

Background: SpyA is a streptoccocal ADP-ribosyltransferase that modifies vimentin.
Results: Vimentin is modified in the regulatory N-terminal head domain, and treatment with SpyA results in a defect in filamentation.
Conclusion: Vimentin is a target substrate of SpyA, and ADP-ribosylation affects polymerization dynamics.
Significance: Vimentin filaments are disrupted by a bacterial ADP-ribosyltransferase, SpyA, and ADP-ribosylation of vimentin regulates filament formation.
Keywords: ADP-ribosylation, Bacterial Toxins, Cytoskeleton, Enzyme Kinetics, Intermediate Filaments, Mass Spectrometry (MS), Streptococcus pyogenes, Vimentin, SpyA, ADP-ribosyltransferase
Abstract
The Gram-positive bacterial pathogen Streptococcus pyogenes produces a C3 family ADP-ribosyltransferase designated SpyA (S. pyogenes ADP-ribosyltransferase). Our laboratory has identified a number of eukaryotic protein targets for SpyA, prominent among which are the cytoskeletal proteins actin and vimentin. Because vimentin is an unusual target for modification by bacterial ADP-ribosyltransferases, we quantitatively compared the activity of SpyA on vimentin and actin. Vimentin was the preferred substrate for SpyA (kcat, 58.5 ± 3.4 min−1) relative to actin (kcat, 10.1 ± 0.6 min−1), and vimentin was modified at a rate 9.48 ± 1.95-fold greater than actin. We employed tandem mass spectrometry analysis to identify sites of ADP-ribosylation on vimentin. The primary sites of modification were Arg-44 and -49 in the head domain, with several additional secondary sites identified. Because the primary sites are located in a domain of vimentin known to be important for the regulation of polymerization by phosphorylation, we investigated the effects of SpyA activity on vimentin polymerization, utilizing an in vitro NaCl-induced filamentation assay. SpyA inhibited vimentin filamentation, whereas a catalytic site mutant of SpyA had no effect. Additionally, we demonstrated that expression of SpyA in HeLa cells resulted in collapse of the vimentin cytoskeleton, whereas expression in RAW 264.7 cells impeded vimentin reorganization upon stimulation of this macrophage-like cell line with LPS. We conclude that SpyA modification of vimentin occurs in an important regulatory region of the head domain and has significant functional effects on vimentin assembly.
Introduction
Streptococcus pyogenes (group A streptococcus) is a Gram-positive bacterial pathogen responsible for a number of human diseases, the most common being mild skin infections and pharyngitis, though more serious infections, such as necrotizing fasciitis, can occur (1). S. pyogenes produces numerous toxins, including superantigens, proteases, and potent cytolysins. Recently, we described a novel NAD+ glycohydrolase and mono-ADP-ribosyltransferase (ADPRT),2 SpyA (2), and demonstrated a role for SpyA in streptococcal pathogenesis (3).
Although ADPRTs serve diverse functions, they all maintain a similar mechanism of action, the covalent transfer of an ADP-ribose moiety, donated from NAD+, onto a target protein, modifying target activity. Endogenous eukaryotic ADPRTs function as regulatory enzymes and can modify both intra- and extracellular proteins. Intracellular protein targets include the intermediate filament desmin, heterotrimeric G protein βγ subunit, and elongation factor 2 (4–10). A member of the ectoenzyme family of vertebrate mono-ADPRTs, ART-1, modifies the defensin human neutrophil peptide-1, regulating its antimicrobial and cytotoxic activities (11, 12).
ADPRTs also represent a class of potent bacterial toxins. Among them are cholera, diphtheria, and pertussis toxins. The clostridial C2 and C3 toxin families represent a class of bacterial ADPRTs that target cytoskeletal proteins. The actin cytoskeleton is a well characterized target of the C2 toxins; ADP-ribosylated actin subunits are sterically unable to form filaments and furthermore act as capping proteins on existing filaments, leading to the eventual collapse of the actin cytoskeleton (13–16). There is also evidence of C2 family toxin-mediated microtubular reorganization (17). The C3 family of toxins, including the Staphylococcus aureus EDIN (also termed C3stau) toxin, are small enzymes that lack a known translocation domain. These toxins inactivate Rho GTPases, resulting in a downstream massive and lethal reorganization of the actin cytoskeleton (18–24). Finally, the promiscuous bacterial effector protein ExoS of Pseudomonas aeruginosa has been shown to ADP-ribosylate numerous targets, including the intermediate filament vimentin (25).
Previously, we identified vimentin as a substrate for SpyA (2). Vimentin is an intermediate filament (IF) protein found in cells of mesenchymal origin and forms filaments 10 nm in diameter. Like other IF proteins, vimentin is composed of a globular “head” domain at the N terminus followed by two coiled-coil regions and a globular “tail” domain. Although the role of vimentin is currently being fully elucidated, it is known to be important in cellular stability to mechanical stress, cell movement during wound healing, and leukocyte adhesion and migration (26–29). There is also evidence for an important role of vimentin in organelle positioning and membrane protein trafficking (30). Like other cytoskeletal proteins, vimentin appears to have a role in cell signal transduction, potentially as a scaffold for signaling molecules, and caspase-cleaved vimentin promotes apoptosis (31–34). Vimentin production has also been implicated in the maturation and full bactericidal function of macrophages (35, 36).
The effects of SpyA on vimentin function have not been described; however, vimentin was recently identified as a target in a screen of endogenously ADP-ribosylated proteins (37). Although the effect of ADP-ribosylation on actin by bacterial ADPRTs has been studied extensively, the effect of this modification on vimentin has not been previously elucidated. However, ADP-ribosylation of the related IF protein desmin by an endogenous muscle ADPRT has been reported and was found to cause impairment in IF formation (4, 5, 38). The two major sites of modification of desmin, determined by MALDI analysis, were located in the N-terminal head domain in the same region containing regulatory phosphorylation sites, important for the regulation of polymerization (6, 39, 40).
The current study seeks to characterize the SpyA-mediated ADP-ribosylation of vimentin. Although SpyA was shown to be a promiscuous ADPRT, modifying a number of proteins in a two-dimensional gel analysis, vimentin appeared to be a major target (2). We present enzyme kinetic data for SpyA modification of vimentin and actin, which supports the hypothesis that vimentin is an important target of SpyA. To understand the nature of the ADP-ribosylation of vimentin, we determined primary sites of modification using tandem mass spectrometry. We demonstrate the effect of SpyA-mediated ADP-ribosylation on the ability of vimentin to form filaments in vitro and the in vivo effects of SpyA on the vimentin cytoskeleton in HeLa cells and activated RAW 264.7 macrophages.
EXPERIMENTAL PROCEDURES
Materials
Restriction enzymes, T4 DNA ligase, Taq DNA polymerase, and Pfu Turbo DNA polymerase were purchased from New England Biolabs. [32P]NAD+, etheno-NAD (ϵ-NAD+), and biotinylated NAD+ were purchased from American Radiolabeled Chemicals (St. Louis, MO), Biolog Inc. (Hayward, CA), and Trevigen (Gaithersburg, MD), respectively. All tissue culture supplies were purchased from Invitrogen, and all chemicals were acquired from Sigma-Aldrich unless indicated otherwise. Bovine cardiac actin and Syrian hamster vimentin were purchased from Cytoskeleton (Denver, CO). Chemicon (Temecula, CA) produced all antibodies except where indicated. Protective antigen was acquired from List Biologicals (Campbell, CA), and Slow Fade light Antifade reagent was from Molecular Probes, Inc. (Grand Island, NY). Ultrapure Salmonella minnesota R595 lipopolysaccharide (LPS) was a gift from Dr. Brad Cookson and Dr. Tessa Bergsbaken (List Biologicials).
Bacterial Strains and Growth Conditions
S. pyogenes SF370 was purchased from the ATCC (The Global Bioresource CenterTM). Escherichia coli strains BL21(DE3), DH5α, and Rosetta (DE3) were acquired from Novagen (Philadelphia, PA). E. coli DH5α was the host strain for all recombinant plasmids, unless otherwise noted. E. coli cultures were grown in Luria broth (LB) with aeration at 37 °C, in the presence of either kanamycin (30 μg/ml) or carbenicillin (50 μg/ml) when needed.
Construction of Plasmid for Expression of Human Vimentin
Human cDNA (I.M.A.G.E. Clone ID 4823475) was used to amplify a 1,401-bp PCR product consisting of the gene encoding vimentin. The primer pair GCCATGGGATCCACCAGGTCCGTGTCCT (forward, NcoI site in boldface type) and GAAGCTTCTTCAAGGTCATCGTGATG (reverse, HindIII site in boldface type) was employed for PCR. The PCR product was digested with NcoI and HindIII and ligated into NcoI/HindIII-digested pET21d (Novagen). The resulting plasmid was designated pET21Vim and transformed into competent E. coli Rosetta (DE3) using the manufacturer's protocol.
Construction of Plasmids for Transfection of Eukaryotic Cells
For transfection of HeLa cells, transfection plasmids pCMV-myc-SpyA and pCMV-myc-SpyA-E187A were constructed by PCR-amplifying the spyA and spyA-E187A using pET15b-SpyA and pET15b-SpyAE187A template, respectively, and primers GAGAGAGAGAATTCTGGTCTGTGAGCACTATGAGCGGGC (forward primer) and GAGAGAGACTCGAGTTACAAACTGCCCTTGAAATACGCTTC (reverse primer). The PCR product was digested with EcoRI and XhoI and ligated into the eukaryotic expression vector pCMV-myc (Clontech, Mountain View, CA) also digested with EcoRI and XhoI and transformed into E. coli. A second SpyA expression plasmid utilizing the bicistronic expression plasmid pEF1α-IRES-DsRed-Express2 (Clontech) was created for use in the RAW 264.7 cells. The spyA and spyA-E187A inserts were created as described above, using forward primer GAGAGAGAGAATTCGCCACCATGGTCTGTGAGCACTATGAGCGG and reverse primer GAGAGAGAGGATCCTCACAAACTGCCCTTGAAATACGCTTCTATG. The resulting PCR product was digested with EcoRI and BamHI, ligated into similarly digested pEF1α-IRES-DsRed-Express2, and transformed into E. coli. The resulting plasmids were pEF1α-IRES-DsRed-Express2-SpyA and pEF1α-IRES-DsRed-Express2-SpyAE187A.
Recombinant Protein Expression, Purification, and Activity
For expression and purification of SpyA and the catalytic site mutant SpyAE187A, the expression plasmids pET15bSpyA and pET15bSpyA-E187A were utilized as described (2).
All enzymes were subsequently assayed for activity via a vimentin modification assay. 877 nm vimentin, 100 mm biotinylated NAD, 0.5 μm SpyA, 100 μm biotinylated NAD+ in 100 mm HEPES, pH 7.2, was reacted for 1 h at 37 °C. The samples were then resolved on a 15% SDS-polyacrylamide gel. Biotinylated samples were analyzed via Western blot against biotinylated ADP-ribose using streptavidin-HRP (1:10,000) (Pierce).
To express human vimentin, E. coli BL21(DE3) Rosetta was transformed with pET21Vim and grown in LB with carbenicillin (50 μg/ml) overnight with aeration at 37 °C. The transformed cells were subcultured and grown to an A600 of 0.6. Isopropyl β-d-thiogalactoside was added to a final concentration of 1 mm. Vimentin was expressed for 6 h at 37 °C. Cells were then centrifuged for 10 min at 4,400 × g at 4 °C and then frozen at −20 °C. The pellets were defrosted and passed through a French press to lyse cells. Lysates were centrifuged for 10 min at 4,400 × g at 4 °C, and pelleted inclusion bodies were collected. The inclusion bodies were then washed twice in 0.1% Triton X-100 in PBS. The final pellet was dissolved overnight in 8 m urea in vimentin subunit buffer (10 mm Tris, pH 8.4). This and all subsequent steps were done at 4 °C with stirring. The urea was removed by stepwise dialysis, beginning with dialysis against 4 m urea in vimentin subunit buffer for 4 h. The vimentin solution was subsequently dialyzed against 2, 1, and 0.5 m and finally 0 m urea in vimentin subunit buffer, each for a minimum of 4 h.
Determination of Kinetic Constants
Unless otherwise stated, all constants were expressed as averaged values ± S.D. of three independent experiments, each done in triplicate. To determine the kinetic constants, the fluorescent NAD+ analog, ϵ-NAD+, was used as described (41), with the following modifications. The ADP-ribosylation reaction was performed as follows: 8.77 μm human vimentin or actin was added to 0.085 μm SpyA in 100 mm HEPES buffer at 37 °C. Nine concentrations of ϵ-NAD+ were added for final concentrations of 300, 200, 100, 50, 25, 12.5, 6.25, 1, or 0 μm. Control experiments were also performed in the absence of vimentin or actin to determine the rate of NAD+-glycohydrolysis. To create a standard calibration curve, concentrations of ϵ-AMP identical to those used for ϵ-NAD+ were also analyzed under the same conditions. Reactions were then run on an Envision Multilabel plate reader (PerkinElmer Life Sciences) at 300-nm excitation and 415-nm emission. Readings were taken for a minimum of 30 min. Prior to data analysis, the background glycohydrolase activity was subtracted from the ADP-ribosylation reactions. Fluorescence intensity was converted to micromolar of product formed using the ϵ-AMP calibration curve as previously described (42, 43). The linear range, 0–5 min, was determined, and the rates of the reactions were measured for each concentration of ϵ-NAD+. These rates were subsequently analyzed using nonlinear regression analysis by Prism software. The data were fit to a standard Michaelis-Menten model. Statistical significance was measured by an unpaired t test, and the two-tailed p value was reported as less than 0.0001 (GraphPad).
Competition Assay
Equimolar amounts of hamster vimentin and actin (877 nm) were added to 0.5 μm SpyA, 100 μm biotinylated NAD+ or [32P]NAD+ in 100 mm HEPES. Reactions were incubated at 37 °C, and 20-μl aliquots were taken at 0, 0.25, 5, 10, 30, and 90 min. Proteins were then resolved via SDS-PAGE, and biotinylated samples were assessed by Western blot as described under “Recombinant Protein Expression, Purification, and Activity.” Radiolabeled samples, in triplicate, were subjected to phosphorimaging using a Storm 840 PhosphorImager (GE Healthcare) and analyzed using ImageJ software (44).
Chemical Sensitivity Assay
Hydroxylamine (NH2OH) and mercuric chloride (HgCl2) were used to probe the chemical sensitivity of SpyA-modified vimentin. SpyA (0.5 μm) was reacted with 4.5 μm hamster vimentin and 100 μm biotinylated NAD+ in 100 mm HEPES for 1 h at 37 °C. The reaction was terminated by boiling for 5 min, and samples were incubated with either 0.5 m NaCl (control), 1 mm HgCl2, or 0.5 m NH2OH for 90 min at 30 °C. Samples were then resolved by 15% SDS-PAGE and analyzed via Western blot as describe under “Competition Assay.”
Stoichiometric Analysis
A time course, with time points at 0, 5, 10, 20, 30, 60, 90, 120, 150, and 180 min, was run to determine ADP-ribose incorporation into vimentin. 2.3 μm hamster vimentin was incubated at 37 °C with 0.3 μm SpyA and a 100 μm mixture of NAD+ and [32P]NAD+ in 100 mm HEPES. Reactions were terminated by the addition of 6× SDS-PAGE loading buffer with β-mercaptoethanol. Proteins were resolved by 15% SDS-PAGE. The gel was fixed and dried, and vimentin bands were excised and subjected to scintillation counting. A calibration curve was created using serial dilutions of the NAD+/[32P]NAD+ mixture, and counts/min were converted to μm radiolabeled ADP-ribose incorporated/μm vimentin.
Mass Spectrometry Analysis
An 877 nm concentration of either hamster or human vimentin was incubated with SpyA (1, 0.5, or 0.2 μm) and 100 μm NAD+ in 100 mm HEPES buffer for 5, 15, or 30 min. Following protein incubation with SpyA, urea was added to a final concentration of 6 m, and 1.5 μl of 1.5 m Tris, pH 8.8, was added to maintain a basic pH during reduction. Proteins were reduced for 1 h at 37 °C with 5 mm tris(2-carboxyethyl)phosphine. Alkylation of cysteine residues was performed with 30 mm iodoacetamide for 1 h in the dark at room temperature, followed by the addition of dithiothreitol (DTT) to a final concentration of 30 mm and a 1-h incubation at room temperature. Samples were then diluted with 900 μl of 50 mm ammonium bicarbonate, and sequencing grade trypsin (Promega, Madison, WI) or GluC (Roche Applied Science) was added at a protein/enzyme ratio of 1:50. Digestion proceeded overnight at room temperature. Samples were desalted using a Vydac silica C18 macrospin column (The Nest Group, Southborough, MA).
ADP-ribosylated peptides were analyzed using the “marker ion” approach previously described by Hengel et al. (45, 46). Briefly, peptides were analyzed using electrospray ionization in a linear ion trap Fourier transform ion cyclotron resonance mass spectrometer or linear ion trap Orbitrap (Thermo Electron Corp., San Jose, CA). HPLC columns were packed in house (0.75-μm inner diameter × 11 cm; 100 Å Magic C18AQ: Michrom Bioresources, Auburn, CA), and separations were performed with an inline Agilent 1100 binary HPLC pump or nanoAcquity (Waters, Milford, MA), using a linear gradient of 5–35% acetonitrile over 60 min. Electrospray ionization voltage was applied with a liquid junction prior to the analytical column using a gold wire and a micro-tee junction (47). All ions were measured in positive ion mode, and precursor ion masses were detected in the Fourier transform mass analyzer. Source parameters were set as follows: electrospray ionization voltage, 2.5 kV; capillary temperature, 265 °C; capillary voltage, 13 V; tube lens, 35 V. For all fragmentation methods, the resolution was set to 5.0e4 and 2.0e4 for MS and MS2, respectively. Automatic gain control was used to maintain constant ion populations at 5e5 and 2e5 for MS and MS2, respectively. For linear ion trap tandem mass spectrometric acquisitions, the top five most intense multiply charged ions per MS spectrum were selected for fragmentation. When fragment ion data were collected in the Fourier transform ion cyclotron resonance mass spectrometer or Orbitrap, the top four most intense multiply charged ions per MS spectrum were selected for fragmentation. Fragment ion spectra acquired with collision-induced dissociation were produced using 35% collision energy and a 2.0 Da isolation window. Electron capture dissociation (ECD) tandem mass spectra were acquired using a 5-eV fragmentation energy for 70 ms with a delay of 0.3 ms and a 3.0 Da isolation window. Data were analyzed using SEQUEST, specifying an optional arginine modification of 541 daltons (48). For ECD data searches, SEQUEST was modified to search for both c and z ions. Proteins were identified from the complete IPI database for human or Syrian hamster depending on the origin of vimentin.
To identify precursor ions of peptides containing m6, m8, and p5, ADP-ribose, PERL scripts were used to search for ions assuming singly charged ADP-ribose fragments in collision-induced dissociation data as previously described (45). To reduce false positive identifications using the marker ion strategy, scans that correlated to an unmodified peptide identification with a probability greater than or equal to 0.9 from the SEQUEST/Interact files were removed from the marker ion scan lists. For ECD targeting experiments, marker ions were required to be observed at an ion intensity of greater than 1,000 with the associated scan containing p5 and m6, m8, or both m6 and m8. Approximately 10% of scans used to generate ECD target lists were manually validated for marker ions prior to ECD analysis.
In Vitro Polymerization of Vimentin
Polymerization of vimentin was accomplished as described (49) with the following modifications. Hamster vimentin at 0.02 μg/μl (350 nm) was incubated with 40 nm SpyA, SpyAE187A, or SpyA buffer and 100 μm NAD+ in HEPES buffer for 3 h at 37 °C. The reactions were split into two equal aliquots; to induce polymerization, NaCl to 150 mm was added to one, water to the other. Polymerization was allowed to proceed for 60 min at room temperature. Filaments were separated from soluble subunits via ultracentrifugation (100,000 × g, 30 min, 20 °C). A 20-μl volume of supernatant was then carefully pipetted from the reactions and subjected to 15% SDS-PAGE. Proteins were transferred to nitrocellulose membrane and probed for vimentin using goat anti-human vimentin and bovine anti-goat HRP.
Analysis of Eukaryotic Cells Expressing SpyA
HeLa cells obtained from ATCC were grown in DMEM supplemented with 4 mm l-glutamine, 4,500 mg/liter glucose, 10% FCS, and 1% streptomycin/penicillin. They were maintained in 75-cm2 cell culture flasks at 37 °C, 5% CO2. Cells were seeded at 5 × 104 cells/ml and left overnight on coverslips in 24-well tissue culture plates. Murine macrophage-like cells RAW 264.7 were a gift from Dr. Brad Cookson and were grown as described for the HeLa cells.
HeLa cells were transfected with 0.3 g of DNA using Fugene 6 (Roche Applied Science) according to the manufacturer's directions and left for 20 h. The BD cytofix/cytoperm kit (BD Biosciences) was used for fixation and permeabilization of the cells. The cells were then stained with rabbit anti-human vimentin (Stemgent) and monoclonal mouse anti-Myc (Sigma) for 1 h. The cells were washed three times with BD Perm/Wash buffer and then incubated for 1 h with cy5 anti-rabbit antibody (Jackson Immunoresearch Laboratories, Inc.) and Dylight 488 goat anti-mouse IgG (KPL). The cells were washed three times with BD Perm/Wash buffer. The coverslips were then mounted in VECTASHIELD (Vector Laboratories). Fluorescence was visualized using a deconvolution microscope (Applied Precision Deltavision/Softworx, Universal Imaging Metamorph, University of Washington Keck Imaging Center). Cells were then visually assessed for an intact vimentin cytoskeleton, and the percentage of transfected cells with collapsed vimentin filaments was calculated (cells undergoing cell death, as determined by membrane blebbing and disrupted nuclei, were not counted). Results were subjected to an unpaired t test, two-tailed p value.
RAW 264.7 cells were transfected with either pEF1α-IRES-DsRed-Express2-SpyA or pEF1α-IRES-DsRed-Express2-SpyAE187A as described for HeLa cells. After 18 h of transfection, the cells were then activated by treatment with 100 ng/ml LPS for 4 h and fixed using 10% formalin solution. After fixation, the cells were gently permeabilized with Tween 20 detergent and stained for 1 h with rabbit anti-vimentin antibodies followed by anti-rabbit FITC-conjugated IgG. Cells were mounted in VECTASHIELD and analyzed using a Nikon spinning disk confocal microscope. Transfected cells were identified by expression of dsRed-Express2, assessed for the presence of extracellular vimentin, and analyzed via NIS Elements Analysis software. Cells displaying gross morphological changes under differential interference contrast microscopy, such as loss of volume, membrane blebbing, or rupture, or other signs of cell death were excluded from analysis. The number of transfected cells with abnormal vimentin staining was determined, with the observer blinded to the nature (mutant versus wild type) of the transfected spyA. A Fisher exact test with a two-tailed p value was used to interpret the results.
RESULTS
SpyA Preferentially ADP-ribosylates Vimentin
Previously, our laboratory identified the cytoskeletal proteins vimentin and actin as SpyA substrates (2). Actin is a well known target of cytotoxic ADP-ribosylation by the C2 bacterial ADPRT toxins; however, modification of vimentin is relatively rare and has not been characterized (25, 51). To delineate the relative rates of activity of SpyA on vimentin and actin, Michaelis-Menten kinetic analyses were used to determine the turnover number, kcat, and the Michaelis-Menten constant, Km. In our studies, NAD+ was the varied substrate because actin and vimentin polymerize at concentrations necessary to determine their kinetic constants.
ADPRT activity against vimentin and actin was determined using the ϵ-NAD+ fluorescence assay (41). The release of nicotinamide during catalysis abolished the intrinsic fluorescent quenching, leading to an increase in fluorescence, and was used as a proxy to measure the formation of ADP-ribosylated vimentin and actin. The increase in fluorescence from the NAD+-glycohydrolase activity of SpyA, in the absence of modification substrate, was low and subtracted as background. The ϵ-ADP-ribose analog ϵ-AMP was used to create a calibration curve as a standard for ADP-ribosylated product (42, 43). After defining a time point in the linear range, 5 min, the initial velocities were measured and plotted against ϵ-NAD+ concentrations, and the data were fit to a standard Michaelis-Menten curve (Fig. 1). The measured Km values were 76.2 ± 9.3 and 79.6 ± 3.5 μm for vimentin and actin, respectively, comparable with other bacterial ADPRT toxins (Table 1). The close Km values indicate that NAD+ binding is similar during the catalysis of NAD+ to ADP-ribose for both vimentin and actin. The kcat for vimentin was found to be 58.5 ± 3.4 min−1, whereas this value for actin was lower, 10.1 ± 0.6 min−1 (Table 1). The kcat values for vimentin and actin were found to be significantly different (p ≥ 0.0001). The turnover number for vimentin was comparable with, although slightly higher than, than reported for the Rho-targeting C3 enzymes (Table 1). Unlike the other actin-targeting ADPRTs, iota and Photox toxins, the SpyA kcat value for actin was considerably lower. The SpyA catalytic rate for vimentin was nearly 6 times greater that than for actin, and the kcat for vimentin was within the same order of magnitude as for other characterized ADPRTs and their substrates.
FIGURE 1.
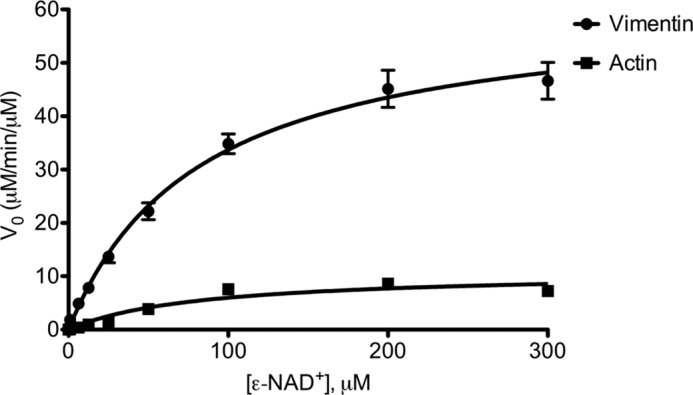
Rate of substrate modification by SpyA. Recombinant human vimentin or bovine actin (8.77 μm) was incubated with SpyA (0.085 μm) at the indicated concentrations of ϵ-NAD+ for 5 min at 37 °C. The increase in fluorescence was measured and plotted to determine initial velocity (V0) as a function of μm product formed/min/μm SpyA. Initial velocity was plotted against [ϵ-NAD+] and analyzed by Prism statistical software using the nonlinear regression analysis fit to a standard Michaelis-Menten model. Data presented are for a representative experiment of three independent experiments done in triplicate (vimentin) or duplicate (actin). Error bars, S.D.
Table I.
Km and kcat values for bacterial ADPRTs
Values for kinetic parameters of SpyA were determined as described in the legend to Fig. 1. All values were determined in the presence of excess substrate and variable [ϵ-NAD+]. Data represent the mean ±S.D. of at least three independent experiments. ND, not determined. DT, diphtheria toxin; PT, pertussis toxin; SBTI, soybean trypsin inhibitor; eEF-2, eukaryotic elongation factor 2.
| Bacteria | Toxin | Substrate(s) | Km | kcat | Reference/Source |
|---|---|---|---|---|---|
| μm | min−1 | ||||
| S. pyogenes | SpyA | Vimentin | 76.2 ± 9.3 | 58.5 ± 3.4 | This paper |
| Actin | 79.6 ± 3.5 | 10.1 ± 0.6 | |||
| Photorhabdus luminescens | Photox | Actin | 27.4 ± 4.4 | 1,680 ± 75 | Ref. 41 |
| Clostridium botulinum | C2Ia | Actin | 21.3 ± 1.2 | ND | Ref. 74 |
| Clostridium perfringens | Iota aa | Actin | 3.8–6.0 | 2,024 ± <15% | Refs. 61 and 74 |
| Salmonella enterica | C/SpvBa | Actin | 4.4 ± 0.6 | ND | Ref. 74 |
| C. botulinum | C3bot | RhoA | 8 | 15b | Ref. 75 |
| Clostridium limosum | C3lim | RhoA | 52 ± <15% | 0.28 ± <15% | Ref. 76 |
| Bacillus cereus | C3cer | RhoA | 20.8 ± 1.6 | 22.25 ± 0.9 | Ref. 77 |
| RhoB | 56.4 ± 3.6 | 4.19 ± 0.65 | |||
| RhoC | 17.1 ± 3.3 | 0.99 ± 0.08 | |||
| P. aeruginosa | ExoS | SBTI | 30 ± 10 | 25 ± 20 | Refs. 78 and 79 |
| Ras | 9 | ND | |||
| P. aeruginosa | ExoA | eEF-2 | 275 ± 52 | 675 ± 85 | Ref. 42 |
| Corynebacterium diphtheriae | DT | eEF-2 | 2–9 | 58.8–182 | Refs. 80–82 |
| Bordetella pertussis | PT | αi3C20c | 27.0 | 3.0 | Refs. 83 and 84 |
| Giαd | 23.0 | 40.0 |
a C2I and ι a are the enzymatic components of C2 and ι toxin, respectively. C/SpvB is the C terminal enzyme domain of SpvB.
b Calculated from Vmax from Ref. 75.
c C-terminal 20 amino acids of the α-subunit of the pertussis toxin target G-protein Gi3.
d α-Subunit of the pertussis toxin target Gi1 protein.
Because the kcat values suggested that vimentin was the preferred target for SpyA-mediated ADP-ribosylation, a modified competitive assay was performed. SpyA was reacted with equimolar amounts of actin and vimentin in the presence of radiolabeled NAD+, and the relative rates of modification of vimentin and actin were determined for the linear range (Fig. 2). SpyA modified vimentin at a rate 9.48 ± 1.95 times faster than actin. Taken together, these results suggest that vimentin is a relevant and preferred target of SpyA.
FIGURE 2.
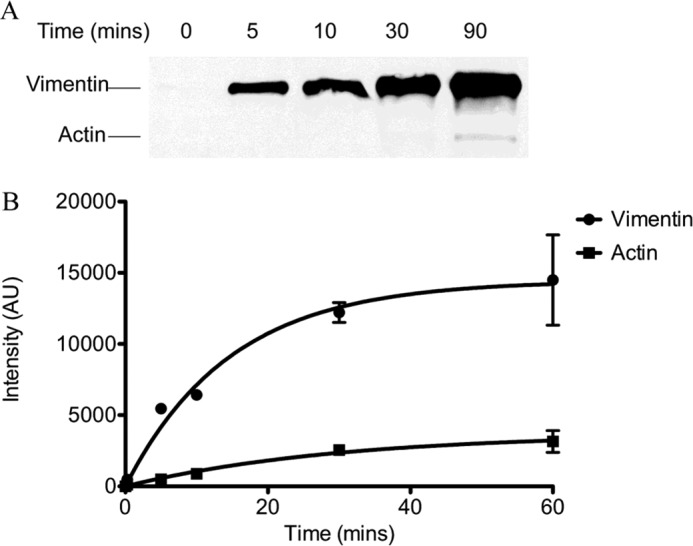
SpyA preferentially modifies vimentin. Equimolar amounts of recombinant hamster vimentin and bovine actin (877 nm) were incubated with SpyA (0.5 μm) and biotinylated NAD+ (A) or [32P]NAD+ (B) for the indicated time. Reactions were subjected to 8–16% SDS-PAGE. A, the gel was transferred to a nitrocellulose membrane and probed for biotin with streptavidin-HRP. B, the gel was subjected to phosphorimaging, and the resulting bands were quantitatively analyzed with ImageJ software. The band intensities in arbitrary units (AU) were plotted against time. Data presented are for a representative experiment. Error bars, S.D. Three independent experiments were conducted.
Vimentin Is Modified on Arginines in Serine-rich N-terminal Head Domain
SpyA contains a biglutamate catalytic site (EXE), a feature specific to arginine-targeting ADPRTs (2, 52). To verify the amino acid involved in the linkage between vimentin and ADP-ribose, the linkage was chemically challenged with hydroxylamine. Hydroxylamine has been shown to specifically cleave the ADP-ribose moiety from arginine residues while leaving intact bonds between ADP-ribose and other known acceptor residues, asparagine and cysteine (53). Treatment with hydroxylamine resulted in the release of labeled ADP-ribose from modified vimentin, whereas treatment with neither mercuric chloride, which cleaves linkages to cysteine, nor the NaCl control resulted in a loss of signal (Fig. 3). Based on the specific chemical sensitivity, the amino acid residues modified were concluded to be arginines.
FIGURE 3.
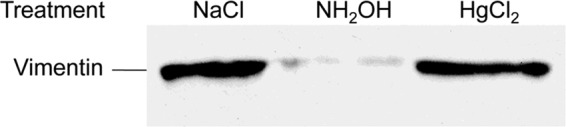
Vimentin is modified on arginine residues. Recombinant hamster vimentin (4.5 μm) was incubated with biotinylated NAD+ (100 μm) and SpyA (0.5 μm) at 37 °C. The reactions were terminated by boiling for 10 min. The indicated chemical was added (hydroxylamine (0.5 m), mercuric chloride (1 mm), or NaCl (1 m)) and incubated for 60 min at room temperature. The vimentin was subjected to SDS-PAGE, transferred, and probed with streptavidin.
The amount of SpyA-mediated incorporation of ADP-ribose into vimentin was quantitatively analyzed by determining the amount of radiolabeled APD-ribose transferred to vimentin. Analysis of the stoichiometry of ADP-ribosylation of vimentin by SpyA indicated that there are multiple modification sites (Fig. 4). Based on these calculations, SpyA transferred 2 mol of ADP-ribose/mol of vimentin, suggesting an average of two ADP-ribosylation events per vimentin subunit. Tandem mass spectrometric analysis was subsequently undertaken to identify the modified amino acids. Identification of ADP-ribose-modified peptides relied upon detection of ADP-ribose-specific marker ions from collision-induced dissociation analysis followed by peptide sequencing with ECD (46). All ions were observed within the mass analyzer mass accuracy specifications (i.e. >10 ppm for Orbitrap acquisitions). After analysis of multiple preparations, a single peptide was consistently identified at the limit of detection (SpyA ≤1 μm, time point ≤30 min), containing arginines at positions 44 and 49, termed P44/49 (Table 2). Although both Arg-44 and Arg-49 were modified, only a single arginine was modified on any given peptide (i.e. we did not observe any peptides modified with two ADP-ribose moieties). Three additional peptides were identified less frequently than P44/49; these peptides contained Arg-63, -303, and -449. Arg-44, -49, and -63 reside within the N-terminal head domain, which contains multiple regulatory serine phosphorylation sites (Fig. 5). Peptides containing these ADP-ribose-modified arginines were identified in both human and hamster vimentin preparations.
FIGURE 4.
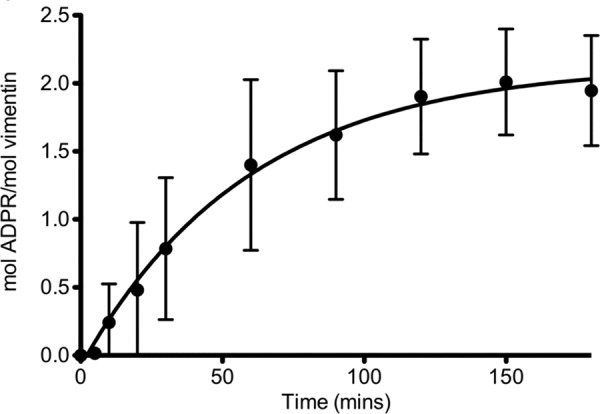
SpyA modifies vimentin at two sites. Recombinant wild type SpyA (0.3 μm) was incubated with vimentin (2.3 μm) in the presence of [32P]NAD+ (100 μm) in buffer (100 μm HEPES) for the indicated amount of time. The reaction was stopped by the addition of 5× SDS-protein sample buffer. Reactions were subjected to an 8–16% gradient SDS-PAGE, and the radioactive vimentin bands were excised and quantified via scintillation counting. [32P]ADP-ribose incorporation into vimentin was determined as a percentage of the total radioactivity per reaction volume and converted into moles. Mol of ADP-ribose (ADPR)/mol of vimentin were calculated and plotted against time. Approximately 2 mol of [32P]ADP-ribose ware incorporated/mol of vimentin. Data represent the averages of four experiments presented as means ± S.D. (error bars).
TABLE 2.
Sites of modification on vimentin
Modified arginines are in boldface type and denoted by R*. Peptides were identified using the mass spectrometry ion marker identification method (46). All peptides were identified at the lowest limit of detection for each independent assay. Numbering refers to the human vimentin sequence.
| Peptide name | Peptide | Modified arginine(s) |
|---|---|---|
| P44/49 | TYSLGSALR*PSTSR*SLYASSPGGVYATR | Arginines 44 and 49 |
| P63 | SLYASSPGGVYATR*SSAVR | Arginine 63 |
| P303 | FADLSEAANR*NNDALR | Arginine 303 |
| P449 | TVETR*DGQVINETSQHHDDLE | Arginine 449 |
FIGURE 5.

SpyA modifies arginines in the head domain of vimentin. The sequence represents the primary sequence of the N-terminal head domain of vimentin. Modified arginines are marked in boldface type as R*, and phosphorylation sites are denoted by underlining (40, 54, 71–73).
ADP-ribosylated Vimentin Exhibits Defect in in Vitro Polymerization
The modifications of vimentin in the serine-rich head domain suggested that SpyA-mediated ADP-ribosylation of vimentin may cause a defect in polymerization of vimentin into filaments, similar to that caused by phosphorylation of regulatory serines (54). The ability of modified vimentin to form filaments was assayed utilizing the inherent nature of vimentin to polymerize in vitro in high NaCl buffer (150 mm). Controls included the SpyA catalytic site mutant, SpyAE187A, and buffer (0.5 m NaCl, 0.1 m MES). Filamentation was assayed using the well established sedimentation assay. Vimentin polymerization was induced by the addition of NaCl, and ultracentrifugation was utilized to separate polymers from soluble subunits (49). When subjected to SpyA treatment prior to NaCl-induced filamentation, soluble vimentin subunits were retained in the supernatants, whereas vimentin treated with SpyAE187A or buffer completely polymerized into filaments (Fig. 6, lanes 1–3). Minimal background filamentation occurs due to the presence of the NaCl in the buffer used. Although all three treatments of vimentin (SpyA, SpyAE187A, and buffer) maintained soluble subunits prior to the addition of additional NaCl, SpyA-treated vimentin retained more soluble subunits than either SpyAE187A or buffer-treated vimentin (Fig. 6, lanes 4–6), recapitulating the results observed in the high salt condition (lanes 1–3).
FIGURE 6.
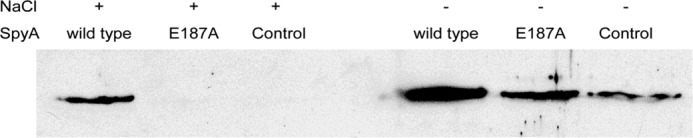
Treatment with SpyA causes a defect in filament formation. Vimentin (0.02 μg/μl, 350 nm) was treated with wild type SpyA (40 nm), inactive mutant SpyAE187A (40 nm), or buffer (25 mm NaCl and 5 mm MES) in the presence of NAD+. NaCl was added to a final concentration of 150 μm where indicated. Polymerization was allowed to occur for 1 h at 22 °C. Because SpyA buffer contains some NaCl, background filamentation occurred in the absence of additional NaCl. Filaments were separated from soluble subunits by ultracentrifugation (100,000 × g) for 30 min at 22 °C. Soluble subunits were then subjected to SDS-PAGE analysis, transferred to nitrocellulose, and probed with goat anti-vimentin antibody and secondary mouse-anti-goat HRP antibody. Soluble subunits were absent upon induction of filamentation unless modified by wild type SpyA.
Effect of SpyA on Vimentin Cytoskeleton of HeLa and RAW 264.7 Cells
Studies on the effects of C3 family ADPRTs are complicated by the absence of known binding and translocation domains and poor uptake of the toxins by cells. A variety of modifications to the toxins have therefore been employed to allow efficient access of the toxin to intracellular targets for functional studies (55–57). To allow expression of SpyA within cells, the eukaryotic expression vector pCMV-myc-SpyA was used. This construct expresses SpyA, lacking the N-terminal 30-amino acid putative signal sequence (2), fused to a 13-amino acid c-Myc epitope at the N terminus. Upon transfection of HeLa cells with pCMV-myc-SpyA, a pronounced collapse of the vimentin cytoskeleton around the nucleus (Fig. 7, D and F) was observed in transfected cells expressing SpyA (indicated by green immunofluorescence staining of myc-SpyA; Fig. 7, E and F), similar to the perinuclear collapse of vimentin observed after treatment of fibroblasts with the PKC activator phorbol 12-myristate 13-acetate (58). Transfection with plasmid containing catalytically inactive SpyA, pCMV-myc-SpyAE187A, had no apparent effect on the vimentin cytoskeleton (Fig. 7, A and C). 72.9 ± 7.3% of cells transfected with wild type SpyA showed vimentin collapse, as defined by retraction of the cytoskeleton from the cell membrane, whereas 24.9 ± 8.5% of cells in the presence of the catalytic mutant showed some collapse (p = 0.0018). Extensive and complete vimentin collapse around the nucleus was observed only in wild type SpyA transfectants. For this analysis, cells appearing to be undergoing cell death, characterized by cells appearing to produce apoptotic bodies and nuclear fragmentation, were not enumerated.
FIGURE 7.

SpyA disrupts vimentin filaments. Catalytic mutant SpyAE187A (A–C) or wild type SpyA (D–F) was expressed from the pCMV-myc-SpyAE187A or pCMV-myc-SpyA plasmid, respectively. Visualization of vimentin was accomplished by staining with anti-vimentin antibody and immunofluorescence (A and D). The presence of SpyA was detected by immunofluorescence staining of the N-terminal c-Myc tag (B and E). Merged images show both SpyA and vimentin (C and F). Expression of SpyA in cells transfected with pCMV-myc-SpyA (C), but not SpyAE187A (F), showed a marked collapse of the vimentin cytoskeletal filaments. Cells were observed for retraction of the vimentin cytoskeleton and statistically analyzed (72.9 ± 7.3% of cells transfected with pCMV-myc-SpyA showed vimentin retraction, and 24.9 ± 8.5% of cells transfected with pCMV-myc-SpyAE187A showed some retraction (p = 0.0018), with complete collapse observed only in cells transfected with the gene encoding active SpyA). Scale bar, 10 μm.
It has previously been shown that up-regulation and secretion of vimentin are important steps in the proper differentiation and activation of macrophages (35, 36, 59, 60). To test the effect of SpyA on the vimentin cytoskeleton of the murine macrophage-like cell line RAW 264.7, these cells were transfected with the bicistronic expression vector pEF1α-IRES-DsRed-Express2-SpyA or pEF1α-IRES-DsRed-Express2-SpyAE187A. After allowing protein expression for 18 h, LPS was then added (100 ng/ml) to differentiate and activate the macrophages. The cells were then analyzed for the presence of an intact vimentin cytoskeleton via immunofluorescent microscopy. Although inactive (no LPS treatment) cells contained nearly unobservable levels of vimentin, activated, untransfected cells exhibited extensive vimentin polymerization, forming a network of filament-like structures within the cell. Cells transfected with DNA encoding the catalytic mutant SpyAE187A were predominantly indistinguishable from the control cells (Fig. 8, A–C). The majority of cells transfected with DNA encoding the wild type SpyA exhibited either a complete loss or significant reduction of vimentin filaments (Fig. 8, D–I), with a minority of cells exhibiting an intact vimentin network (Fig. 8, J–L). Complete loss of the vimentin cytoskeleton was never observed in the cells expressing the catalytic mutant SpyAE187A. In a blinded test, 80.0% of wild type SpyA-transfected cells were identified as having either reduced or no vimentin filaments, whereas 10.3% of cells transfected with the catalytic mutant were identified as having reduced staining compared with untransfected controls (p < 0.0001). These results indicated that expression of the wild type SpyA, but not the active site mutant toxin, caused a defect in vimentin polymerization.
FIGURE 8.

Vimentin filamentation in SpyA-transfected RAW 264.7 cells. RAW cells were transfected with the mammalian expression vector pEF1α-IRES-DsRed-Express2-SpyAE187A, expressing dsRed-Express2 and either wild type SpyA or SpyAE187A bicistronically. 18 h after expression, the cells were stimulated with 100 ng/ml LPS to induce vimentin expression and filamentation, fixed, permeabilized, and stained for vimentin. Transfectants were identified by the expression of intracellular fluorescent protein dsRed-Express2. A–C, transfected cell expressing dsRedExpress/SpyAE187A stained for vimentin. Staining was identical to that of control (non-transfected, mock-transfected, or empty vector) cells. D–F, a subset of SpyA-transfected cells had only partial vimentin staining. G–I, the majority of wild type SpyA-transfected cells exhibited little to no vimentin staining, some of which exhibited a flattened morphology. J–L, a minority of cells transfected with the wild type SpyA expression vector pEF1α-IRES-DsRed-Express2-SpyA were consistent with and indistinguishable from control cells. 28 of 35 cells transfected with the wild type SpyA exhibited decreased or no vimentin staining compared with 4 of 39 cells transfected with the SpyAE187A catalytic mutant (p < 0.0001).
DISCUSSION
SpyA has been shown to be a promiscuous ADPRT, modifying a number of proteins in eukaryotic cell extracts (2). Prominent among these were actin, a previously described target of other bacterial ADPRTs, and vimentin, an uncharacterized bacterial ADPRT target (2, 25). The actin cytoskeleton is a common target of bacterial toxins, and SpyA was shown to cause depolymerization of the actin cytoskeleton in vivo (2). In this report, to resolve the affinity of SpyA for vimentin relative to actin, we quantitatively analyzed the ADP-ribosylation of vimentin and actin by SpyA by determining the enzyme kinetics (Km and kcat) of SpyA for NAD+ in the presence of actin and vimentin. We observed a significantly higher kcat value of SpyA for vimentin and noted that the activity of SpyA for vimentin was in the range of activities of other bacterial ADPRTs for their targets. The Km values for NAD+ in the presence of both actin and vimentin were similar, indicating that SpyA was able to bind NAD+ with comparable affinity in the presence of both actin and vimentin. Thus, the difference in kcat values was not due to alteration of NAD+ affinity upon binding of different substrates, although the nature of substrate specificity was not determined in this study.
Although both kcat values were within the same order of magnitude as a number of other known ADPRT substrates, the turnover number for vimentin was nearly 6 times that of actin. When exposed to equimolar amounts of vimentin and actin within the same reaction, SpyA modified vimentin at a nearly 10-fold greater rate than actin. Taken together, the higher vimentin kcat value and the faster rate of modification of vimentin relative to actin suggested that vimentin was the preferred target of SpyA.
In comparison with other bacterial ADPRT toxins, the kcat values were similar to those of the C3 exoenzymes (Table 1). The kcat value of actin for the actin-targeting toxins Photox and iota was relatively high, whereas for SpyA, the actin kcat was significantly lower (41, 61). The greater SpyA kcat value for vimentin indicated that vimentin was a more highly modified substrate than actin.
Vimentin is a dynamic structure that is regulated by a complex cell cycle-dependent endogenous phosphorylation. Phosphorylation generally occurs in the N-terminal head domain, resulting in disassembly of vimentin filaments, and is mediated by a number of kinases, including PKA, PKC, CaM-kinase II, PAK, and Cdc2 kinase (for reviews, see Refs. 62 and 63). There is also precedence for bacterial dephosphorylation of the vimentin cytoskeleton; the Salmonella typhimurium SptP effector tyrosine phosphatase protein binds to, and may dephosphorylate, vimentin, leading to disruption of the vimentin cytoskeleton (64).
Similar to phosphorylation events, we find the vimentin head domain to be a target of modification by SpyA, with the primary site of modification found on Arg-44/49 and a secondary modification on Arg-63. The additional two ADP-ribosylation sites were Arg-303 and Arg-449, which lie at the beginning of the second (2b) coiled-coil region and the tail domain, respectively. Relatively little is known about the region surrounding Arg-303, and although two phosphorylation sites (Thr-456 and Ser-457) have been identified, they are currently uncharacterized (65).
The head domain of all intermediate filaments is crucial for filament assembly, and a recent study of vimentin structure suggests phosphorylation of this domain prevents the association of tetramers into higher order structures (66). This disruption was mediated by movement of the head domain upon phosphorylation, potentially caused by steric hindrance or a local change in charge due to the addition of a phosphate group. ADP-ribosylation of arginines in the head domain, including Arg-44, -49, and -63, may cause dissociation of subunits via a similar mechanism of action as phosphorylation on vimentin tetramer association.
Vimentin was recently shown to be endogenously ADP-ribosylated, although little is currently known about the nature of that modification (37). However, desmin, a related intermediate filament, was shown to be modified by mammalian muscle ADPRT at several positions, resulting in the disruption of desmin assembly (4, 5, 38). The major site of modification was determined to be Arg-48, with Arg-68 as a secondary site, and both lie in the head domain, similar to arginines 44, 49, and 63 of vimentin (6). Similar to endogenous ADP-ribosylation of desmin, SpyA-mediated ADP-ribosylation of soluble vimentin subunits resulted in a defect in filamentation (Fig. 6). It is highly likely that SpyA functions similarly to the desmin-specific endogenous ADPRT, where the modification of one or more of the identified head domain arginine residues results in the inhibition of filament formation. This study presents further evidence that ADP-ribosylation of the head domain of intermediate filaments may represent a novel form of cytoskeletal regulation, beyond phosphorylation.
Although its role in bacterial infection has not been elucidated, the vimentin cytoskeleton may play an important role. The effect of modification of the vimentin cytoskeleton by SpyA was investigated in the context of live cells and led to a collapse of the vimentin filaments around the nucleus (Fig. 7). Although this study cannot rule out the effect of other SpyA ADP-ribosylated targets on this collapse, it is possible that SpyA-mediated modification was directly responsible for the collapse in light of the in vitro effect of ADP-ribosylation on vimentin filamentation. The vimentin cytoskeletal network has been found to be an important component in fibroblast motility and contractile ability, both important for wound closure (26). When subjected to mechanical injury, vimentin−/− mice were shown to have a defect in the wound healing process (27). Recently, in a mouse skin infection model, we found that mice infected with spyA− S. pyogenes produced smaller lesions than those infected with wild type S. pyogenes, (3). It is an intriguing possibility that modification and disruption of the vimentin cytoskeleton may be a molecular mechanism by which S. pyogenes inhibits the healing of lesions. Furthermore, reorganization of the vimentin cytoskeleton, via phosphorylation of the head domain, has been shown to be involved in proper neutrophil granule secretion, suggesting an important regulatory role of vimentin that potentially could be affected by ADP-ribosylation of the head domain (67, 68).The ADP-ribosylation of vimentin presented here, resulting in an in vitro defect in polymerization and probably responsible for the collapse of the in vivo cytoskeleton, may aid in preventing wound healing and proper neutrophil function at sites destroyed by the numerous toxins secreted by S. pyogenes, leading to further dissemination of the bacteria.
ADP-ribosylation and/or disruption of normal vimentin regulation may also affect the reorganization of the vimentin cytoskeleton in LPS-activated RAW 264.7 cells. Endogenous vimentin regulation has recently been shown to be involved in macrophage maturation, and vimentin secretion has been shown to be important for intracellular survival of phagocytized E. coli (35, 36). Utilizing the murine macrophage cell line RAW 264.7, we showed transfection of these cells by wild type SpyA resulted in a reduction or absence of vimentin intra- and extracellular staining in a majority of transfected cells. Because intracellular vimentin is up-regulated during monocyte differentiation and extracellular vimentin has been shown to have an important role in intracellular killing of phagocytized bacteria, an intriguing possibility is that SpyA may have a role in reducing the microbicidal activity of macrophages. Although SpyA appeared to reduce filamentous vimentin of a macrophage-like cell line, it is unknown what effect SpyA may have on other cell types. Interestingly, S. pyogenes has been shown to bind to vimentin secreted by injured skeletal muscle cells, potentially allowing “homing” of disseminated S. pyogenes to areas of soft tissue injury, which are associated with necrotizing fasciitis (69, 70). The effect of ADP-ribosylation of vimentin on this activity of S. pyogenes remains to be seen.
The in vivo disorganization of the vimentin cytoskeleton suggests that SpyA may affect numerous cellular functions, potentially including macrophage maturation and generation of the oxidative burst, neutrophil granule secretion, and fibroblast contraction and motility involved in wound healing. Future studies will focus on the elucidation of the effects of SpyA on these cellular processes.
Acknowledgments
We thank Dr. Michael Starnbach for the plasmid gift and Dr. Itay Levin for technical support as well as Dr. Natalia Korotkova for the expression and purification of human vimentin.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grants A1064515 and 1U54 AI57141–02, NIH Pharmacological Sciences Training Grant T32 GM07750, and NIH, NIGMS, National Research Service Award T32 GM07270. This work was also supported by the University of Washington Proteomics Resource.
This work is dedicated to the memory of Carleen M. Collins, 1955–2008.
- ADPRT
- ADP-ribosyltransferase
- ϵ-NAD+
- etheno-NAD+
- ϵ-AMP
- etheno-AMP
- ECD
- electron capture dissociation
- IF
- intermediate filament.
REFERENCES
- 1. Martin J. M., Green M. (2006) Group A streptococcus. Semin. Pediatr. Infect. Dis. 17, 140–148 [DOI] [PubMed] [Google Scholar]
- 2. Coye L. H., Collins C. M. (2004) Identification of SpyA, a novel ADP-ribosyltransferase of Streptococcus pyogenes. Mol. Microbiol. 54, 89–98 [DOI] [PubMed] [Google Scholar]
- 3. Hoff J. S., DeWald M., Moseley S. L., Collins C. M., Voyich J. M. (2011) SpyA, a C3-like ADP-ribosyltransferase, contributes to virulence in a mouse subcutaneous model of Streptococcus pyogenes infection. Infect. Immun. 79, 2404–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang H. Y., Graves D. J., Robson R. M., Huiatt T. W. (1993) ADP-ribosylation of the intermediate filament protein desmin and inhibition of desmin assembly in vitro by muscle ADP-ribosyltransferase. Biochem. Biophys. Res. Commun. 197, 570–577 [DOI] [PubMed] [Google Scholar]
- 5. Huang H. Y., Zhou H., Huiatt T. W., Graves D. J. (1996) Target proteins for arginine-specific mono(ADP-ribosyl) transferase in membrane fractions from chick skeletal muscle cells. Exp. Cell Res. 226, 147–153 [DOI] [PubMed] [Google Scholar]
- 6. Zhou H., Huiatt T. W., Robson R. M., Sernett S. W., Graves D. J. (1996) Characterization of ADP-ribosylation sites on desmin and restoration of desmin intermediate filament assembly by de-ADP-ribosylation. Arch. Biochem. Biophys. 334, 214–222 [DOI] [PubMed] [Google Scholar]
- 7. Lupi R., Corda D., Di Girolamo M. (2000) Endogenous ADP-ribosylation of the G protein β subunit prevents the inhibition of type 1 adenylyl cyclase. J. Biol. Chem. 275, 9418–9424 [DOI] [PubMed] [Google Scholar]
- 8. Lupi R., Dani N., Dietrich A., Marchegiani A., Turacchio S., Berrie C. P., Moss J., Gierschik P., Corda D., Di Girolamo M. (2002) Endogenous mono-ADP-ribosylation of the free Gβγ prevents stimulation of phosphoinositide 3-kinase-γ and phospholipase C-β2 and is activated by G-protein-coupled receptors. Biochem. J. 367, 825–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fendrick J. L., Iglewski W. J. (1989) Endogenous ADP-ribosylation of elongation factor 2 in polyoma virus-transformed baby hamster kidney cells. Proc. Natl. Acad. Sci. U.S.A. 86, 554–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jäger D., Werdan K., Müller-Werdan U. (2011) Endogenous ADP-ribosylation of elongation factor-2 by interleukin-1β. Mol. Cell. Biochem. 348, 125–128 [DOI] [PubMed] [Google Scholar]
- 11. Paone G., Wada A., Stevens L. A., Matin A., Hirayama T., Levine R. L., Moss J. (2002) ADP ribosylation of human neutrophil peptide-1 regulates its biological properties. Proc. Natl. Acad. Sci. U.S.A. 99, 8231–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paone G., Stevens L. A., Levine R. L., Bourgeois C., Steagall W. K., Gochuico B. R., Moss J. (2006) ADP-ribosyltransferase-specific modification of human neutrophil peptide-1. J. Biol. Chem. 281, 17054–17060 [DOI] [PubMed] [Google Scholar]
- 13. Margarit S. M., Davidson W., Frego L., Stebbins C. E. (2006) A steric antagonism of actin polymerization by a salmonella virulence protein. Structure 14, 1219–1229 [DOI] [PubMed] [Google Scholar]
- 14. Wegner A., Aktories K. (1988) ADP-ribosylated actin caps the barbed ends of actin filaments. J. Biol. Chem. 263, 13739–13742 [PubMed] [Google Scholar]
- 15. Weigt C., Just I., Wegner A., Aktories K. (1989) Nonmuscle actin ADP-ribosylated by botulinum C2 toxin caps actin filaments. FEBS Lett. 246, 181–184 [DOI] [PubMed] [Google Scholar]
- 16. Perieteanu A. A., Visschedyk D. D., Merrill A. R., Dawson J. F. (2010) ADP-ribosylation of cross-linked actin generates barbed-end polymerization-deficient F-actin oligomers. Biochemistry 49, 8944–8954 [DOI] [PubMed] [Google Scholar]
- 17. Schwan C., Stecher B., Tzivelekidis T., van Ham M., Rohde M., Hardt W. D., Wehland J., Aktories K. (2009) Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 5, e1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chardin P., Boquet P., Madaule P., Popoff M. R., Rubin E. J., Gill D. M. (1989) The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 8, 1087–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paterson H. F., Self A. J., Garrett M. D., Just I., Aktories K., Hall A. (1990) Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 111, 1001–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wiegers W., Just I., Müller H., Hellwig A., Traub P., Aktories K. (1991) Alteration of the cytoskeleton of mammalian cells cultured in vitro by Clostridium botulinum C2 toxin and C3 ADP-ribosyltransferase. Eur. J. Cell Biol. 54, 237–245 [PubMed] [Google Scholar]
- 21. Wilde C., Chhatwal G. S., Schmalzing G., Aktories K., Just I. (2001) A novel C3-like ADP-ribosyltransferase from Staphylococcus aureus modifying RhoE and Rnd3. J. Biol. Chem. 276, 9537–9542 [DOI] [PubMed] [Google Scholar]
- 22. Wilde C., Aktories K. (2001) The Rho-ADP-ribosylating C3 exoenzyme from Clostridium botulinum and related C3-like transferases. Toxicon 39, 1647–1660 [DOI] [PubMed] [Google Scholar]
- 23. Wilde C., Chhatwal G. S., Aktories K. (2002) C3stau, a new member of the family of C3-like ADP-ribosyltransferases. Trends Microbiol. 10, 5–7 [DOI] [PubMed] [Google Scholar]
- 24. Wilde C., Just I., Aktories K. (2002) Structure-function analysis of the Rho-ADP-ribosylating exoenzyme C3stau2 from Staphylococcus aureus. Biochemistry 41, 1539–1544 [DOI] [PubMed] [Google Scholar]
- 25. Coburn J., Dillon S. T., Iglewski B. H., Gill D. M. (1989) Exoenzyme S of Pseudomonas aeruginosa ADP-ribosylates the intermediate filament protein vimentin. Infect. Immun. 57, 996–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eckes B., Dogic D., Colucci-Guyon E., Wang N., Maniotis A., Ingber D., Merckling A., Langa F., Aumailley M., Delouvée A., Koteliansky V., Babinet C., Krieg T. (1998) Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 111, 1897–1907 [DOI] [PubMed] [Google Scholar]
- 27. Eckes B., Colucci-Guyon E., Smola H., Nodder S., Babinet C., Krieg T., Martin P. (2000) Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 113, 2455–2462 [DOI] [PubMed] [Google Scholar]
- 28. Nieminen M., Henttinen T., Merinen M., Marttila-Ichihara F., Eriksson J. E., Jalkanen S. (2006) Vimentin function in lymphocyte adhesion and transcellular migration. Nat. Cell Biol. 8, 156–162 [DOI] [PubMed] [Google Scholar]
- 29. Ivaska J., Pallari H. M., Nevo J., Eriksson J. E. (2007) Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313, 2050–2062 [DOI] [PubMed] [Google Scholar]
- 30. Styers M. L., Kowalczyk A. P., Faundez V. (2005) Intermediate filaments and vesicular membrane traffic. The odd couple's first dance? Traffic 6, 359–365 [DOI] [PubMed] [Google Scholar]
- 31. Pallari H. M., Eriksson J. E. (2006) Sci. STKE 2006, pe53- [DOI] [PubMed] [Google Scholar]
- 32. Coulombe P. A., Wong P. (2004) Cytoplasmic intermediate filaments revealed as dynamic and multipurpose scaffolds. Nat. Cell Biol. 6, 699–706 [DOI] [PubMed] [Google Scholar]
- 33. Byun Y., Chen F., Chang R., Trivedi M., Green K. J., Cryns V. L. (2001) Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 8, 443–450 [DOI] [PubMed] [Google Scholar]
- 34. Schietke R., Bröhl D., Wedig T., Mücke N., Herrmann H., Magin T. M. (2006) Mutations in vimentin disrupt the cytoskeleton in fibroblasts and delay execution of apoptosis. Eur. J. Cell Biol. 85, 1–10 [DOI] [PubMed] [Google Scholar]
- 35. Mor-Vaknin N., Punturieri A., Sitwala K., Markovitz D. M. (2003) Vimentin is secreted by activated macrophages. Nat. Cell Biol. 5, 59–63 [DOI] [PubMed] [Google Scholar]
- 36. Benes P., Macecková V., Zdráhal Z., Konecná H., Zahradnícková E., Muzík J., Smarda J. (2006) Role of vimentin in regulation of monocyte/macrophage differentiation. Differentiation 74, 265–276 [DOI] [PubMed] [Google Scholar]
- 37. Dani N., Stilla A., Marchegiani A., Tamburro A., Till S., Ladurner A. G., Corda D., Di Girolamo M. (2009) Combining affinity purification by ADP-ribose-binding macro domains with mass spectrometry to define the mammalian ADP-ribosyl proteome. Proc. Natl. Acad. Sci. U.S.A. 106, 4243–4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan J., Huiatt T. W., Liao C. X., Robson R. M., Graves D. J. (1999) The effects of mono-ADP-ribosylation on desmin assembly-disassembly. Arch. Biochem. Biophys. 363, 314–322 [DOI] [PubMed] [Google Scholar]
- 39. Geisler N., Weber K. (1988) Phosphorylation of desmin in vitro inhibits formation of intermediate filaments. Identification of three kinase A sites in the amino-terminal head domain. EMBO J. 7, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geisler N., Hatzfeld M., Weber K. (1989) Phosphorylation in vitro of vimentin by protein kinases A and C is restricted to the head domain. Identification of the phosphoserine sites and their influence on filament formation. Eur. J. Biochem. 183, 441–447 [DOI] [PubMed] [Google Scholar]
- 41. Visschedyk D. D., Perieteanu A. A., Turgeon Z. J., Fieldhouse R. J., Dawson J. F., Merrill A. R. (2010) Photox, a novel actin-targeting mono-ADP-ribosyltransferase from Photorhabdus luminescens. J. Biol. Chem. 285, 13525–13534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Armstrong S., Merrill A. R. (2001) Application of a fluorometric assay for characterization of the catalytic competency of a domain III fragment of Pseudomonas aeruginosa exotoxin A. Anal. Biochem. 292, 26–33 [DOI] [PubMed] [Google Scholar]
- 43. Klebl B. M., Pette D. (1996) A fluorometric assay for measurement of mono-ADP-ribosyltransferase activity. Anal. Biochem. 239, 145–152 [DOI] [PubMed] [Google Scholar]
- 44. Abramoff M. D., Magalhaes P. J., Ram S. J. (2004) Image processing with ImageJ. Biophotonics International 11, 36–42 [Google Scholar]
- 45. Hengel S. M., Shaffer S. A., Nunn B. L., Goodlett D. R. (2009) Tandem mass spectrometry investigation of ADP-ribosylated kemptide. J. Am. Soc. Mass Spectrom. 20, 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hengel S. M., Icenogle L., Collins C., Goodlett D. R. (2010) Sequence assignment of ADP-ribosylated peptides is facilitated as peptide length increases. Rapid Commun. Mass Spectrom. 24, 2312–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yi E. C., Lee H., Aebersold R., Goodlett D. R. (2003) A microcapillary trap cartridge-microcapillary high-performance liquid chromatography electrospray ionization emitter device capable of peptide tandem mass spectrometry at the attomole level on an ion trap mass spectrometer with automated routine operation. Rapid Commun. Mass Spectrom. 17, 2093–2098 [DOI] [PubMed] [Google Scholar]
- 48. Eng J. K., McCormack A. L., Yates J. R., III (1994) J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 49. Herrmann H., Kreplak L., Aebi U. (2004) Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol. 78, 3–24 [DOI] [PubMed] [Google Scholar]
- 50. Ballard J. D., Collier R. J., Starnbach M. N. (1996) Anthrax toxin-mediated delivery of a cytotoxic T-cell epitope in vivo. Proc. Natl. Acad. Sci. U.S.A. 93, 12531–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aktories K., Lang A. E., Schwan C., Mannherz H. G. (2011) Actin as target for modification by bacterial protein toxins. FEBS J. 278, 4526–4543 [DOI] [PubMed] [Google Scholar]
- 52. Vogelsgesang M., Aktories K. (2006) Exchange of glutamine-217 to glutamate of Clostridium limosum exoenzyme C3 turns the asparagine-specific ADP-ribosyltransferase into an arginine-modifying enzyme. Biochemistry 45, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 53. Aktories K., Just I., Rosenthal W. (1988) Different types of ADP-ribose protein bonds formed by botulinum C2 toxin, botulinum ADP-ribosyltransferase C3 and pertussis toxin. Biochem. Biophys. Res. Commun. 156, 361–367 [DOI] [PubMed] [Google Scholar]
- 54. Eriksson J. E., He T., Trejo-Skalli A. V., Härmälä-Braskén A. S., Hellman J., Chou Y. H., Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 55. Liu X., Hu Y., Filla M. S., Gabelt B. T., Peters D. M., Brandt C. R., Kaufman P. L. (2005) The effect of C3 transgene expression on actin and cellular adhesions in cultured human trabecular meshwork cells and on outflow facility in organ cultured monkey eyes. Mol. Vis. 11, 1112–1121 [PubMed] [Google Scholar]
- 56. Genth H., Gerhard R., Maeda A., Amano M., Kaibuchi K., Aktories K., Just I. (2003) Entrapment of Rho ADP-ribosylated by Clostridium botulinum C3 exoenzyme in the Rho-guanine nucleotide dissociation inhibitor-1 complex. J. Biol. Chem. 278, 28523–28527 [DOI] [PubMed] [Google Scholar]
- 57. Aullo P., Giry M., Olsnes S., Popoff M. R., Kocks C., Boquet P. (1993) A chimeric toxin to study the role of the 21-kDa GTP-binding protein Rho in the control of actin microfilament assembly. EMBO J. 12, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nery F. C., Zeng J., Niland B. P., Hewett J., Farley J., Irimia D., Li Y., Wiche G., Sonnenberg A., Breakefield X. O. (2008) TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J. Cell Sci. 121, 3476–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garg A., Barnes P. F., Porgador A., Roy S., Wu S., Nanda J. S., Griffith D. E., Girard W. M., Rawal N., Shetty S., Vankayalapati R. (2006) Vimentin expressed on Mycobacterium tuberculosis-infected human monocytes is involved in binding to the NKp46 receptor. J. Immunol. 177, 6192–6198 [DOI] [PubMed] [Google Scholar]
- 60. Tsuru A., Nakamura N., Takayama E., Suzuki Y., Hirayoshi K., Nagata K. (1990) Regulation of the expression of vimentin gene during the differentiation of mouse myeloid leukemia cells. J. Cell Biol. 110, 1655–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nagahama M., Sakaguchi Y., Kobayashi K., Ochi S., Sakurai J. (2000) Characterization of the enzymatic component of Clostridium perfringens ι-toxin. J. Bacteriol. 182, 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Minin A. A., Moldaver M. V. (2008) Intermediate vimentin filaments and their role in intracellular organelle distribution. Biochemistry 73, 1453–1466 [DOI] [PubMed] [Google Scholar]
- 63. Omary M. B., Ku N. O., Tao G. Z., Toivola D. M., Liao J. (2006) “Heads and tails” of intermediate filament phosphorylation. Multiple sites and functional insights. Trends Biochem. Sci. 31, 383–394 [DOI] [PubMed] [Google Scholar]
- 64. Murli S., Watson R. O., Galán J. E. (2001) Role of tyrosine kinases and the tyrosine phosphatase SptP in the interaction of Salmonella with host cells. Cellular Microbiology 3, 795–810 [DOI] [PubMed] [Google Scholar]
- 65. Chou Y. H., Opal P., Quinlan R. A., Goldman R. D. (1996) The relative roles of specific N- and C-terminal phosphorylation sites in the disassembly of intermediate filament in mitotic BHK-21 cells. J. Cell Sci. 109, 817–826 [DOI] [PubMed] [Google Scholar]
- 66. Aziz A., Hess J. F., Budamagunta M. S., Voss J. C., Fitzgerald P. G. (2010) Site-directed spin labeling and electron paramagnetic resonance determination of vimentin head domain structure. J. Biol. Chem. 285, 15278–15285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pryzwansky K. B., Merricks E. P. (1998) Chemotactic peptide-induced changes of intermediate filament organization in neutrophils during granule secretion. Role of cyclic guanosine monophosphate. Mol. Biol. Cell 9, 2933–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee K. Y., Liu L., Jin Y., Fu S. B., Rosales J. L. (2012) Cdk5 mediates vimentin Ser-56 phosphorylation during GTP-induced secretion by neutrophils. J. Cell. Physiol. 227, 739–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bryant A. E., Bayer C. R., Huntington J. D., Stevens D. L. (2006) Group A streptococcal myonecrosis. Increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J. Infect. Dis. 193, 1685–1692 [DOI] [PubMed] [Google Scholar]
- 70. Hamilton S. M., Bayer C. R., Stevens D. L., Lieber R. L., Bryant A. E. (2008) Muscle injury, vimentin expression, and nonsteroidal anti-inflammatory drugs predispose to cryptic group A streptococcal necrotizing infection. J. Infect. Dis. 198, 1692–1698 [DOI] [PubMed] [Google Scholar]
- 71. Tsujimura K., Ogawara M., Takeuchi Y., Imajoh-Ohmi S., Ha M. H., Inagaki M. (1994) Visualization and function of vimentin phosphorylation by cdc2 kinase during mitosis. J. Biol. Chem. 269, 31097–31106 [PubMed] [Google Scholar]
- 72. Ando S., Tanabe K., Gonda Y., Sato C., Inagaki M. (1989) Domain- and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 28, 2974–2979 [DOI] [PubMed] [Google Scholar]
- 73. Ogawara M., Inagaki N., Tsujimura K., Takai Y., Sekimata M., Ha M. H., Imajoh-Ohmi S., Hirai S., Ohno S., Sugiura H. (1995) Differential targeting of protein kinase C and CaM kinase II signalings to vimentin. J. Cell Biol. 131, 1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hochmann H., Pust S., von Figura G., Aktories K., Barth H. (2006) Salmonella enterica SpvB ADP-ribosylates actin at position arginine 177. Characterization of the catalytic domain within the SpvB protein and a comparison with binary clostridial actin-ADP-ribosylating toxins. Biochemistry 45, 1271–1277 [DOI] [PubMed] [Google Scholar]
- 75. Miyaoka T., Tsuchiya M., Hara N., Ishino H., Shimoyama M. (1996) Activation of Clostridium botulinum C3 exoenzyme-catalyzed ADP-ribosylation of RhoA by K+ in a Mg2+-dependent manner. J. Biochem. 119, 200–207 [DOI] [PubMed] [Google Scholar]
- 76. Böhmer J., Jung M., Sehr P., Fritz G., Popoff M., Just I., Aktories K. (1996) Active site mutation of the C3-like ADP-ribosyltransferase from Clostridium limosum. Analysis of glutamic acid 174. Biochemistry 35, 282–289 [DOI] [PubMed] [Google Scholar]
- 77. Wilde C., Vogelsgesang M., Aktories K. (2003) Rho-specific Bacillus cereus ADP-ribosyltransferase C3cer cloning and characterization. Biochemistry 42, 9694–9702 [DOI] [PubMed] [Google Scholar]
- 78. Knight D. A., Barbieri J. T. (1999) Expression of FAS-independent ADP-ribosyltransferase activity by a catalytic deletion peptide of Pseudomonas aeruginosa exoenzyme S. Biochemistry 38, 5858–5863 [DOI] [PubMed] [Google Scholar]
- 79. Arnoldo A., Curak J., Kittanakom S., Chevelev I., Lee V. T., Sahebol-Amri M., Koscik B., Ljuma L., Roy P. J., Bedalov A., Giaever G., Nislow C., Merrill R. A., Lory S., Stagljar I. (2008) Identification of small molecule inhibitors of Pseudomonas aeruginosa exoenzyme S using a yeast phenotypic screen. PLoS Genet. 4, e1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dolan K. M., Lindenmayer G., Olson J. C. (2000) Functional comparison of the NAD binding cleft of ADP-ribosylating toxins. Biochemistry 39, 8266–8275 [DOI] [PubMed] [Google Scholar]
- 81. Parikh S. L., Schramm V. L. (2004) Transition state structure for ADP-ribosylation of eukaryotic elongation factor 2 catalyzed by diphtheria toxin. Biochemistry 43, 1204–1212 [DOI] [PubMed] [Google Scholar]
- 82. Wilson B. A., Reich K. A., Weinstein B. R., Collier R. J. (1990) Active-site mutations of diphtheria toxin. Effects of replacing glutamic acid 148 with aspartic acid, glutamine, or serine. Biochemistry 29, 8643–8651 [DOI] [PubMed] [Google Scholar]
- 83. Scheuring J., Schramm V. L. (1997) Pertussis toxin. Transition state analysis for ADP-ribosylation of G-protein peptide αi3C20. Biochemistry 36, 8215–8223 [DOI] [PubMed] [Google Scholar]
- 84. Scheuring J., Berti P. J., Schramm V. L. (1998) Transition state structure for the ADP-ribosylation of recombinant Giα1 subunits by pertussis toxin. Biochemistry 37, 2748–2758 [DOI] [PubMed] [Google Scholar]