDNA containing a C-terminally linked HA tag was PCR-amplified from a mouse cDNA library and cloned into a pAAV vector containing the PGK promoter. For the generation of the PICALM shRNA construct, the human U6 promoter region and the shRNA sequence 5′-GGAAATGGAACCACTAAGATTCAAGAGATCTTAGTGGTTCCATTTCCTTTTTTACGCGT-3′ were inserted into the pAAV vector containing the eGFP gene driven by the PGK promoter. N2a-APP695 cells were transfected with the above mentioned constructs or the control vectors for 24 h and harvested for further analysis.
APP Internalization Assay
N2a-APP695 cells were plated on 8-well chamber slides, washed with cold PBS, and incubated at 4 °C with 6E10 antibody (Covance; 1:200 in PBS) for 30 min to label surface APP. Cells were carefully washed with ice-cold PBS and incubated at 37 °C for 0, 5, and 10 min to permit internalization. Cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized in 0.1% Triton X-100, and blocked in 1% bovine serum albumin in PBS/Triton X-100. Cell preparations were incubated with goat anti-PICALM primary antibody (C-18) (Santa Cruz Biotechnology, sc6433), washed, and then incubated with donkey Alexa Fluor 488-conjugated anti-mouse IgG (Invitrogen) and donkey Cy3-conjugated anti-goat IgG (Jackson ImmunoResearch Laboratories). Cells were mounted and examined using a confocal microscope (Zeiss LSM).
Cell Surface Biotinylation and Internalization Assays
Cell surface biotinylation and internalization assays were performed as described (22) with modifications. Briefly, cells in 6-well plates were washed with cold PBS, and surface proteins were labeled with a non-membrane-permeant, cleavable biotin derivative, sulfo-NHS-SS-biotin (1 mg/ml in PBS). Cells were kept at 4 °C for 30 min in the dark and gently rocked during the incubation period. The biotin reagent was quenched by treating the cells with two 15-min washes of 0.1 m glycine in PBS. Cells were rinsed with PBS and lysed in radioimmune precipitation assay buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) deoxycholate, and 1× protease inhibitor mixture. Lysates were incubated with Dynabeads MyOne Streptavidin T1 (Invitrogen) for 2 h in a rotary mixer to isolate biotin-labeled proteins. For the internalization assay, surface biotin-labeled cells were incubated for varying times at 37 °C to allow internalization. Internalization was stopped by rapid cooling on ice. To cleave biotin exposed at the cell surface, cells were incubated three times for 20 min at 4 °C with 50 mm 2-mercaptoethanesulfonic acid (Sigma) in 50 mm Tris-HCl, pH 8.7, 100 mm NaCl, and 2.5 mm CaCl2. After thorough rinsing with PBS containing 20 mm Hepes, cells were lysed in radioimmune precipitation assay buffer, and internalized biotinylated proteins were immunoprecipitated with streptavidin and subjected to immunoblotting for APP (CT695; Invitrogen).
To analyze transferrin uptake, cells were rinsed with DMEM twice and cultured in serum-free medium for 2 h. Cells were incubated with biotin-labeled transferrin (20 μg/ml) in serum-free medium for various time periods at 37 °C. Cells were then placed on ice, washed with ice-cold PBS twice, and then subsequently incubated three times for 10 min on ice in 25 mm MesNa, pH 5.0 containing 150 mm NaCl and 50 μm deferoxamine mesylate and for 10 min in PBS. Cells were lysed with radioimmune precipitation assay buffer, and the lysates were fractionated by SDS-PAGE followed by immunoblotting with anti-biotin antibody (Cell Signaling Technology) to detect transferrin uptake.
Transgenic Mice with Viral Injection
Animal care and surgical procedures were approved by the Animal Studies Committee of Washington University School of Medicine in accordance with guidelines of the United States National Institutes of Health. APPswe/PS1ΔE9 transgenic mice (APP/PS1; The Jackson Laboratory) (23) were injected with AAV8 viral particles (generated by Hope Center Viral Core at Washington University) using defined stereotaxic coordinates for the hippocampus. APP/PS1 mice at 6 months of age were divided into four groups. Each group was injected with 2 μl of AAV8 viral particles (1.5 × 1012 viral genomes/ml) into hippocampi bilaterally. Four different viral injections, balancing male and female mice, were performed: 1) AAV-shPICALM-eGFP (five males and seven females), 2) AAV-shScrambled-eGFP (five males and seven females), 3) AAV-eGFP (six males and 12 females), and 4) AAV-PICALM-HA (six males and 12 females). Virus was slowly infused through a 30-gauge needle (0.2 μl/min) over 10 min; the syringe was left in place for an additional 10 min before it was slowly withdrawn. Mice were sacrificed 4 month later.
Brain Tissue Preparation
Mice were deeply anesthetized with isoflurane and transcardially perfused with PBS. Brains were extracted and cut into two hemispheres. Right hemispheres were immediately dissected, snap frozen on dry ice, and stored at −80 °C for biochemical analysis. Left hemispheres were fixed in 4% paraformaldehyde for 24 h followed by 30% sucrose in PBS at 4 °C. Coronal sections (50 μm) were cut on a freezing-sliding microtome. Collected slices were stored in cryoprotectant solution (0.2 m phosphate-buffered saline, 30% sucrose, and 30% ethylene glycol) at −20 °C.
Immunohistochemistry
Sections were incubated in 0.3% H2O2 in Tris-buffered saline for 10 min, washed with TBS, blocked with 3% dry milk in TBS-X (0.25% Triton X-100 in TBS) for 1 h, and incubated with primary antibody overnight. A solution from the Vectastain ABC kit (1:400) was applied to brain slices for 1 h followed by 0.025% 3,3′-diaminobenzidine tetrachloride in 0.25% NiCl2 and 0.05% H2O2 for 10–15 min. The slices were placed on glass slides, dried overnight, dehydrated, and mounted.
X-34 Plaque Staining
Brain slices were mounted on Superfrost Plus slides, permeabilized with 0.25% Triton X-100 for 30 min, and stained with X-34 (a generous gift from Robert Mach) dissolved in 40% ethanol and 60% water, pH 10 for 20 min. Tissue was then thoroughly rinsed in PBS and mounted with Fluoromount mounting medium.
Plaque Quantification
Brain sections (50 μm) were collected every 300 μm from rostral anterior commissure to caudal hippocampus. Sections were stained with X-34 or immunostained with HJ3.4 antibodies (n = 12–18 per group). Four slices per animal were used. A NanoZoomer Digital Scanner (Hamamatsu Photonics) was used to create high resolution digital images of the stained brain slices. The total area of plaque coverage in the hippocampus and cortex was measured using NIH ImageJ software and expressed as percent total area for each slice. The results for an animal included four different percent total area values, which were averaged to represent each animal. Plaque number was also determined using ImageJ, averaged from four sections from each animal, and expressed as number of plaques/section.
Immunofluorescence
The immunofluorescence double labeling method has been described (24). A mixture of goat anti-PICALM (C-18; 1:100) and a cell-specific antibody was applied to the sections overnight at 4 °C. The cell-specific antibodies included mouse anti-NeuN antibody (Sigma; 1:1000) to identify neurons, mouse anti-GFAP antibody (Sigma; 1:100) to identify astrocytes, and rabbit anti-Iba1 (Wako Chemicals; 1:1000) to identify microglia. The APP C-terminal antibody (CT695) (Invitrogen; 1:200) was used to detect APP. A secondary antibody mixture of Cy3-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratories) and Alexa Fluor 488-conjugated donkey anti-mouse IgG or anti-rabbit IgG (Invitrogen) was applied. Sections were examined for staining and co-localization by confocal microscopy.
Aβ ELISA
Aβ in the conditioned medium and cell lysates from transfected N2a-APP695 cells was detected by sandwich ELISA. To detect Aβ in the hippocampus, dissected hippocampus was sequentially homogenized in PBS and then in 5 m guanidine in TBS, pH 8.0. All ELISA samples were diluted in a final buffer of 1.5% BSA, 300 mm Tris-HCl, pH 8.0, 0.05% azide, and protease inhibitors. Aβx–40 and Aβx–42 were assessed with mouse monoclonal coating antibodies HJ2 (anti-Aβ35–40) and HJ7.4 (anti-Aβ37–42), respectively, and a biotinylated central domain antibody, HJ5.1 (anti-Aβ13–28), as the detecting antibody followed by streptavidin-poly-HRP-40 (Fitzgerald Industries). All ELISAs were developed using Super Slow ELISA 3,3′,5,5′-tetramethylbenzidine (Sigma), and absorbance was read on a Bio-Tek Epoch plate reader (Winooski, VT) at 650 nm. Standard curves were generated from synthetic human Aβ1–40 or Aβ1–42 peptides (American Peptide).
Western Blot
Equal amounts of protein for each sample were run on a 4–12% Bis-Tris gel (Invitrogen) and transferred to PVDF membranes. Blots were probed with the following antibodies: PICALM (Santa Cruz Biotechnology; 1:200), APP-CT695 (1:500), Biotin (Cell Signaling Technology; 1:500), and actin (Sigma; 1:2000). Normalized band intensity was quantified using Labworks software.
Statistical Analysis
All data were analyzed by two-tailed Student's t test and expressed as mean ± S.E. Differences between two sets of data were deemed significant at p < 0.05 (*) and p < 0.01 (**).
RESULTS
PICALM and APP Co-localize during Endocytosis in Vitro
To examine the cellular localization of PICALM and APP, N2a neuroblastoma cells stably overexpressing APP695 (N2a-APP695) were incubated at 4 °C (to inhibit endocytosis) while the cell surface APP was labeled using the 6E10 antibody. The cells were subsequently warmed to 37 °C for varying times (0, 5, and 10 min) to permit endocytosis, then fixed, and immunostained with PICALM antibodies to permit double staining of internalized APP and PICALM (Fig. 1). The specificity of the PICALM antibody has been demonstrated in previous knockdown studies (25). Prior to endocytosis (0 min), surface-labeled APP was observed exclusively on the cell surface, separate from PICALM immunostaining largely found on vesicles in the cytosol (Fig. 1), as would be expected in the absence of endocytosis. After warming cells to 37 °C, cell surface-labeled APP was observed in intracellular vesicles double labeled with PICALM, clearly demonstrating that the two proteins co-localize after initiation of endocytosis. PICALM and APP immunostaining during steady-state conditions was identical to the images obtained 5 and 10 min after rewarming the cells to 37 °C (data not shown).
FIGURE 1.

APP co-localizes with PICALM after endocytosis. Live N2a-APP695 cells were surface-labeled with 6E10 antibodies at 4 °C to prevent endocytosis. The cells were then warmed to 37 °C for varying times to allow internalization, fixed, and immunostained for PICALM and surface-labeled APP. Prior to endocytosis (0 min), no co-localization of surface APP and PICALM was observed. Only after endocytosis was permitted was co-localization of surface-labeled APP and PICALM seen in intracellular vesicles.
Manipulation of PICALM Expression Alters APP Internalization, Processing, and Aβ Production in Vitro
To investigate the effect of PICALM on APP internalization and processing, we developed several constructs to knock down or overexpress PICALM. An shRNA construct directed against PICALM was generated; a scrambled shRNA (shScrambled) served as a control. To overexpress PICALM, a construct carrying PICALM linked to an HA tag driven by a PGK promoter (PICALM-HA) was generated; the empty vector served as a negative control. N2a-APP695 cells were transfected with these constructs. Twenty-four hours later, cells were incubated at 4 °C with sulfo-NHS-SS-biotin to biotinylate cell surface proteins. After warming to 37 °C for varying times to permit endocytosis, surface biotin was removed, cells were lysed, and internalized biotinylated proteins were precipitated with streptavidin beads and immunoblotted with anti-APP antibodies to quantify internalized APP.
Cells transfected with shPICALM showed a 70% decrease in PICALM expression compared with cells transfected with shScrambled (Fig. 2, A and B), demonstrating successful knockdown. PICALM knockdown resulted in a significant decrease in APP internalization and concomitant accumulation of steady-state cell surface APP (Fig. 2, A and C). In addition, there was a trend toward a decrease in β-CTF/total CTF ratio and an increase in α-CTF/total CTF (Fig. 2, A and C). Conversely, cells transfected with PICALM-HA demonstrated greater than a 4-fold increase in expression compared with cells transfected with empty vector (Fig. 2, A and B). PICALM overexpression resulted in a significant increase in internalized APP with a decrease in steady-state surface APP (Fig. 2, A and D). Again, there was a trend toward an increase in β-CTF/total CTF and a decrease in α-CTF/total CTF (Fig. 2, A and D). The expression of full-length APP in cells was unchanged by PICALM knockdown or overexpression (Fig. 2, A, C, and D). Collectively, these results demonstrate that PICALM is involved in APP internalization.
FIGURE 2.
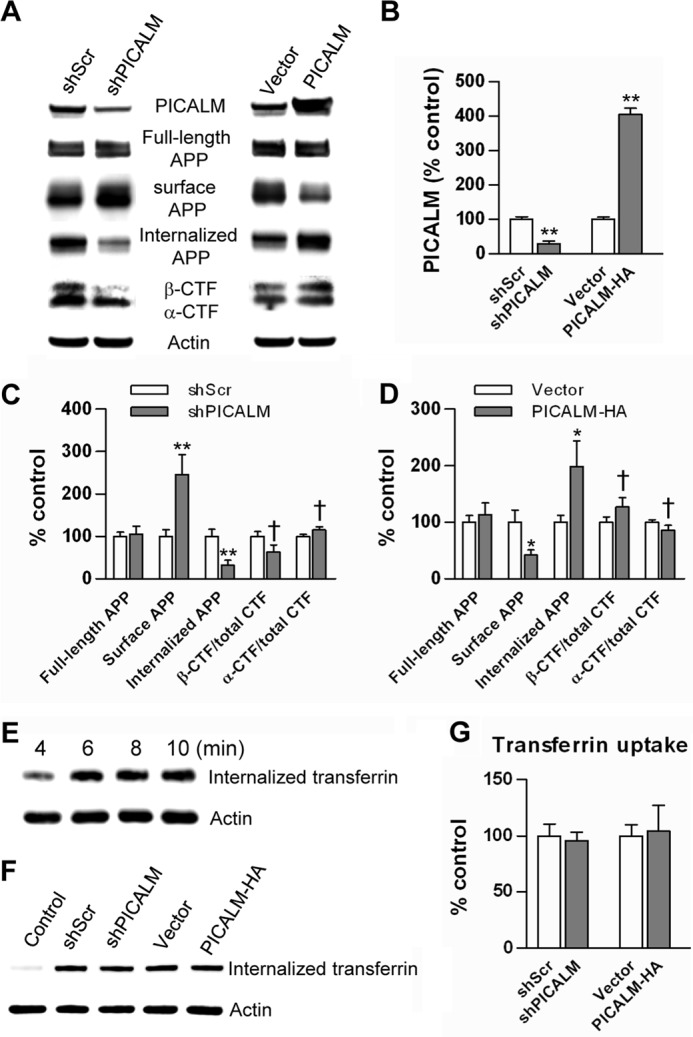
PICALM manipulation alters cell surface APP and APP internalization. N2a-APP695 cells were transfected with shPICALM, shScrambled (shScr), PICALM-HA, or empty vector. Twenty-four hours after transfection, cell surface proteins were biotinylated at 4 °C. To examine cell surface APP, cells were lysed, immunoprecipitated with streptavidin beads, and immunoblotted with APP antibody (CT695). To determine APP internalization, surface-biotinylated cells were returned to 37 °C for 10 min to permit endocytosis. The remaining cell surface biotin was cleaved, and internalized biotinylated proteins were extracted, immunoprecipitated, and then immunoblotted for APP (CT695). PICALM knockdown reduced PICALM expression by greater than 70%, whereas overexpression increased PICALM by 4–5-fold (A and B). PICALM knockdown resulted in significant increases in steady-state surface APP accumulation while decreasing internalized APP (C). Likewise, PICALM overexpression decreased surface APP accumulation while increasing internalized APP (D). PICALM knockdown resulted in a trend toward a decrease in β-CTF/total CTF (p = 0.107) and an increase in α-CTF/total CTF (p = 0.122) (C). Conversely, PICALM overexpression increased β-CTF/total CTF (p = 0.146) and decreased α-CTF/total CTF (p = 0.142), but these changes did not reach statistical significance (D). Full-length APP expression was unchanged by the manipulations (C and D). A time course of transferrin internalization showed saturation by 6–8 min after the initiation of endocytosis (E). Neither knockdown nor overexpression of PICALM resulted in any change in transferrin internalization (F and quantified in G); the control consisted of cells permitted to bind transferrin but prevented from internalizing it. Values are mean ± S.E. (error bars); n = 3 each. *, p < 0.05; **, p < 0.01 by Student's t test.
To determine the specificity of the effect of PICALM on endocytosis, we examined its effect on transferrin internalization. The transferrin receptor is ubiquitously expressed and is known to undergo clathrin-mediated endocytosis with rapid recycling to the cell surface (26). PICALM, however, can selectively internalize some cargoes over others (20, 27). A time course of transferrin uptake performed in N2a cells revealed rapid uptake saturating at ∼6–8 min after initiation of endocytosis (Fig. 2E). Therefore, for the remaining experiments, transferrin internalization was determined 4 min after initiation of endocytosis in N2a cells transfected with shPICALM, shScrambled, or PICALM-HA. Neither knockdown nor overexpression of PICALM altered transferrin uptake (Fig. 2, F and G), consistent with prior reports (20, 27). These results suggest that the effect of PICALM on endocytosis is cargo-specific. APP endocytosis was regulated by PICALM, but transferrin endocytosis is not.
To examine the effect of PICALM on Aβ peptide production, we transfected N2a-APP695 cells with the four constructs described above. Conditioned media from cells and cell lysates were collected for quantification of Aβ using ELISA. PICALM knockdown resulted in a decrease in Aβ40 and Aβ42 in conditioned media compared with cells transfected with shScrambled (Fig. 3, A and B). In addition, Aβ40 levels in cell lysates were decreased (Aβ42 was undetectable in cell lysates from both groups) (Fig. 3C). Conversely, overexpression of PICALM resulted in increases in Aβ in conditioned media and in cell lysates (Fig. 3, A–C). These results indicate that PICALM influences Aβ peptide production and release from the cell.
FIGURE 3.
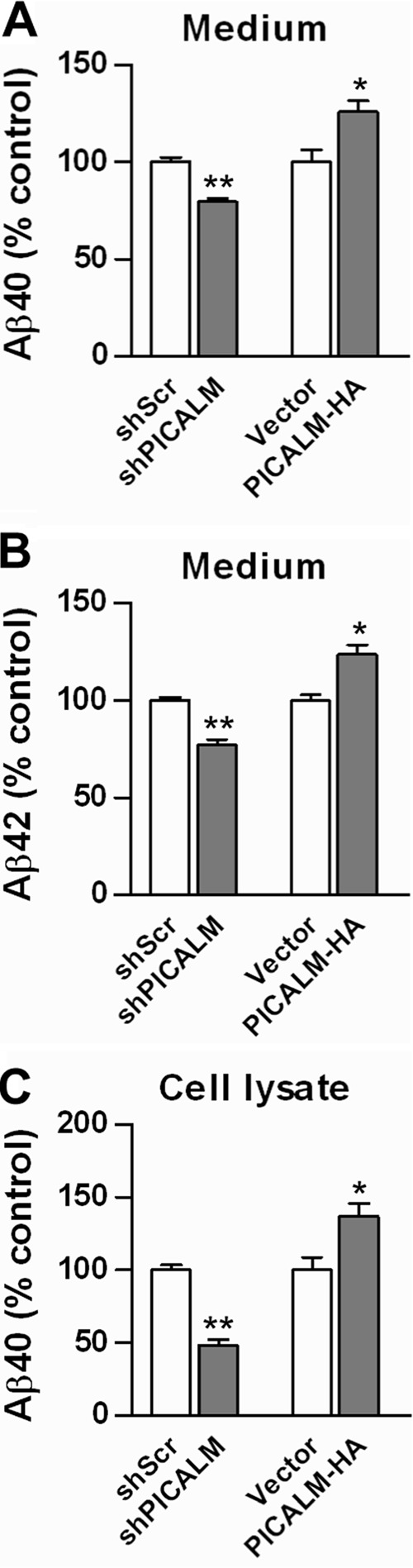
Manipulation of PICALM expression alters Aβ synthesis and release. Conditioned media from N2a-APP695 cells transfected with knockdown or overexpression vectors were collected, and Aβ levels were quantified using ELISA. PICALM knockdown decreased Aβ40, whereas PICALM overexpression increased Aβ40 levels in conditioned medium (A). Similar changes in Aβ42 levels were found after PICALM manipulation (B). Cell lysates from N2a-APP695 cells showed similar changes in Aβ40 levels (C). Aβ42 levels were undetectable in cell lysates. Values are mean ± S.E. (error bars); n = 3 each. *, p < 0.05; **, p < 0.01. shScr, shScrambled.
PICALM Is Expressed in Neurons and Co-localizes with APP/Aβ in APP/PS1 Mice
To examine the localization of PICALM in mouse brain, we performed immunostaining on APP/PS1 brain sections using PICALM, NeuN, GFAP, Iba1, and APP antibodies. In cortex and hippocampus, PICALM was expressed primarily in neurons (NeuN+ cells) (Fig. 4, A–F) with no co-labeling in astrocytes (GFAP+ cells) or microglia (Iba1+ cells) (data not shown). Furthermore, PICALM+ cells also were co-labeled with the APP antibody, suggest that PICALM is expressed in neurons, which also express APP/Aβ in the brains of APP/PS1 mice (Fig. 4, G–L).
FIGURE 4.

Neuronal localization of PICALM in APP/PS1 mice. Double labeling of PICALM (red) and neuronal cell-specific marker NeuN (green) (A–F) in the cortex (left panel) and hippocampus (right panel) of 3-month-old APP/PS1 mice was performed. PICALM (A and D) co-localizes with NeuN (B and E) in neurons (best seen in the merged images (C and F)). The boxed insets (upper right corner) show higher power images of individual double labeled cells. PICALM and APP co-localize in neurons of the cortex and hippocampus (G–L). Scale bar, 20 μm.
Manipulation of PICALM Expression in APP/PS1 Mice Alters APP Processing and Aβ Generation in Vivo
Picalm knock-out mice have recently been generated; however, these mice are embryonic lethal due to defects in brain development.3 Thus, to examine the influence of PICALM on amyloid plaque pathogenesis in the absence of developmental defects, we used a viral gene transfer approach to alter PICALM expression in the hippocampus of mature APP/PS1 mice. To knock down PICALM, we used the shRNA construct described above packaged in an AAV8 vector driven by a U6 promoter (also expressing eGFP as a reporter). A scrambled shRNA packaged in the same AAV8 vector served as a control. To overexpress PICALM, the same PICALM-HA construct described above (inserted into an AAV8 viral vector driven by a PGK promoter) was used. The viral vector carrying eGFP served as a control. The viral vectors were injected into the hippocampus of 6-month-old APP/PS1 mice. The hippocampus was chosen for viral targeting because of its easily accessible location and discrete containment for viral gene transfer. In addition, it was chosen for its characteristic and representative deposition of amyloid plaques in APP/PS1 mice. Four months after injection (at 10 months of age), mice were sacrificed and assessed for viral gene transfer using a variety of criteria. Sections from brains injected with knockdown vectors were examined for GFP fluorescence. Brains demonstrating uniform expression of GFP throughout the hippocampus were selected for further analysis. In these brains, GFP was observed throughout the anterior hippocampus, spanning 3–4 mm in the anterior-posterior direction (Fig. 5A). The major cell type expressing GFP was neurons (based on cellular morphology) and appeared in several layers of the hippocampus.
FIGURE 5.

Manipulation of PICALM expression in vivo using viral gene transfer. Fluorescence images of GFP (green) and PICALM-HA (red) expression in the hippocampus of APP/PS1 mice injected with AAV8-shPICALM-GFP vectors are shown. Sequential images of brain sections confirm high transduction efficiency throughout the anterior hippocampus; expression is primarily in neurons based on cell morphology (A). Extracts from hippocampi transduced with knockdown and overexpression viral vectors were subjected to Western blotting (B). PICALM expression was decreased after transduction with knockdown viral vectors and increased with overexpressing viral vectors (C). PICALM knockdown revealed a trend toward a decrease in β-CTF relative to total CTF (p = 0.121), whereas α-CTF relative/total CTF showed an increasing trend (p = 0.111) (D). Likewise, PICALM overexpression resulted in an increasing trend in β-CTF/total CTF (p = 0.114), and a trend toward a decrease in α-CTF/total CTF (p = 0.114) (E). Full-length APP after PICALM knockdown and overexpression demonstrated no change (D and E). Values are mean ± S.E. (error bars); n = 5 mice/group. **, p < 0.01. shScr, shScrambled.
To confirm knockdown, hippocampal extracts were subjected to Western blotting using anti-PICALM antibodies, which demonstrated ∼50% knockdown in overall hippocampal PICALM protein levels (Fig. 5, B and C). PICALM knockdown altered APP processing but not APP expression. The expression of full-length APP was unchanged by PICALM knockdown; however, similar to the N2a studies, β-CTF/total CTF demonstrated a trend toward a decrease with a concomitant trend toward an increase in α-CTF/total CTF (Fig. 5, B and D).
The overexpression vectors also successfully transduced neurons in the hippocampus as assessed by HA immunostaining (Fig. 5A) and increased PICALM expression by almost 3-fold (Fig. 5, B and C). PICALM overexpression resulted in a trend toward increased β-CTF/total CTF expression and a decrease in α-CTF/total CTF (Fig. 5, B and E). PICALM overexpression did not alter expression of full-length APP (Fig. 5, B, D, and E).
To determine whether these manipulations in PICALM expression in the hippocampus impacted Aβ production, transduced hippocampi from the 10-month-old mice above were serially homogenized in PBS and then in guanidine to extract PBS-soluble and -insoluble fractions. Each fraction was quantified using ELISAs to measure both Aβ40 and Aβ42. PICALM knockdown resulted in a decrease in both PBS-soluble and -insoluble fractions of Aβ40 and Aβ42 (Fig. 6, A and B). These findings are consistent with the in vitro studies, which demonstrated decreases in Aβ production following PICALM knockdown in N2a-APP695 cells. PICALM overexpression resulted in an increase in hippocampal Aβ40 and Aβ42 levels in both PBS-soluble and -insoluble fractions (Fig. 6, C and D). Again, these findings were consistent with the in vitro studies above.
FIGURE 6.
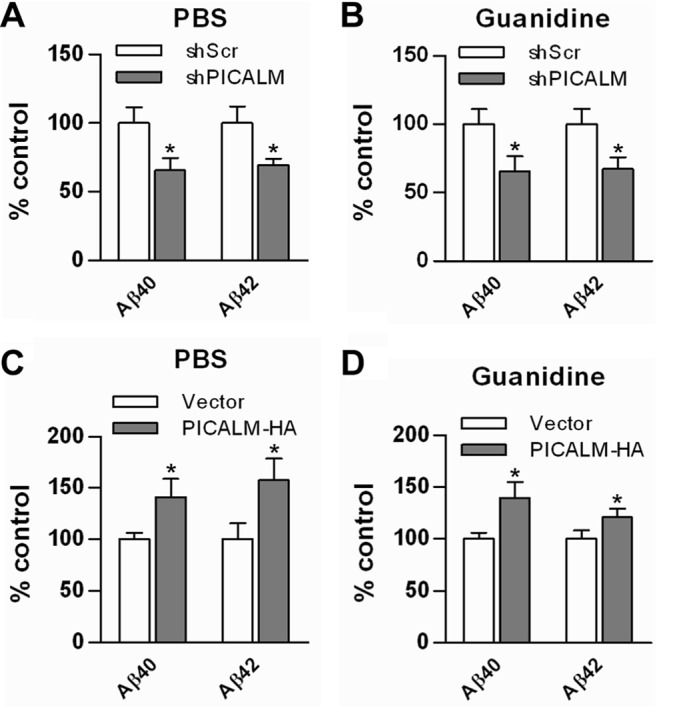
PICALM manipulation via viral gene transfer alters Aβ synthesis in hippocampus of APP/PS1 mice. Dissected hippocampi from PICALM knockdown and overexpression animals were homogenized first in PBS and then in 5 mm guanidine, and Aβ was quantified using ELISA. PICALM knockdown resulted in an ∼30% decrease in PBS-soluble Aβ40 and Aβ42 levels (A). In addition, insoluble guanidine fractions showed a similar decrease in Aβ40 and Aβ42 levels (B). Conversely, PICALM overexpression resulted in a 40–50% increase in PBS-soluble Aβ40 and Aβ42 levels (C) and a 20–40% increase in insoluble Aβ40 and Aβ42 (D). Values are mean ± S.E. (error bars); n = 7–9 mice/group. *, p < 0.05. shScr, shScrambled.
Manipulation of PICALM Expression Alters Amyloid Plaque Load in Vivo
To determine whether PICALM manipulations influenced amyloid plaque pathogenesis, we stained brains with X-34 to assess compact plaques and immunostained with HJ3.4 antibodies to assess Aβ plaque load. PICALM knockdown in the hippocampus resulted in significant decreases in both X-34-stained and Aβ-immunostained plaque number and load (Fig. 7, A, B, and D). As expected, both X-34-stained and Aβ-immunostained plaques did not change in the corresponding cortex (Fig. 7, A, C, and E). Conversely, PICALM overexpression in the hippocampus resulted in increases in plaque number and load defined by both X-34 staining and Aβ immunostaining (Fig. 7, F, G, and I), but no increases were seen in the cortex (Fig. 7, H and J). These data are consistent with the changes in the insoluble Aβ concentrations from the hippocampal homogenates (above). Collectively, these results indicate that PICALM expression influences amyloid plaque accumulation likely by regulating brain Aβ levels via APP metabolism.
FIGURE 7.

PICALM manipulation alters amyloid plaque number and load in hippocampus of APP/PS1 mice. Representative images of Aβ-immunostained plaques in shPICALM and shScrambled (shScr) injected mice are shown (A). Quantifications of plaque burden showed significant decreases (Aβ-immunostained plaques and X-34-stained compact plaques) in the hippocampus where shRNA was targeted but not in the overlying cortex (B–E). Representative Aβ-immunostained images from PICALM-HA and empty vector injection are shown (F). PICALM-HA produced a significantly higher plaque number and burden in the hippocampus but not overlying cortex (G–J). Values are mean ± S.E. (error bars); n = 12–18 mice/group. *, p < 0.05. IHC, immunohistochemistry.
DISCUSSION
Endocytosis of APP from neuronal plasma membranes is a key event leading to generation of Aβ. Therefore, understanding the factors that regulate APP uptake, trafficking, and processing may be critical for designing therapeutic strategies for AD. Several recent genome-wide association studies have reported that a PICALM SNP is associated with late onset AD (9, 11, 12, 28); however, its role in disease pathogenesis is unknown. In the present study, we demonstrate that PICALM co-localizes with APP/Aβ in N2a-APP cells in vitro and in neurons in APP/PS1 mice. Furthermore, PICALM manipulation alters both cell surface and internalized APP, suggesting a regulatory role of PICALM in APP internalization. This regulatory role appears somewhat cargo-specific as transferrin uptake was unaltered by PICALM manipulation. We also present evidence that PICALM influences processing of APP and resultant Aβ production. These in vitro findings are consistent with the in vivo experiments. Knockdown of PICALM in the hippocampus of APP/PS1 transgenic mice decreased Aβ production and amyloid plaque deposition, whereas overexpression of PICALM increased hippocampal Aβ concentration and plaque load. Therefore, our studies provide evidence that PICALM-mediated endocytosis may play a critical role in the pathogenesis of amyloid plaques.
Endocytic pathways have been known to be involved in Aβ generation for some time. Deletion of the internalization motif YENPTY within the cytoplasmic domain of APP or disruption of the clathrin lattice by potassium depletion resulted in reduced APP internalization and diminished Aβ release (5). Furthermore, inhibition of clathrin-dependent endocytosis by overexpression of the dominant negative mutant dynamin I reduced Aβ production and release into the extracellular space (29). PICALM is an adaptor protein that plays a critical role in clathrin-mediated endocytosis (13, 14). Depletion of PICALM affects clathrin coat formation (15) and inhibits EGF receptor internalization (27). Consistent with these findings, we found that inhibition of PICALM expression in N2a-APP695 cells resulted in the reduction of APP internalization. However, PICALM appears to show specificity for its effects on internalized cargo. Consistent with previous reports (14, 20, 27), we found that transferrin uptake was unaffected by PICALM manipulation. This specificity suggests that PICALM may be a potential target for intervention to prevent disease pathogenesis; however, much more characterization of the effects of PICALM on internalization of other cargo is needed.
Many adaptor proteins with a phosphotyrosine binding domain, including Fe65, X11/Mint, Disabled-1 (Dab-1), and JNK-interacting protein family members, have been shown to bind to the YENPTY internalization motif within the cytoplasmic domain of APP (30). Overexpression or knock-out of some of these adaptors affects Aβ generation and deposition in the brains of transgenic mice, suggesting a physiological role for these proteins in regulating APP processing. Of interest, Fe65 has been shown to act as a functional linker, which brings lipoprotein receptor-related protein and APP into a protein complex, in modulating APP trafficking and Aβ production (31). JNK-interacting protein-1, a scaffolding protein for the JNK kinase cascade, has also been suggested to serve as a bridge between the kinesin light chain subunit of kinesin-1 and APP to mediate the intracellular trafficking of APP (32). Although PICALM clearly co-localizes with APP in the endocytosed vesicles of the cell and modulation of PICALM expression alters Aβ generation, we have been unable to demonstrate a direct interaction between PICALM and APP as assayed by co-immunoprecipitation (data not shown), suggesting a low affinity or indirect interaction of these two proteins. Future studies will determine whether a bridge protein links PICALM in modulating APP trafficking.
In contrast to our finding that PICALM knockdown reduced Aβ production in vitro, a previous study demonstrated that PICALM knockdown had no effect on Aβ (33). Differences in cell lines, APP expression, efficiency of transduction, and efficacy of knockdown may account for this discrepancy. Although we used N2a-APP695 cells (mouse neuroblastoma cells expressing wild type APP695), Wu et al. (33) used SHSY5Y-APPsw (human neuroblastoma cells expressing APP with the K670N/M671L Swedish mutation). Previous studies have shown that the cellular sites for processing Swedish APP may be different from those of wild type APP (34–36).
PICALM manipulation resulted in significant changes in Aβ levels both in vitro and in vivo; however, statistically significant changes in α- and β-CTF were not observed. Although β-CTF is generated in concert with the initial cleavage of APP on the path toward Aβ generation, steady-state levels of these peptides depend not only on production but also on clearance, which are very different between CTFs and Aβ. β-CTF undergoes further cleavages mediated by a variety of proteases, including the γ-secretase complex (3). Aβ is one of the possible products of β-CTF cleavage. In addition, cellular release of the two peptides is very different. Thus, a one-to-one stoichiometry between β-CTF and Aβ is not expected. The discrepancy between Aβ and β-CTF might also be due to limited numbers of samples. Additionally, the detection HJ5.1 antibody (Aβ13–28) used in the ELISA might react with the α- and γ-secretase-cleaved p3 peptide (Aβ17–40/42) in addition to Aβ, which is likely to underestimate the effect of PICALM gene manipulation in both the in vitro and in vivo experiments.
We have shown that PICALM manipulation also affects plaque burden in APP/PS1 transgenic mice. These results demonstrate the importance of clathrin-mediated endocytosis of APP in Aβ production and plaque accumulation. Inhibition of PICALM decreased soluble Aβ in the hippocampus as reflected by the PBS-soluble fraction, suggesting that the production of Aβ in these mice is reduced. Insoluble Aβ as reflected by guanidine extraction was also altered following PICALM manipulation, suggesting that PICALM may play a role in promoting amyloid plaque formation and Aβ deposition in AD. This is in agreement with the concept that conversion from normal soluble Aβ to insoluble amyloid plaques appears to be concentration-dependent such that high levels of peptide are more likely to be converted into multimeric oligomers or plaques (37).
We have found that PICALM is predominantly expressed in neurons in both the cortex and hippocampus in agreement with others (38, 39). APP is also mainly expressed in neurons, and Aβ is primarily produced by neurons (40). In our studies, viral gene transfer with AAV8 led to predominantly neuronal transduction: AAV8 is known to have a propensity for neurons in brain (41). Indeed, examination of GFP (or the HA tag) driven by the PGK promoter in the viral vectors consistently demonstrates labeling in neurons within the hippocampus (cornu ammonis layers and dentate). Thus, the manipulation of PICALM is likely to have occurred in neurons. These results add further support to the idea that at least a significant portion of Aβ in plaques originates in neurons.
PICALM is localized throughout a neuron, including at the presynaptic terminal (42). Synaptic transmission involves synaptic vesicle exocytosis followed by clathrin-mediated endocytosis of the vesicular membrane. This endocytosis is associated with increased APP internalization and elevated Aβ generation (6, 43). Given that PICALM mediates APP endocytosis in neurons, it may play a particular role in synapse-dependent Aβ generation.
The SNP initially shown to have linkage to AD risk (rs3851179), which is far upstream (∼88 kb) of the PICALM coding region (9), has been independently confirmed by several other groups (11, 28, 44). Moreover, significant associations with several other SNPs closer to PICALM have been demonstrated, including rs561655 (19 kb upstream) (45) and rs541458 (7 kb upstream) (10, 45–47). Strong linkage disequilibrium spanning rs385119 to the 5′ region of PICALM adds support to the linkage to PICALM. No other gene resides in this linkage disequilibrium region, indicating that rs385119 or any SNP in linkage disequilibrium with it could be the genetic variant driving the association. This linkage disequilibrium structure points to PICALM as the most likely candidate gene. The results in the current study add support to this contention.
Our cell model of APP processing and animal model of plaque pathogenesis rely on overexpression of human APP as endogenous mouse Aβ expressed in N2a cells is undetectable by our ELISA and does not result in amyloid plaque pathogenesis in wild type mice. Thus, our conclusions are based on APP-overexpressing models. Future studies will need to examine the effect of PICALM manipulation on endogenously expressed APP.
Although our cell and animal model study suggests that PICALM plays an important role in APP endocytosis, processing, and Aβ production, two recent studies report conflicting results regarding the influence of PICALM SNPs on CSF Aβ levels (47, 48). Our study did not assess the specific role of the SNP but rather examined the effect of gross changes in PICALM expression. Further studies will be required to understand the precise functional consequence of SNP rs3851179.
Another recent study using a yeast model of Aβ toxicity revealed that PICALM may play a role in modulating Aβ toxicity by impairing endocytosis of plasma membrane receptors (42). In this model, Aβ expression was directly targeted to the endoplasmic reticulum unlike in mammalian cells where APP in the plasma membrane is endocytosed and proteolytically processed into Aβ in endosomes (2, 5, 6). Although this report focused on Aβ-induced cytotoxicity, the authors did not directly examine the effect of PICALM on APP endocytosis and processing. It is possible that PICALM may be involved in multiple mechanisms in AD pathogenesis.
In summary, our study demonstrates that PICALM plays an important role in APP endocytosis with a resulting impact on Aβ production and release in cell culture models and APP/PS1 mice. Manipulation of PICALM expression also results in a change in amyloid plaque load in these mice. Therefore, PICALM appears to be a critical regulator of APP endocytosis, processing, Aβ production, and amyloid plaque pathogenesis. These findings are consistent with the initial discovery that PICALM SNPs are associated with AD risk and suggest a molecular mechanism for this effect. Although blocking clathrin-mediated endocytosis is not a viable therapeutic option given its ubiquitous role in cellular function, further definition of the specific pathways involved in APP endocytosis and processing may reveal other potential targets for intervention.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 NS67905, P01 NS32636 (to J. M. L.), and K01 AG029524 (to J. R. C). This work was also supported by a grant from the Charles F. and Joanne Knight Alzheimer's Disease Research Center at Washington University (to J. R. C).
T. Maeda, personal communication.
- AD
- Alzheimer disease
- PICALM
- phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia
- APP
- amyloid precursor protein
- Aβ
- amyloid β peptide
- CTF
- C-terminal fragment
- AAV
- adeno-associated virus
- PGK
- phosphoglycerate kinase
- eGFP
- enhanced GFP
- NHS
- N-hydroxysuccinimide
- GFAP
- glial fibrillary acidic protein
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 2. Vetrivel K. S., Thinakaran G. (2006) Amyloidogenic processing of β-amyloid precursor protein in intracellular compartments. Neurology 66, Suppl. 1, S69–S73 [DOI] [PubMed] [Google Scholar]
- 3. Thinakaran G., Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nordstedt C., Caporaso G. L., Thyberg J., Gandy S. E., Greengard P. (1993) Identification of the Alzheimer β/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J. Biol. Chem. 268, 608–612 [PubMed] [Google Scholar]
- 5. Koo E. H., Squazzo S. L. (1994) Evidence that production and release of amyloid β-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389 [PubMed] [Google Scholar]
- 6. Cirrito J. R., Kang J. E., Lee J., Stewart F. R., Verges D. K., Silverio L. M., Bu G., Mennerick S., Holtzman D. M. (2008) Endocytosis is required for synaptic activity-dependent release of amyloid-β in vivo. Neuron 58, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marsh M., McMahon H. T. (1999) The structural era of endocytosis. Science 285, 215–220 [DOI] [PubMed] [Google Scholar]
- 8. Maycox P. R., Link E., Reetz A., Morris S. A., Jahn R. (1992) Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. J. Cell Biol. 118, 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Williams A., Jones N., Thomas C., Stretton A., Morgan A. R., Lovestone S., Powell J., Proitsi P., Lupton M. K., Brayne C., Rubinsztein D. C., Gill M., Lawlor B., Lynch A., Morgan K., Brown K. S., Passmore P. A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A. D., Love S., Kehoe P. G., Hardy J., Mead S., Fox N., Rossor M., Collinge J., Maier W., Jessen F., Schürmann B., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frölich L., Hampel H., Hüll M., Rujescu D., Goate A. M., Kauwe J. S., Cruchaga C., Nowotny P., Morris J. C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P. P., Van Broeckhoven C., Livingston G., Bass N. J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C. E., Tsolaki M., Singleton A. B., Guerreiro R., Mühleisen T. W., Nöthen M. M., Moebus S., Jöckel K. H., Klopp N., Wichmann H. E., Carrasquillo M. M., Pankratz V. S., Younkin S. G., Holmans P. A., O'Donovan M., Owen M. J., Williams J. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambert J. C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M. J., Tavernier B., Letenneur L., Bettens K., Berr C., Pasquier F., Fiévet N., Barberger-Gateau P., Engelborghs S., De Deyn P., Mateo I., Franck A., Helisalmi S., Porcellini E., Hanon O., European Alzheimer's Disease Initiative Investigators, de Pancorbo M. M., Lendon C., Dufouil C., Jaillard C., Leveillard T., Alvarez V., Bosco P., Mancuso M., Panza F., Nacmias B., Bossù P., Piccardi P., Annoni G., Seripa D., Galimberti D., Hannequin D., Licastro F., Soininen H., Ritchie K., Blanché H., Dartigues J. F., Tzourio C., Gut I., Van Broeckhoven C., Alpérovitch A., Lathrop M., Amouyel P. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41, 1094–1099 [DOI] [PubMed] [Google Scholar]
- 11. Seshadri S., Fitzpatrick A. L., Ikram M. A., DeStefano A. L., Gudnason V., Boada M., Bis J. C., Smith A. V., Carassquillo M. M., Lambert J. C., Harold D., Schrijvers E. M., Ramirez-Lorca R., Debette S., Longstreth W. T., Jr., Janssens A. C., Pankratz V. S., Dartigues J. F., Hollingworth P., Aspelund T., Hernandez I., Beiser A., Kuller L. H., Koudstaal P. J., Dickson D. W., Tzourio C., Abraham R., Antunez C., Du Y., Rotter J. I., Aulchenko Y. S., Harris T. B., Petersen R. C., Berr C., Owen M. J., Lopez-Arrieta J., Varadarajan B. N., Becker J. T., Rivadeneira F., Nalls M. A., Graff-Radford N. R., Campion D., Auerbach S., Rice K., Hofman A., Jonsson P. V., Schmidt H., Lathrop M., Mosley T. H., Au R., Psaty B. M., Uitterlinden A. G., Farrer L. A., Lumley T., Ruiz A., Williams J., Amouyel P., Younkin S. G., Wolf P. A., Launer L. J., Lopez O. L., van Duijn C. M., Breteler M. M. (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., Abraham R., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Jones N., Stretton A., Thomas C., Richards A., Ivanov D., Widdowson C., Chapman J., Lovestone S., Powell J., Proitsi P., Lupton M. K., Brayne C., Rubinsztein D. C., Gill M., Lawlor B., Lynch A., Brown K. S., Passmore P. A., Craig D., McGuinness B., Todd S., Holmes C., Mann D., Smith A. D., Beaumont H., Warden D., Wilcock G., Love S., Kehoe P. G., Hooper N. M., Vardy E. R., Hardy J., Mead S., Fox N. C., Rossor M., Collinge J., Maier W., Jessen F., Rüther E., Schürmann B., Heun R., Kölsch H., van den Bussche H., Heuser I., Kornhuber J., Wiltfang J., Dichgans M., Frölich L., Hampel H., Gallacher J., Hüll M., Rujescu D., Giegling I., Goate A. M., Kauwe J. S., Cruchaga C., Nowotny P., Morris J. C., Mayo K., Sleegers K., Bettens K., Engelborghs S., De Deyn P. P., Van Broeckhoven C., Livingston G., Bass N. J., Gurling H., McQuillin A., Gwilliam R., Deloukas P., Al-Chalabi A., Shaw C. E., Tsolaki M., Singleton A. B., Guerreiro R., Mühleisen T. W., Nöthen M. M., Moebus S., Jöckel K. H., Klopp N., Wichmann H. E., Pankratz V. S., Sando S. B., Aasly J. O., Barcikowska M., Wszolek Z. K., Dickson D. W., Graff-Radford N. R., Petersen R. C., van Duijn C. M., Breteler M. M., Ikram M. A., DeStefano A. L., Fitzpatrick A. L., Lopez O., Launer L. J., Seshadri S., Berr C., Campion D., Epelbaum J., Dartigues J. F., Tzourio C., Alpérovitch A., Lathrop M., Feulner T. M., Friedrich P., Riehle C., Krawczak M., Schreiber S., Mayhaus M., Nicolhaus S., Wagenpfeil S., Steinberg S., Stefansson H., Stefansson K., Snaedal J., Björnsson S., Jonsson P. V., Chouraki V., Genier-Boley B., Hiltunen M., Soininen H., Combarros O., Zelenika D., Delepine M., Bullido M. J., Pasquier F., Mateo I., Frank-Garcia A., Porcellini E., Hanon O., Coto E., Alvarez V., Bosco P., Siciliano G., Mancuso M., Panza F., Solfrizzi V., Nacmias B., Sorbi S., Bossù P., Piccardi P., Arosio B., Annoni G., Seripa D., Pilotto A., Scarpini E., Galimberti D., Brice A., Hannequin D., Licastro F., Jones L., Holmans P. A., Jonsson T., Riemenschneider M., Morgan K., Younkin S. G., Owen M. J., O'Donovan M., Amouyel P., Williams J. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dreyling M. H., Martinez-Climent J. A., Zheng M., Mao J., Rowley J. D., Bohlander S. K. (1996) The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc. Natl. Acad. Sci. U.S.A. 93, 4804–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tebar F., Bohlander S. K., Sorkin A. (1999) Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell 10, 2687–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meyerholz A., Hinrichsen L., Groos S., Esk P. C., Brandes G., Ungewickell E. J. (2005) Effect of clathrin assembly lymphoid myeloid leukemia protein depletion on clathrin coat formation. Traffic 6, 1225–1234 [DOI] [PubMed] [Google Scholar]
- 16. Sorkina T., Bild A., Tebar F., Sorkin A. (1999) Clathrin, adaptors and eps15 in endosomes containing activated epidermal growth factor receptors. J. Cell Sci. 112, 317–327 [DOI] [PubMed] [Google Scholar]
- 17. Wendland B., Emr S. D. (1998) Pan1p, yeast eps15, functions as a multivalent adaptor that coordinates protein-protein interactions essential for endocytosis. J. Cell Biol. 141, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang B., Koh Y. H., Beckstead R. B., Budnik V., Ganetzky B., Bellen H. J. (1998) Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 19. Nonet M. L., Holgado A. M., Brewer F., Serpe C. J., Norbeck B. A., Holleran J., Wei L., Hartwieg E., Jorgensen E. M., Alfonso A. (1999) UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol. Biol. Cell 10, 2343–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harel A., Wu F., Mattson M. P., Morris C. M., Yao P. J. (2008) Evidence for CALM in directing VAMP2 trafficking. Traffic 9, 417–429 [DOI] [PubMed] [Google Scholar]
- 21. Miller S. E., Sahlender D. A., Graham S. C., Höning S., Robinson M. S., Peden A. A., Owen D. J. (2011) The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 147, 1118–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lakshmana M. K., Yoon I. S., Chen E., Bianchi E., Koo E. H., Kang D. E. (2009) Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid β peptide generation. J. Biol. Chem. 284, 11863–11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., Younkin S. G., Borchelt D. R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 13, 159–170 [DOI] [PubMed] [Google Scholar]
- 24. Yan P., Xu J., Li Q., Chen S., Kim G. M., Hsu C. Y., Xu X. M. (1999) Glucocorticoid receptor expression in the spinal cord after traumatic injury in adult rats. J. Neurosci. 19, 9355–9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bushlin I., Petralia R. S., Wu F., Harel A., Mughal M. R., Mattson M. P., Yao P. J. (2008) Clathrin assembly protein AP180 and CALM differentially control axogenesis and dendrite outgrowth in embryonic hippocampal neurons. J. Neurosci. 28, 10257–10271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudinskiy N., Grishchuk Y., Vaslin A., Puyal J., Delacourte A., Hirling H., Clarke P. G., Luthi-Carter R. (2009) Calpain hydrolysis of α- and β2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J. Biol. Chem. 284, 12447–12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang F., Khvorova A., Marshall W., Sorkin A. (2004) Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J. Biol. Chem. 279, 16657–16661 [DOI] [PubMed] [Google Scholar]
- 28. Jun G., Naj A. C., Beecham G. W., Wang L. S., Buros J., Gallins P. J., Buxbaum J. D., Ertekin-Taner N., Fallin M. D., Friedland R., Inzelberg R., Kramer P., Rogaeva E., St George-Hyslop P., Alzheimer's Disease Genetics Consortium, Cantwell L. B., Dombroski B. A., Saykin A. J., Reiman E. M., Bennett D. A., Morris J. C., Lunetta K. L., Martin E. R., Montine T. J., Goate A. M., Blacker D., Tsuang D. W., Beekly D., Cupples L. A., Hakonarson H., Kukull W., Foroud T. M., Haines J., Mayeux R., Farrer L. A., Pericak-Vance M. A., Schellenberg G. D. (2010) Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carey R. M., Balcz B. A., Lopez-Coviella I., Slack B. E. (2005) Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid β protein. BMC Cell Biol. 6, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taru H., Suzuki T. (2009) Regulation of the physiological function and metabolism of AβPP by AβPP binding proteins. J. Alzheimers Dis. 18, 253–265 [DOI] [PubMed] [Google Scholar]
- 31. Pietrzik C. U., Yoon I. S., Jaeger S., Busse T., Weggen S., Koo E. H. (2004) FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J. Neurosci. 24, 4259–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuda S., Matsuda Y., D'Adamio L. (2003) Amyloid β protein precursor (AβPP), but not AβPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J. Biol. Chem. 278, 38601–38606 [DOI] [PubMed] [Google Scholar]
- 33. Wu F., Matsuoka Y., Mattson M. P., Yao P. J. (2009) The clathrin assembly protein AP180 regulates the generation of amyloid-β peptide. Biochem. Biophys. Res. Commun. 385, 247–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haass C., Lemere C. A., Capell A., Citron M., Seubert P., Schenk D., Lannfelt L., Selkoe D. J. (1995) The Swedish mutation causes early-onset Alzheimer's disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1, 1291–1296 [DOI] [PubMed] [Google Scholar]
- 35. Perez R. G., Squazzo S. L., Koo E. H. (1996) Enhanced release of amyloid β-protein from codon 670/671 “Swedish” mutant β-amyloid precursor protein occurs in both secretory and endocytic pathways. J. Biol. Chem. 271, 9100–9107 [DOI] [PubMed] [Google Scholar]
- 36. Sodhi C. P., Rampalli S., Perez R. G., Koo E. H., Quinn B., Gottardi-Littell N. R. (2004) The endocytotic pathway is required for increased Aβ42 secretion during apoptosis. Brain Res. Mol. Brain Res. 128, 201–211 [DOI] [PubMed] [Google Scholar]
- 37. Lomakin A., Teplow D. B., Kirschner D. A., Benedek G. B. (1997) Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. U.S.A. 94, 7942–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao P. J., Zhang P., Mattson M. P., Furukawa K. (2003) Heterogeneity of endocytic proteins: distribution of clathrin adaptor proteins in neurons and glia. Neuroscience 121, 25–37 [DOI] [PubMed] [Google Scholar]
- 39. Baig S., Joseph S. A., Tayler H., Abraham R., Owen M. J., Williams J., Kehoe P. G., Love S. (2010) Distribution and expression of picalm in Alzheimer disease. J. Neuropathol. Exp. Neurol. 69, 1071–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. LeBlanc A. C., Papadopoulos M., Bélair C., Chu W., Crosato M., Powell J., Goodyer C. G. (1997) Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J. Neurochem. 68, 1183–1190 [DOI] [PubMed] [Google Scholar]
- 41. Klein R. L., Dayton R. D., Tatom J. B., Henderson K. M., Henning P. P. (2008) AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol. Ther. 16, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Treusch S., Hamamichi S., Goodman J. L., Matlack K. E., Chung C. Y., Baru V., Shulman J. M., Parrado A., Bevis B. J., Valastyan J. S., Han H., Lindhagen-Persson M., Reiman E. M., Evans D. A., Bennett D. A., Olofsson A., DeJager P. L., Tanzi R. E., Caldwell K. A., Caldwell G. A., Lindquist S. (2011) Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science 334, 1241–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marquez-Sterling N. R., Lo A. C., Sisodia S. S., Koo E. H. (1997) Trafficking of cell-surface β-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J. Neurosci. 17, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carrasquillo M. M., Belbin O., Hunter T. A., Ma L., Bisceglio G. D., Zou F., Crook J. E., Pankratz V. S., Dickson D. W., Graff-Radford N. R., Petersen R. C., Morgan K., Younkin S. G. (2010) Replication of CLU, CR1, and PICALM associations with Alzheimer disease. Arch. Neurol. 67, 961–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., Gallins P. J., Buxbaum J. D., Jarvik G. P., Crane P. K., Larson E. B., Bird T. D., Boeve B. F., Graff-Radford N. R., De Jager P. L., Evans D., Schneider J. A., Carrasquillo M. M., Ertekin-Taner N., Younkin S. G., Cruchaga C., Kauwe J. S., Nowotny P., Kramer P., Hardy J., Huentelman M. J., Myers A. J., Barmada M. M., Demirci F. Y., Baldwin C. T., Green R. C., Rogaeva E., St George-Hyslop P., Arnold S. E., Barber R., Beach T., Bigio E. H., Bowen J. D., Boxer A., Burke J. R., Cairns N. J., Carlson C. S., Carney R. M., Carroll S. L., Chui H. C., Clark D. G., Corneveaux J., Cotman C. W., Cummings J. L., DeCarli C., DeKosky S. T., Diaz-Arrastia R., Dick M., Dickson D. W., Ellis W. G., Faber K. M., Fallon K. B., Farlow M. R., Ferris S., Frosch M. P., Galasko D. R., Ganguli M., Gearing M., Geschwind D. H., Ghetti B., Gilbert J. R., Gilman S., Giordani B., Glass J. D., Growdon J. H., Hamilton R. L., Harrell L. E., Head E., Honig L. S., Hulette C. M., Hyman B. T., Jicha G. A., Jin L. W., Johnson N., Karlawish J., Karydas A., Kaye J. A., Kim R., Koo E. H., Kowall N. W., Lah J. J., Levey A. I., Lieberman A. P., Lopez O. L., Mack W. J., Marson D. C., Martiniuk F., Mash D. C., Masliah E., McCormick W. C., McCurry S. M., McDavid A. N., McKee A. C., Mesulam M., Miller B. L., Miller C. A., Miller J. W., Parisi J. E., Perl D. P., Peskind E., Petersen R. C., Poon W. W., Quinn J. F., Rajbhandary R. A., Raskind M., Reisberg B., Ringman J. M., Roberson E. D., Rosenberg R. N., Sano M., Schneider L. S., Seeley W., Shelanski M. L., Slifer M. A., Smith C. D., Sonnen J. A., Spina S., Stern R. A., Tanzi R. E., Trojanowski J. Q., Troncoso J. C., Van Deerlin V. M., Vinters H. V., Vonsattel J. P., Weintraub S., Welsh-Bohmer K. A., Williamson J., Woltjer R. L., Cantwell L. B., Dombroski B. A., Beekly D., Lunetta K. L., Martin E. R., Kamboh M. I., Saykin A. J., Reiman E. M., Bennett D. A., Morris J. C., Montine T. J., Goate A. M., Blacker D., Tsuang D. W., Hakonarson H., Kukull W. A., Foroud T. M., Haines J. L., Mayeux R., Pericak-Vance M. A., Farrer L. A., Schellenberg G. D. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corneveaux J. J., Myers A. J., Allen A. N., Pruzin J. J., Ramirez M., Engel A., Nalls M. A., Chen K., Lee W., Chewning K., Villa S. E., Meechoovet H. B., Gerber J. D., Frost D., Benson H. L., O'Reilly S., Chibnik L. B., Shulman J. M., Singleton A. B., Craig D. W., Van Keuren-Jensen K. R., Dunckley T., Bennett D. A., De Jager P. L., Heward C., Hardy J., Reiman E. M., Huentelman M. J. (2010) Association of CR1, CLU and PICALM with Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum. Mol. Genet. 19, 3295–3301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schjeide B. M., Schnack C., Lambert J. C., Lill C. M., Kirchheiner J., Tumani H., Otto M., Tanzi R. E., Lehrach H., Amouyel P., von Arnim C. A., Bertram L. (2011) The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch. Gen. Psychiatry 68, 207–213 [DOI] [PubMed] [Google Scholar]
- 48. Kauwe J. S., Cruchaga C., Karch C. M., Sadler B., Lee M., Mayo K., Latu W., Su'a M., Fagan A. M., Holtzman D. M., Morris J. C., Alzheimer's Disease Neuroimaging Initiative, and Goate A. M. (2011) Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer's disease. PLoS One 6, e15918. [DOI] [PMC free article] [PubMed] [Google Scholar]