

Background: DNA double-stranded break (DSB) repair is critical for the maintenance of genome stability and prevention of cancer.
Results: Dimerization of CtIP, a critical DSB repair protein, is important for its localization to chromosomal DSBs in mammalian cells.
Conclusion: CtIP dimerization is required for DSB repair.
Significance: These studies help to understand the molecular mechanisms of DSB repair in mammalian cells.
Keywords: DNA Damage Response, DNA Repair, Genomic Instability, Homologous Recombination, Mammal, CtIP, Dimerization, End Resection
Abstract
CtIP (CtBP-interacting protein) associates with BRCA1 and the Mre11-Rad50-Nbs1 (MRN) complex and plays an essential role in homologous recombination (HR)-mediated DNA double-stranded break (DSB) repair. It has been described that CtIP forms dimers in mammalian cells, but the biological significance is not clear. In this study, we identified a conserved motif in the N terminus of CtIP, which is required for dimer formation. We further showed that CtIP mutants impaired in forming dimers are strongly defective in HR, end resection, and activation of the ataxia telangiectasia and Rad3-related pathway, without notable change of CtIP interactions with BRCA1 or Nbs1. In addition to HR, CtIP dimerization is also required for microhomology-mediated end joining. Live cell imaging of enhanced GFP-tagged CtIP demonstrates that the CtIP dimerization mutant fails to be localized to DSBs, whereas placing a heterologous dimerization motif to the dimerization mutant restores CtIP recruitment to DSBs. These studies suggest that CtIP dimer formation is essential for its recruitment to DSBs on chromatin upon DNA damage. Furthermore, DNA damage-induced phosphorylation of CtIP is significantly reduced in the CtIP dimerization mutants. Therefore, in addition to the C-terminal conserved domains critical for CtIP function, the dimerization motif on the N terminus of CtIP is also conserved and essential for its function in DNA damage responses. The severe repair defects of CtIP dimerization mutants are likely due to the failure in localization to chromosomal DSBs upon DNA damage.
Introduction
In response to DNA DSBs,2 the Mre11-Rad50-Nbs1 (MRN) complex is immediately recruited to DNA ends, and this recruitment is critical for ataxia telangiectasia mutated (ATM) activation to initiate various DNA damage responses, including activating homologous recombination (HR)-mediated DSB repair (1, 2). CtIP was originally identified as a transcriptional co-repressor and a tumor suppressor, and more recent work suggests that it plays a critical role in DNA damage responses (3, 4). CtIP is recruited to DNA damage sites and cooperates with MRN to promote HR-mediated DSB repair (5–7). CtIP binds to BRCA1 in a CDK-dependent manner, and formation of the functional complex MRN-CtIP-BRCA1 is important for end resection to initiate HR-mediated DSB repair (5, 6, 8–11). Consistent with its role in end resection, CtIP is also required for DSB-induced replication protein A (RPA) foci formation and ATR activation. CtIP deficiency causes sensitivity to various DNA-damaging agents such as IR and camptothecin (CPT) (11, 12).
CtIP is a homologue of DNA repair protein Sae2 in Saccharomyces cerevisiae and Ctp1 in Schizosaccharomyces pombe (6, 12). The sequence homology with Sae2 and Ctp1 is poor, and the most conserved region is located at the C terminus of CtIP, described as the Sae2-like domain (6). However, CtIP shares many functional similarities with Sae2 and Ctp1 (6, 12, 13). Like Sae2 and Ctp1, CtIP interacts with MRN and functions together with MRN to promote end resection for HR-mediated DSB repair (14). CtIP and Sae2 are both phosphorylated by CDKs, and these CDK-mediated phosphorylation events are important for regulating CtIP activity in end resection (6, 11, 15). In a genetic screening of sae2 mutants that fail to activate the Tel1/Mre11-dependent DNA damage signaling pathway, a sae2 mutant allele L25P/E171G was identified with a strong phenotype in various DNA damage responses to a level similar to that of the sae2 null mutant (16). Sae2-L25P/E171G is highly sensitive to methyl methanesulfonate and CPT, shows synthetic lethality with rad27Δ and exhibits meiosis defect. Subsequent analysis revealed that this mutant allele is defective in forming Sae2 oligomers (16). It was also described that human CtIP forms a homodimer through the N terminus of CtIP (17), but the biological function of CtIP dimerization remains unknown in mammalian cells.
In this study, we identified a conserved motif present at the N terminus of human CtIP, which is critical for CtIP dimerization. We showed that the CtIP dimerization mutants are severely defective in HR-mediated DSB repair and are sensitive to DNA-damaging agents. Furthermore, DNA end resection is defective in the CtIP dimerization mutants, and consequently ATR activation is also impaired. In addition to HR, we demonstrated that these mutants are also deficient in using microhomology-mediated end joining (MMEJ) to repair DSBs. Although CtIP is not required for ATM activation, IR-induced CtIP phosphorylation is impaired in the dimerization mutants. By using live cell imaging of fluorescent-tagged proteins, we found that CtIP dimerization is required for its recruitment to DSBs on chromatin. The defect in localizing CtIP dimerization mutants to DSBs thus underlies the severe defects of these mutants in mediating DNA damage responses.
EXPERIMENTAL PROCEDURES
Antibodies and Plasmids Generated
CtIP polyclonal antibodies were generated by immunizing rabbits with GST-fused CtIP fragments covering amino acids 1–220 or 631–897 and were affinity-purified. Other antibodies used include anti-FLAG-M2 (Sigma), anti-Myc-9E10 (Novus Biologicals), anti-HA-11 (Covance), anti-Ku70 (Santa Cruz Biotechnology), anti-RPA2 (Oncogene), anti-H2AX-S319-p, anti-Chk1-S317-p (Cell Signaling Technology), anti-Chk1 (R & D Systems), anti-Chk2-T68 (GeneTex), and anti-Chk2 (Santa Cruz Biotechnology).
All CtIP point or deletion mutants were generated by using the QuikChange site-directed mutagenesis kit (Stratagene) or by PCR amplification and ligation into pcDNA3 or Babe-puro vectors containing HA-, Myc-, or FLAG-tagged epitopes. Recombinant baculoviruses expressing GST- or FLAG-tagged CtIP or Nbs1 were generated by using Bac-to-Bac baculovirus expression systems (Invitrogen). EGFP-tagged CtIP or indicated mutants were generated using EGFP-C1 expression vector (Clontech). To make GCN4-CtIP LKEX4Emut, two pairs of annealed oligomers (GCN4BamHIF 5′-GATCCAGAATGAAACAACTTGAAGACAAGGTTGAAGAATTGCTTTCGAAAAATTATCACTTGGAA, GCN4BamHIR 5′-ACCTCATTTTCCAAGTGATAATTTTTCGAAAGCAATTCTTCAACCTTGTCTTCAAGTTGTTTCATTCTG and GCN4BglIIF 5′-AATGAGGTTGCCAGATTAAAGAAATTAGTTGGCGAACGCAACGCCGCCATACGTAGTA, GCN4BglIIR 5′-GATCTACTACGTATGGCGGCGTTGCGTTCGCCAACTAATTTCTTTAATCTGGCA) were inserted into the BamHI site of HA-CtIP LKEX4Emut plasmid. The final construct encodes for a 33-amino acid GCN4 dimerization domain and a six-amino acid linker NAAIRS (18) between HA tag and CtIP (supplemental Fig. S4).
Cell Culture and RNA Interference
Human U2OS and 293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum in the presence of antibiotics. Sf21 insect cells were cultured in Grace's insect medium supplemented with 10% fetal bovine serum. Silencing of endogenous CtIP was performed as described (5, 9). Exogenous CtIP wild type (1–897) or indicated mutants generated above also contained shRNA and siRNA-resistant silent mutation sites, which were introduced by site-directed mutagenesis.
Immunoprecipitation and Immunofluorescence
Immunoprecipitation and Western blot analysis were performed as described (9, 19). The cells were lysed with NETN buffer (20 mm Tris, pH 8.0, 1 mm EDTA, 150 mm NaCl, and 0.5% Nonidet P-40) for 30 min. Cleared cell lysates were then collected for immunoprecipitation or boiled in 2× SDS loading buffer and subjected to SDS-PAGE. For immunostaining, the cells were fixed with 4% paraformaldehyde, followed by immunostaining analysis as described (9, 19). Fluorescence microscopy was performed using a Nikon Eclipse E800 fluorescence microscope and SPOT Advanced imaging software.
HR and MMEJ Assay
EGFP-based repair substrates to assay for DSB repair mediated by HR (EGFP-HR, shown in Fig. 3B) or by MMEJ (EGFP-MMEJ, shown in Fig. 5A) were generated as follows. For EGFP-HR construct, a full-length EGFP cassette was disrupted by insertion of the 18-bp I-SceI cleavage recognition site containing two in-frame stop codons in EGFP ORF (supplemental Fig. S1A), followed by insertion of an inactive, truncated EGFP donor fragment downstream of hygromycin resistance marker. For EGFP-MMEJ, a full-length EGFP cassette was inactivated by inserting a 27-bp oligonucleotide containing an I-SceI cleavage site flanked on both sides by 9-bp microhomology sequence. This was achieved by duplicating the 9-bp sequence present in EGFP. U2OS cells were stably integrated with EGFP-HR and EGFP-MMEJ repair substrates by transfection using FuGENE (Roche Applied Science) followed by hygromycin drug selection, and single clones were selected and confirmed by Southern blotting for single-copy integration.
FIGURE 3.

Dimerization of CtIP is important for HR-mediated DSB repair. A, cell survival assay after exposing cells expressing HA-CtIP 1–897 or indicated mutant to various doses of camptothecin. Averages and standard deviations (error bars) of three independent experiments are shown. B, schematic representation of the EGFP-based HR repair assay substrate, EGFP-HR (left panel). U2OS EGFP-HR cells were stably expressed with HA-CtIP wild type or indicated mutants by retroviral infection, with endogenous CtIP silenced by siRNA and shRNA. I-SceI was induced by transfection of I-SceI-IRES-dsRedNLS, and 72 h after transfection, the cells were trypsinized, and FACS analysis was performed. The data shown represent the means of three independent experiments; the error bars indicate the standard deviation (middle panel). Western blot shows expression of HA-CtIP wild type and mutants, with Ku70 used as a loading control (right panel). C, U2OS cells stably expressing HA-CtIP wild type or indicated mutants, with endogenous CtIP silenced by RNAi, were treated with DMSO (indicated as No) or with 1 μm CPT for 2 h and then fixed for co-immunostaining with γH2AX and RPA2 antibodies (left). γH2AX signal was used as a marker for CPT-induced DSB damage. RPA2 foci-positive cells were identified as cells with noticeable γH2AX and RPA2 foci formation (>30 foci/nuclei with bright, punctate staining) after damage treatment. In the wild type CtIP cells (CtIP 1–897), the red arrows indicate typical, representative foci-positive cells (enlarged in top left panel) compared with foci-negative cells, shown by white arrows (enlarged in top left panel). In the mutant CtIP-LKEX4Emut, one RPA2 foci-positive cell is also marked with a red arrow. The percentage of RPA2 foci-positive cells among γH2AX-foci-positive cells for each sample is shown in right top panel. The data represent the means ± S.D. of three independent experiments (>100 total cells counted for each experiment). The Western blot shows the expression levels of HA-CtIP 1–897 or indicated mutants, with endogenous CtIP silenced by RNAi (bottom right panel). D, U2OS cells stably expressing HA-CtIP wild type (CtIP 1–897) or indicated mutants, with endogenous CtIP silenced, were treated with IR radiation (10 Gy) and allowed to recover for 1 h. After recovery, the cells were collected and lysed. Immunoblotting was performed with the indicated antibodies.
FIGURE 5.

The conserved LKEX4E motif of CtIP is important for MMEJ-mediated DSB repair. A, schematic of the MMEJ repair assay substrate. B, U2OS EGFP-MMEJ cells stably expressing HA-CtIP 1–897 or indicated mutants, with endogenous CtIP silenced by shRNA and siRNA, were transfected with I-SceI-IRES-dsRedNLS, and 72 h after transfection, the cells were trypsinized, and FACS analysis was performed. The data shown represent the means of three independent experiments; error bars indicate the S.D. Western blot shows expression of HA-CtIP 1–897 and mutants, with Ku70 used as a loading control.
To induce I-SceI for generation of DSBs, we developed a new I-SceI-IRES-dsRedNLS construct designed to control for transfection (supplemental Fig. S1B). The construct contains an internal ribosome entry site (IRES) of the encephalomyocarditis virus (20–22), which permits both I-SceI and dsRedNLS (dsRed with a nuclear localization sequence (NLS)) coding regions to be translated from a single mRNA. Cells expressing I-SceI-IRES-dsRedNLS can be specifically selected and gated for by FACS analysis of EGFP-HR and EGFP-MMEJ repair assay. The percentage of I-SceI-induced DSB repair is determined by the ratio of cells exhibiting both green and red signals to all red cells, as revealed by FACS analysis. Cells with both green and red signals and with red signals only are shown in supplemental Fig. S1C.
U2OS EGFP-HR and EGFP-MMEJ cells were stably expressed with shRNA- and siRNA-resistant wild type HA-tagged CtIP or CtIP indicated mutants for the dimerization motif by retroviral infection. After knockdown of endogenous CtIP by shRNA and siRNA, the cells were transfected with I-SceI-IRES-dsRedNLS and plated in nonselective medium for 72 h after transfection and then recovered by trypsinization, and the collected cells were analyzed using a BD Accuri C6 flow cytometer and the accompanying analysis software (BD Biosciences).
Protein Purification and in Vitro DNA Binding Assay
GST-CtIP wild type and the indicated mutants were expressed in Sf21 insect cells. All of the recombinant proteins were purified using glutathione-agarose resin according to the manufacturer's protocol (GE Healthcare). For in vitro DNA binding assay, DNA fragments (0.5 kb) were generated by PCR amplification of the plasmid pUC19 using a 5′ biotinylated forward oligonucleotide primer and an unlabeled reverse primer. 200 ng of DNA fragments were first incubated with 10 μl of M-270 streptavidin Dynabeads (Invitrogen) in PBS with 0.1% Tween 20. After washing with binding buffer containing 50 mm HEPES, pH 7.5, 50 mm potassium chloride, and 5 mm magnesium chloride. DNA-containing beads were then incubated with purified CtIP wild type or mutants protein in binding buffer for 1.5 h at 4 °C. After washing, the samples were boiled with 2× SDS loading buffer and detected by immunoblotting.
In Vitro Kinase assay
For ATM in vitro kinase assays, FLAG-tagged ATM was transiently transfected into 293T cells and immunoprecipitated using anti-FLAG M2-agarose beads (Sigma). Immunoprecipitates were extensively washed and then incubated with 1.5 μg of purified GST-CtIP or equal amounts of the indicated mutant proteins in the presence of 10 μCi of [γ-32P]ATP in ATM kinase buffer (25 mm HEPES, pH 7.4, 50 mm NaCl, 10 mm MnCl2, 10 mm MgCl2, 1 mm DTT, 5 μm ATP). The kinase reaction was conducted at 30 °C for 30 min. The proteins were separated by SDS-PAGE, and radiolabeled proteins were visualized by gel scanning using a PhophorImage scanner (Typhoon Trio, GE Healthcare).
Laser Microirradiation and Live Cell Imaging
DSBs were generated in live cell nuclei by laser-induced microirradiation using a pulsed nitrogen laser (Laboratory of Dr. David J. Chen, University of Texas Southwestern Medical Center, Dallas, TX; Ref. 23) and a picosecond short pulsed green laser (Laboratory of Dr. Michael W. Berns, University of California, San Diego, CA; Refs. 7 and 24) as previously described. The laser systems were coupled to Zeiss Axiovert microscopes for live cell, time lapse image capture. Fluorescence intensities of the microirradiated area were calculated using Image J software (National Institutes of Health), with the cellular background fluorescence intensity subtracted from the laser-induced damage site intensity. Each data point is the average of 10 independent measurements. The requirement of CtIP dimerization for its recruitment to DSBs on chromatin was revealed by using both the pulsed nitrogen laser and the picosecond short pulsed green laser.
RESULTS
N Terminus of CtIP Is Important for Dimerization
To investigate CtIP dimerization, we tagged CtIP with Myc and HA and expressed them in 293T and U2OS cells. Co-immunoprecipitation showed that HA-CtIP binds to Myc-CtIP, suggesting that CtIP indeed forms dimers in mammalian cells (Fig. 1A and data not shown). Deleting the N-terminal 200 amino acids in both HA-CtIP and Myc-CtIP, or only in HA-CtIP but not Myc-CtIP, completely abolishes the dimer formation, which is consistent with previous findings that the dimerization motifs in CtIP are located at the N terminus of CtIP (Fig. 1A and Ref. 17). These studies also agree with the notion that dimerization motifs need to be present on both alleles to form dimers. Deleting the C terminus conserved Sae2-like domain (CtIP 1–796; Ref. 6) does not impair dimerization (Fig. 1B). CtIP contains two putative coiled-coil domains located near the N and C termini of CtIP, and it was described that the N terminus coiled-coil domain (spanning residues 45–160) is required for CtIP dimer formation (17). We generated a series of CtIP N terminus deletion mutants, and surprisingly we found that deleting the first 45 residues of the full length of CtIP completely abolished the dimer formation as revealed by co-immunoprecipitation of CtIP in 293T cells, whereas deleting residues 45–160 (CtIP 45–160del) containing the N terminus coiled-coil domain does not influence the dimer formation (Fig. 1C). Similar results were obtained when we co-expressed GST-fused CtIP fragments containing different N terminus deletions and FLAG-tagged N terminus CtIP 1–461 fragment in insect cells and examined the interaction by co-immunoprecipitation (Fig. 1D). These studies suggest that the N terminus 45 amino acids of CtIP, but not the N terminus coiled-coil domain, are critical for CtIP dimerization.
FIGURE 1.

CtIP N terminus residues 1–45 mediate dimerization of CtIP. A, 293T cells were co-transfected with HA-CtIP 1–897 (wild type) and Myc-CtIP 1–897 or Myc-CtIP 200–897 or co-transfected with HA-CtIP 200–897 and Myc-CtIP 1–897 or Myc-CtIP 200–897. Anti-HA immunoprecipitation (IP) was performed, followed by anti-Myc Western blotting. B, removal of N terminus but not C terminus prevents binding of Myc-CtIP with HA-CtIP. 293T cells were co-transfected with HA-CtIP and Myc-CtIP, Myc-CtIP 1–796, or Myc-CtIP 200–897. Anti-HA IP was performed, followed by anti-Myc Western blotting. C, the N terminus from amino acids 1–45, but not amino acids 45–160, is important for mediating dimerization of CtIP. HA-CtIP N-terminal deletion mutants were generated as indicated (top panel). 293T cells were co-transfected with Myc-CtIP 1–897 and HA-CtIP 1–897 or the indicated N terminus deletion mutants. Anti-Myc IP was performed, followed by anti-HA Western blotting. D, residues 1–45 are important for dimerization of N terminus CtIP in vitro. GST-CtIP N-terminal deletion mutants were generated as indicated (top panel). FLAG-tagged N-terminal CtIP 1–461 and GST-tagged N-terminal CtIP 1–461 or indicated deletion mutants were expressed in SF21 insect cells by baculovirus infection. Immunoprecipitation was performed using anti-FLAG M2 beads (Sigma) followed by anti-GST immunoblotting.
Conserved LKEX4EV Motif at N Terminus of CtIP Is Important for Dimerization
Further deletion analysis revealed that the N-terminal residues from 20 to 45 in the full length of CtIP (CtIP 20–45del) are required for dimer formation (Fig. 2B). We then performed alignment analysis of the N-terminal CtIP amino acid sequences from different species and identified a conserved LKEX4E(V/L) motif (Fig. 2A). The Sae2-L25P mutant that is defective in oligomerization contains the mutation replacing the Leu residue at the end of this motif (Fig. 2A and Ref. 16). We mutated four conserved amino acids in this motif LKEX4EV of human CtIP to PAAX4AV (Fig. 2A), and co-immunoprecipitation analysis showed that this CtIP mutant, CtIP-LKEX4Emut, completely loses its ability to form dimer (Fig. 2B). These studies suggest that the conserved LKEX4E(V/L) motif at the N terminus of CtIP is important for dimerization.
FIGURE 2.
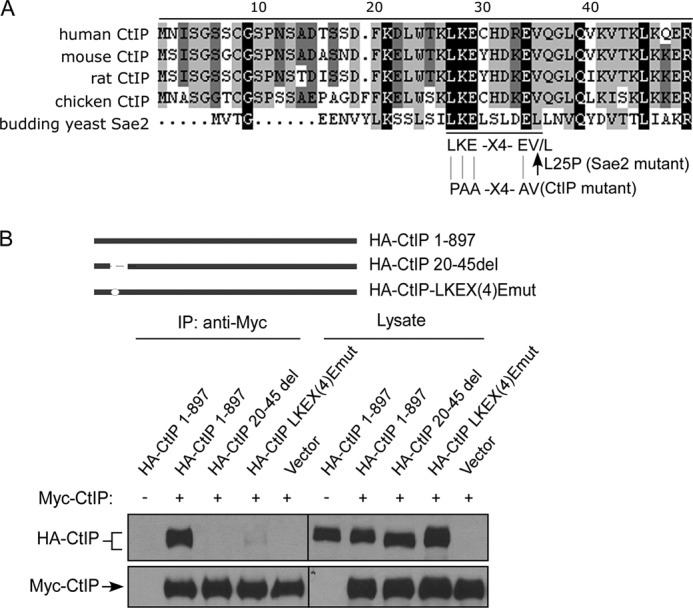
The LKEX4E motif mediates CtIP dimer formation. A, the N terminus of human CtIP is aligned with CtIP from other indicated species including CtIP yeast homologue Sae2 (S. cerevisiae), with black boxes indicating conserved residues. B, 293T cells were co-transfected with Myc-CtIP 1–897 and HA-CtIP 1–897 or the indicated mutants: CtIP 20–45del (amino acids 20–45 deleted) and CtIP LKEX4Emut (conserved residues Leu-27 mutated to proline, and Lys-28, Glu-29, and Glu-34 mutated to alanine). Anti-Myc IP was performed, followed by anti-HA Western blotting. IP, immunoprecipitation.
CtIP Dimerization Mutants Are Impaired in HR-mediated DSB Repair Because of Defects in End Resection
To examine whether CtIP dimerization is important for its repair function, we expressed HA-tagged CtIP 1–897, CtIP 20–45del, and CtIP-LKEX4Emut in U2OS cells, whereas the endogenous CtIP was inactivated by expressing shRNAs. The CtIP dimerization mutants, CtIP 20–45del and CtIP-LKEX4Emut, exhibit strong sensitivity to IR and CPT treatment to a similar level as expressing CtIP shRNAs without reconstitution (Fig. 3A and data not shown). To examine whether these dimerization mutants are defective in HR-mediated DSB repair, we used the U2OS cells carrying the EGFP-based HR substrate stably integrated in chromosomes (Fig. 3B). After inactivating endogenous CtIP, both CtIP 20–45del and CtIP-LKEX4Emut mutants show strong defects in HR-mediated DSB repair.
To further understand the mechanisms underlying the HR defects of the dimerization mutants, we asked whether end resection is defective in these mutants. U2OS cells expressing HA-tagged CtIP 1–897, CtIP 20–45del, CtIP-LKEX4Emut, or the vector control cell line were infected with shRNAs to inactivate endogenous CtIP. End resection was monitored by RPA2 foci formation after CPT treatment for 2 h. Both CtIP 20–45del and CtIP-LKEX4Emut mutants exhibited much reduced RPA2 foci formation compared with CtIP 1–897, but at a level comparable with the vector control U2OS cells expressing CtIP shRNA (Fig. 3C). Similarly, IR-induced RPA2 phosphorylation revealed by RPA2 migration shift is also impaired in the CtIP dimerization mutants CtIP 20–45del and CtIP-LKEX4Emut (supplemental Fig. S2). These studies suggest that the dimerization mutants are defective in end resection, based on RPA2 foci formation and RPA2 phosphorylation, which are used for monitoring single-stranded DNA (ssDNA) accumulation (6, 11). Consistently, ATR activation as revealed by Chk1 phosphorylation is significantly impaired in the CtIP 20–45del and CtIP-LKEX4Emut mutants, likely because of impaired accumulation of ssDNA at DSB ends (Fig. 3D). Despite essential roles of CtIP dimerization in DNA damage responses, CtIP dimer formation is not regulated by DNA damage as revealed by co-immunoprecipitation (Fig. 4A, left panel, and data not shown). Mutating two previously identified ATM phosphorylation sites, Ser-664 and Ser-745 (25), does not affect CtIP dimerization (Fig. 4A, right panel).
FIGURE 4.

The LKEX4EV motif does not affect the interaction between CtIP with BRCA1 and Nbs1. A, CtIP dimerization occurs regardless of DNA damage. 293T cells were co-transfected with HA-CtIP and Myc-CtIP. The cells were treated with IR radiation (20 Gy) and allowed to recover for 1 h. After recovery, the cells were collected and lysed. Anti-HA IP was performed, followed by anti-Myc Western blotting (left panel). 293T cells were co-transfected with Myc-CtIP 1–897 and HA-CtIP 1–897 or ATM targeted sites mutant (S664A/S745A). The cells were treated with IR radiation (20 Gy) and allowed to recover for 1 h. After recovery, the cells were collected and lysed. Anti-Myc IP was performed, followed by anti-HA Western blotting (right panel). B, 293T cells were co-transfected with Myc-BRCA1 and HA-tagged CtIP 1–897 or indicated mutants. Anti-HA IP was performed, followed by anti-Myc Western blotting. C, SF21 insect cells were co-infected with baculoviruses expressing FLAG-Nbs1 and the indicated GST-tagged CtIP constructs. GST pull-down experiments were carried out using GST beads, and immunoblotting was performed using anti-FLAG M2 antibody. IP, immunoprecipitation.
BRCA1 binds to CtIP through its BRCT domains when CtIP is phosphorylated by CDKs at Ser-327, and this interaction is believed to be important for HR (5, 9, 10). Co-immunoprecipitation revealed that the interaction of CtIP dimerization mutants CtIP 20–45del and CtIP-LKEX4Emut with BRCA1 remains at the similar levels as that of CtIP 1–897 (Fig. 4B). Furthermore, the interaction of CtIP with Nbs1 is also not changed when the CtIP dimerization mutants were used (Fig. 4C). These data suggest that CtIP dimerization is not required for its interaction with BRCA1 and Nbs1, and the impaired functions in HR and end resection observed in the CtIP dimerization mutants are probably caused by defects other than their binding to BRCA1 and Nbs1.
CtIP Dimerization Mutants Are Defective in MMEJ
MMEJ is a Ku-independent DSB repair mechanism that uses microhomologous sequences (5–25 bp) for base pairing to mediate end joining (26), and Mre11 and CtIP are both involved in MMEJ (27–30). To test whether CtIP dimerization is needed for MMEJ, we generated an EGFP-based MMEJ substrate with a 9-bp duplication of DNA sequences flanking the I-Sce1 cleavage site (Fig. 5A). Consistent with the previous findings, inactivation of CtIP by shRNAs significantly reduces MMEJ activity (supplemental Fig. S3 and Ref. 28). We further examined MMEJ in U2OS cells expressing the CtIP 20–45del and CtIP-LKEX4Emut mutants, with endogenous CtIP inactivated by CtIP shRNAs. MMEJ efficiency is much reduced in the CtIP dimerization mutants compared with CtIP 1–897 (Fig. 5B). These studies suggest that in addition to HR, the dimerization of CtIP is also required for MMEJ-mediated DSB repair.
CtIP Dimerization Mutants Fail to Be Recruited to Chromosomal DSBs
To further investigate the cause of DSB repair defects in CtIP dimerization mutants, we performed live cell imaging of CtIP recruitment to DSBs by monitoring fluorescent EGFP-tagged CtIP. Because end resection and HR are activated mainly after cells enter the S phase, we specifically monitored the recruitment of CtIP to DSBs in S phase cells. We co-expressed RFP-PCNA with EGFP-CtIP alleles, and S phase cells are marked by PCNA foci (31). A series of N terminus CtIP deletion mutants including CtIP 45–897, CtIP 75–897, and CtIP 135–897, which are defective in CtIP dimerization, were examined, and all of these mutants are significantly impaired in localizing to DSBs (Fig. 6A). To further test whether this recruitment defect is due to failure in forming dimers, we monitored the recruitment of EGFP-tagged CtIP dimerization mutants CtIP 20–45del and CtIP-LKEX4Emut. We found that both mutants exhibit strong defects in localization to DSBs (Fig. 6B). To show that the failure in DSB recruitment is indeed caused by impaired dimerization of CtIP, we inserted a dimeric leucine zipper from the yeast GCN4 transcription factor to the CtIP dimerization mutant CtIP-LKEX4Emut (Refs. 32 and 33 and supplemental Fig. S4). As illustrated in Fig. 6B, the GCN4 heterologous dimerization motif restored the dimer formation of the CtIP-LKEX4Emut and also restored CtIP recruitment to chromosomal DSBs. Meanwhile, the GCN4 dimerization motif rescued the defects of HR and MMEJ impaired in the CtIP dimerization mutant CtIP-LKEX4Emut (Fig. 6, C and D). These studies suggest that CtIP dimerization is required for its recruitment to chromosomal DSBs. Because recruitment to DSBs is essential for CtIP function in DSB repair and checkpoint activation, these studies provide a mechanistic explanation for the severe functional defects of the CtIP dimerization mutants in HR, end resection, and other DNA repair-related activities.
FIGURE 6.

CtIP dimerization is important for its recruitment to DSBs. A, U2OS cells were transiently co-transfected with EGFP-CtIP 1–897 or the indicated N-terminal deletion mutants and mRFP-PCNA, and live cell imaging of S phase cells, marked by PCNA S phase-associated replication foci, was performed on cells following laser-induced microirradiation using a pulsed nitrogen laser. Top panel, absolute intensity of EGFP-CtIP fluorescence signal at damage sites was calculated. The error bars indicate the S.D. Bottom panel, accumulation of EGFP-CtIP WT or indicated N-terminal deletion mutants at laser-induced damage sites are shown by white arrows at the indicated time points. B, U2OS cells were transiently co-transfected with EGFP-CtIP 1–897 or indicated mutants and mRFP-PCNA, and live cell imaging of S phase cells, marked by PCNA S phase-associated replication foci, was performed on cells following laser-induced microirradiation using a picosecond short pulsed green laser. Top panel, absolute intensity of EGFP-CtIP fluorescence signal at damage sites was calculated using Image J software, with an average of 10 independent measurements shown. The error bars indicate the S.D. Middle panel, accumulation of EGFP-CtIP 1–897 or indicated mutants at laser-induced damage sites are shown by white arrows at the indicated time points. Bottom panel, 293T cells were co-transfected with Myc-CtIP 1–897 (wild type) or Myc-GCN4-CtIP-LKEX4Emut and HA-CtIP 1–897 or indicated mutants. Anti-Myc immunoprecipitation (IP) was performed, followed by anti-HA Western blotting. C and D, U2OS EGFP-HR or U2OS EGFP-MMEJ cells stably expressing HA-CtIP 1–897 or indicated mutants, with endogenous CtIP silenced by shRNA and siRNA, were transfected with I-SceI-IRES-dsRedNLS, and 72 h after transfection, the cells were trypsinized, and FACS analysis was performed. The data shown represent the means of three independent experiments; the error bars indicate the S.D. Western blot shows expression of HA-CtIP 1–897 and mutants, with Ku70 used as a loading control.
To examine whether CtIP dimerization mutants may affect CtIP binding to DNA, causing recruitment defects to DSBs on chromatin, we performed DNA binding assays in vitro. GST-fused full-length wild type CtIP and the CtIP mutants CtIP 20–45del and CtIP-LKEX4Emut were expressed in insect cells and affinity-purified (Fig. 7A, right panel). We incubated these purified proteins with a 0.5-kb double-stranded DNA fragment biotinylated at one end. CtIP 1–897, CtIP 20–45del, and CtIP-LKEX4Emut proteins show similar binding to the DNA fragment, as revealed by the pull-down experiments using streptavidin beads (Fig. 7A, left panel). These results suggest that CtIP dimerization mutants do not exhibit noticeable binding defects to double-stranded DNA fragments with one free DSB end in vitro.
FIGURE 7.

DNA damage-induced CtIP phosphorylation is impaired in CtIP dimerization mutants. A, interaction of CtIP 1–897 or indicated mutants with DNA in vitro. Left panel, biotin-labeled DNA fragments (0.5 kb) were used in pull-down assays employing M-270 streptavidin Dynabeads and subjected to Western blotting using anti-CtIP antibody. Right panel, purified recombinants for this assay were resolved in SDS-PAGE (8% gel; Coomassie Blue staining). B, U2OS cells expressing HA-CtIP 1–897 or indicated mutants were treated with or without IR radiation (10 Gy) and allowed to recover for 1 h. After recovery, the cells were collected and lysed. Immunoblotting was performed using anti-HA antibody. C, purified GST-CtIP wild type or dimerization mutants were incubated with [γ-32P]ATP in the presence or absence of ATM kinase and immunoprecipitated from 293T cells by anti-FLAG M2 beads for 30 min at 30 °C. The radiolabeled CtIP was visualized following SDS-PAGE.
CtIP Dimerization Mutants Fail to Be Phosphorylated upon DNA Damage
It was described that CtIP is phosphorylated by ATM upon DNA damage (25). Because CtIP dimerization mutants are defective to be recruited to DSBs, we examined whether these mutants can still be phosphorylated by ATM after DNA damage. HA-tagged CtIP 1–897 and N-terminal deletion mutants HA-CtIP 45–897 and HA-CtIP 200–897 were expressed in U2OS cells. Although CtIP 1–897 exhibited IR-induced phosphorylation as revealed by slower migration shift on SDS-PAGE, the HA-CtIP 45–897 and HA-CtIP 200–897 mutants failed to do so (Fig. 7B, top panel). We also irradiated the U2OS cells expressing CtIP 1–897 or the CtIP 20–45del and CtIP-LKEX4Emut mutants. The dimerization mutants CtIP 20–45del and CtIP-LKEX4Emut are not properly phosphorylated after IR treatment (Fig. 7B, bottom panel). Thus, CtIP dimerization is required for its damage-induced phosphorylation in vivo.
IR-induced CtIP phosphorylation depends on ATM (25). To determine whether CtIP dimerization is required for ATM to phosphorylate CtIP, we performed in vitro ATM kinase assays. We observed that ATM phosphorylates wild type CtIP and CtIP dimerization mutants CtIP 20–45del and CtIP-LKEX4Emut to similar levels (Fig. 7C). These results suggest that CtIP dimerization is not required for ATM to directly phosphorylate CtIP and the defects of damage-induced CtIP phosphorylation in dimerization mutants in vivo are likely caused by their impaired recruitment to chromosomal DSBs.
DISCUSSION
It was described that the conserved sequences in CtIP and its homologues are mainly located at the C terminus, and the identified C terminus Sae2-like domain is critical for DNA damage responses including DSB repair (6, 12). Our studies identified a new conserved motif at the N terminus of CtIP that is required for dimer formation (Fig. 2A). Point mutations in this conserved motif severely impair CtIP function in various DNA damage responses, including defects in HR and MMEJ, end resection, and activation of the ATR pathway upon DSB formation. Since the interaction of the CtIP dimerization mutants with BRCA1, Nbs1, and DNA is unchanged compared to wild type CtIP, this suggests that these mutants largely adopt normal conformation and are active to mediate certain biological functions.
Oligomer formation has been described for Sae2 (16). The Sae2-L25P/E171G mutant identified in budding yeast is defective in forming oligomers and phenocopies the null mutant in DNA repair and checkpoint responses. The point mutant L25P is located at one end of the identified LKEX4EL/V conserved motif, and it will be interesting to examine whether mutating other conserved residues in this motif would impair Sae2 oligomer formation, causing defects in Sae2 function. Similar to the Sae2 oligomer formation mutant, CtIP dimerization mutants are severely defective in various DNA damage responses to the extent of CtIP-deficient cells. Our further analysis demonstrates that loss of CtIP dimer formation ability causes failure in the recruitment of CtIP to DSBs on chromatin, which is essential for mediating various DNA damage responses. The severe defects of CtIP dimerization mutants in DNA repair and checkpoint function are likely due to impaired localization of these mutants to DSB sites. Inserting a heterologous dimerization motif derived from the yeast GCN4 restores the recruitment of CtIP to chromosomal DSBs and also its activity in HR and MMEJ, further supporting that CtIP dimerization is essential for its localization to DSBs and for its DSB repair function. Examining CtIP homologues in other organisms will reveal whether forming dimer/oligomer is a general mechanism to regulate the recruitment of CtIP and its homologues to chromosomal DSBs.
It was described that CtIP recruitment to DSBs on chromatin depends on Nbs1 (7). In fission yeast, the interaction between Ctp1 and Nbs1 through the FHA/BRCT domains of Nbs1 is important for Ctp1 recruitment to DSB ends (12). By co-immunoprecipitation, we showed that CtIP dimerization is not required for its interaction with Nbs1, and thus the chromatin recruitment defect of dimerization mutants is probably not by affecting Nbs1 association-dependent DSB localization in mammalian cells. In vitro DNA binding assays did not reveal noticeable change of the interaction of CtIP dimerization mutants with double-stranded DNA with free DSB ends. However, it remains possible that CtIP dimer formation is required for its binding to DSB ends in the context of chromatin. For instance, CtIP dimerization may create docking sites to favor its interaction with chromosomal DSB ends. It is also possible that dimerization of CtIP is needed for its association with other proteins at DSB ends and/or DSB surrounding chromatin, and these interactions are important for initiating and/or stabilizing CtIP binding to chromosomal DSBs. Because CtIP dimer formation is not changed upon DNA damage, damage-induced protein associations involving CtIP dimers would likely be regulated by additional mechanisms such as damage-induced phosphorylation or other modifications on other proteins to induce their associations with CtIP dimers.
CtIP is phosphorylated by ATM in response to DNA damage. Our studies showed that CtIP dimerization mutants are defective in the recruitment to chromosomal DSBs and are also impaired in DNA damage-induced phosphorylation. This suggests that CtIP may need to be recruited to DSBs or DSB surrounding chromatin to be phosphorylated by ATM. This ATM-mediated phosphorylation of CtIP could contribute to promote CtIP-mediated end resection for MMEJ and HR. It has also been described that ATM deficiency leads to reduced CtIP chromatin recruitment (7). Our in vitro ATM kinase assay shows that ATM phosphorylates wild type CtIP and the CtIP dimerization mutants to similar levels, supporting the notion that impaired damage-induced CtIP phosphorylation is likely due to its failure to be recruited to chromosomal DSBs. However, ATM-dependent phosphorylation of CtIP may further stabilize CtIP binding to the chromosomal DSBs.
It was described that the interaction of CtIP with BRCA1 is important for promoting end resection to initiate HR. We showed that the interaction of CtIP with BRCA1 is not affected by loss of CtIP dimerization. This suggests that CtIP and BRCA1 interaction does not necessarily require CtIP recruitment to DSBs. It also implies that CDK-mediated phosphorylation of CtIP at Ser-327, which mediates CtIP and BRCA1 association, is normal in CtIP dimerization mutants.
We showed that the conserved LKEX4EL/V motif at the N terminus of CtIP is essential for CtIP dimer formation. Although the N terminus coiled-coil domain is implicated in mediating CtIP self-association, we found that this domain is neither necessary nor sufficient for full-length CtIP to form dimers. It remains possible that although the conserved LKEX4EL/V motif is responsible for initiating dimer formation, the coiled-coil domain may contribute to further stabilization of the dimer. It is still unknown whether the LKEX4EL/V motifs are the contact points of two CtIP monomers to form a dimer or whether they are required for initiating dimer formation by orienting other sites on CtIP to interact. Crystal structure analysis will be informative to address how the LKEX4EL/V motif regulates CtIP dimer formation and to clarify the role of the N terminus coiled-coil domain in the dimer formation. Structure analysis of CtIP dimer in the presence of DNA will further illustrate whether dimer formation would modulate the conformation of CtIP in association with DNA, thereby promoting the DNA repair process.
Our studies revealed an important conserved motif at the N terminus of CtIP, which mediates CtIP dimer formation and is essential for various CtIP functions in DNA damage responses. Further analysis of the role of CtIP dimerization in its DNA binding, its biochemical activity with MRN to promote end resection, and its chromatin binding at DSB sites will bring new insights to the molecular mechanisms underlying DSB repair and checkpoint activation.
Acknowledgments
We thank members of the Wu laboratory for helpful discussions. We also thank Drs. Daniel R. Salomon and Stephanie Cherqui (The Scripps Research Institute), Junjie Chen (University of Texas M. D. Anderson Cancer Center), Stephen P. Jackson (University of Cambridge), and Maria Jasin (Memorial Sloan-Kettering Cancer Center) for providing valuable reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants CA102361, GM080677, CA140972, and CA102361-07S1 (to X. W.) and CA162804 (to D. J. C.). This work was also supported by funds from the Beckman Laser Institute (to M. W. B.) and Cancer Prevention Research Institute of Texas Grant RP110465 (to D. J. C.).

This article contains supplemental Figs. S1–S4.
- DSB
- double-stranded break
- MRN
- Mre11-Rad50-Nbs1
- HR
- homologous recombination
- CDK
- cyclin-dependent kinase
- ATR
- ataxia telangiectasia and Rad3-related
- MMEJ
- microhomology-mediated end joining
- EGFP
- enhanced GFP
- ATM
- ataxia telangiectasia mutated
- RPA
- replication protein A
- CPT
- camptothecin
- IRES
- internal ribosome entry site
- NLS
- nuclear localization sequence
- RFP
- red fluorescent protein
- PCNA
- proliferating cell nuclear antigen.
REFERENCES
- 1. Stracker T. H., Petrini J. H. (2011) The MRE11 complex. Starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paull T. T., Lee J. H. (2005) The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 4, 737–740 [DOI] [PubMed] [Google Scholar]
- 3. Chinnadurai G. (2006) CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim. Biophys. Acta 1765, 67–73 [DOI] [PubMed] [Google Scholar]
- 4. You Z., Bailis J. M. (2010) DNA damage and decisions. CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 20, 402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu X., Chen J. (2004) DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 24, 9478–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S. P. (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. You Z., Shi L. Z., Zhu Q., Wu P., Zhang Y. W., Basilio A., Tonnu N., Verma I. M., Berns M. W., Hunter T. (2009) CtIP links DNA double-strand break sensing to resection. Mol. Cell 36, 954–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenberg R. A., Sobhian B., Pathania S., Cantor S. B., Nakatani Y., Livingston D. M. (2006) Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 20, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L., Nievera C. J., Lee A. Y., Wu X. (2008) Cell cycle-dependent complex formation of BRCA1·CtIP·MRN is important for DNA double-strand break repair. J. Biol. Chem. 283, 7713–7720 [DOI] [PubMed] [Google Scholar]
- 10. Yun M. H., Hiom K. (2009) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459, 460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huertas P., Jackson S. P. (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 284, 9558–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Limbo O., Chahwan C., Yamada Y., de Bruin R. A., Wittenberg C., Russell P. (2007) Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 28, 134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paull T. T. (2010) Making the best of the loose ends. Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair 9, 1283–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda S., Nakamura K., Taniguchi Y., Paull T. T. (2007) Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol. Cell 28, 351–352 [DOI] [PubMed] [Google Scholar]
- 15. Huertas P., Cortés-Ledesma F., Sartori A. A., Aguilera A., Jackson S. P. (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455, 689–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim H. S., Vijayakumar S., Reger M., Harrison J. C., Haber J. E., Weil C., Petrini J. H. (2008) Functional interactions between Sae2 and the Mre11 complex. Genetics 178, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dubin M. J., Stokes P. H., Sum E. Y., Williams R. S., Valova V. A., Robinson P. J., Lindeman G. J., Glover J. N., Visvader J. E., Matthews J. M. (2004) Dimerization of CtIP, a BRCA1- and CtBP-interacting protein, is mediated by an N-terminal coiled-coil motif. J. Biol. Chem. 279, 26932–26938 [DOI] [PubMed] [Google Scholar]
- 18. Wilson I. A., Haft D. H., Getzoff E. D., Tainer J. A., Lerner R. A., Brenner S. (1985) Identical short peptide sequences in unrelated proteins can have different conformations. A testing ground for theories of immune recognition. Proc. Natl. Acad. Sci. U.S.A. 82, 5255–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Olson E., Nievera C. J., Liu E., Lee A. Y., Chen L., Wu X. (2007) The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol. Cell. Biol. 27, 6053–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson R. J., Howell M. T., Kaminski A. (1990) The novel mechanism of initiation of picornavirus RNA translation. Trends Biochem. Sci. 15, 477–483 [DOI] [PubMed] [Google Scholar]
- 21. Jang S. K., Kräusslich H. G., Nicklin M. J., Duke G. M., Palmenberg A. C., Wimmer E. (1988) A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62, 2636–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pelletier J., Sonenberg N. (1988) Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334, 320–325 [DOI] [PubMed] [Google Scholar]
- 23. Uematsu N., Weterings E., Yano K., Morotomi-Yano K., Jakob B., Taucher-Scholz G., Mari P. O., van Gent D. C., Chen B. P., Chen D. J. (2007) Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Botvinick E. L., Berns M. W. (2005) Internet-based robotic laser scissors and tweezers microscopy. Microsc. Res. Tech. 68, 65–74 [DOI] [PubMed] [Google Scholar]
- 25. Li S., Ting N. S., Zheng L., Chen P. L., Ziv Y., Shiloh Y., Lee E. Y., Lee W. H. (2000) Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 406, 210–215 [DOI] [PubMed] [Google Scholar]
- 26. McVey M., Lee S. E. (2008) MMEJ repair of double-strand breaks (director's cut). Deleted sequences and alternative endings. Trends Genet 24, 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie A., Kwok A., Scully R. (2009) Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 16, 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bennardo N., Cheng A., Huang N., Stark J. M. (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B. S. (2009) Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 16, 819–824 [DOI] [PubMed] [Google Scholar]
- 30. Dinkelmann M., Spehalski E., Stoneham T., Buis J., Wu Y., Sekiguchi J. M., Ferguson D. O. (2009) Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 16, 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Somanathan S., Suchyna T. M., Siegel A. J., Berezney R. (2001) Targeting of PCNA to sites of DNA replication in the mammalian cell nucleus. J. Cell. Biochem. 81, 56–67 [DOI] [PubMed] [Google Scholar]
- 32. O'Shea E. K., Klemm J. D., Kim P. S., Alber T. (1991) X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254, 539–544 [DOI] [PubMed] [Google Scholar]
- 33. Zgheib O., Pataky K., Brugger J., Halazonetis T. D. (2009) An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol. 29, 1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]