

Background: Serum albumin is a highly abundant circulating protein and may have chaperone-like activity.
Results: Serum albumin titratably inhibits aggregation and amyloid formation of client proteins and maintains chaperone-like activity under physiological conditions.
Conclusion: The chaperone-like activity of serum albumin suggests it protects against protein misfolding and aggregation.
Significance: This may have implications for protein misfolding disorders of the extracellular compartment.
Keywords: Albumin, Amyloid, Molecular Chaperone, Protein Aggregation, Protein Misfolding
Abstract
Although serum albumin has an established function as a transport protein, evidence is emerging that serum albumin may also have a role as a molecular chaperone. Using established techniques to characterize chaperone interactions, this study demonstrates that bovine serum albumin: 1) preferentially binds stressed over unstressed client proteins; 2) forms stable, soluble, high molecular weight complexes with stressed client proteins; 3) reduces the aggregation of client proteins when it is present at physiological levels; and 4) inhibits amyloid formation by both WT and L55P transthyretin. Although the antiaggregatory effect of serum albumin is maintained in the presence of physiological levels of Ca2+ and Cu2+, the presence of free fatty acids significantly alters this activity: stabilizing serum albumin at normal levels but diminishing chaperone-like activity at high concentrations. Moreover, here it is shown that depletion of albumin from human plasma leads to a significant increase in aggregation under physiologically relevant heat and shear stresses. This study demonstrates that serum albumin possesses chaperone-like properties and that this activity is maintained under a number of physiologically relevant conditions.
Introduction
Serum albumin is the most abundant protein in the serum of mammals. Its primary known role is transport: to increase the solubility of various ligands and to courier them around the body. These ligands include Cu2+, Ca2+, and the FFAs, among numerous other hydrophobic molecules (1).
In addition, it has been reported that serum albumin may also have a role as a molecular chaperone; that is, a protein able to prevent the misfolding and/or aggregation of other proteins. Marini et al. (2) observed that BSA is able to protect a number of enzymes from thermal inactivation and thermal aggregation. When compared with α-crystallin (a recognized intracellular chaperone), BSA provided protection against thermal inactivation similar to (and in some cases outperforming) α-crystallin. Significant protection from thermal aggregation was also observed. With this evidence, Marini's group proposed that serum albumin be added to the list of extracellular chaperones, to join just three other bona fide extracellular chaperones: clusterin (3–7), haptoglobin (8, 9), and α2-macroglobulin (9, 10).
Under “stress” situations, such as exposure to heat, UV light, oxidative stress, or shear forces, proteins have an increased tendency to unfold. As proteins misfold and self-associate, they may form either amorphous aggregates or highly structured, protease-resistant fibrils known as amyloid. When proteins form amorphous aggregates, they are not only unable to perform their biological role but may also physically obstruct physiological processes (11, 12). The process of amyloid formation, however, may also produce intermediates that are directly toxic to cells (13). Amyloid formation is understood to be the primary pathogenic mechanism underlying many diseases including Alzheimer's disease, the synucleinopathies, and the systemic amyloidoses, and it also contributes to the pathogenesis of type 2 diabetes mellitus (14). Thus, the potential role of chaperone proteins in the pathogenesis and treatment of such diseases has become a topic of significant interest (15). Groups studying amyloid β peptide (Aβ)2 amyloid formation have shown that serum albumin tightly binds Aβ (16), inhibits Aβ fibril formation (17), and can reduce Aβ-induced lysis of erythrocytes (18). Recently, at concentrations of less than one-tenth of that in blood, serum albumin was shown to markedly inhibit amyloid formation by insulin (19).
Nonetheless, a number of questions remain regarding the chaperone-like activity of serum albumin. Experiments demonstrating the chaperone-like activity of serum albumin to date have used serum albumin concentrations orders of magnitude lower than that found in serum (2, 18, 19). At physiological concentrations, molecular crowding effects become significant, such as effects that favor more compact species, and thus may accelerate or inhibit protein aggregation, the net outcome of which is notoriously difficult to predict (20–30).
Marini et al. (2) reported that Ca2+ at 0.1–0.5 mm abolished the protective action of albumin toward the thermally stressed sorbitol dehydrogenase. Considering that Ca2+ concentrations in serum are in the range 1.1–1.3 mm, the effect of Ca2+ on serum albumin chaperone-like activity is very relevant to its physiological role. The binding of hydrophobic ligands to serum albumin, particularly the free fatty acids, may also alter the activity of this protein (17), because hydrophobic regions are known to play a role in chaperone-client interactions.
In this study, the chaperone-like activity of serum albumin is characterized at physiological concentrations of both serum albumin and important ligands. Using established assays, the antiaggregatory and anti-amyloid properties of serum albumin are demonstrated, and evidence is provided that these properties arise from specific interactions between serum albumin and its client proteins.
EXPERIMENTAL PROCEDURES
Aggregation Assays
Proteins and reagents were purchased from Sigma-Aldrich unless otherwise specified. Defatted, metal-free BSA was used in all alcohol dehydrogenase (ADH) and insulin aggregation assays. To remove metals, BSA was dissolved in NaCl/HEPES buffer (250 mm NaCl, 30 mm HEPES, pH 7.4) with a 5-fold molar excess of EDTA and incubated with rotation at 4 °C overnight. BSA was defatted as described by Chen (31) and then lyophilized and stored at −20 °C. When needed, BSA was dissolved in the desired buffer, and concentration was determined by A280 (ϵ280 = 42,925; Prot-Param tool of SwissProt).
Different concentrations of BSA with ADH or insulin (each 350 μg/ml) were prepared in NaCl/HEPES buffer. To induce aggregation, ADH preparations were heated to 55 °C, insulin preparations were heated to 37 °C in the presence of 15 mm DTT, and turbidity was monitored (A360) as a function of time.
Aggregation data were fit to either Equation 1, 2 (32), or 3 (according to which gave a better fit) by nonlinear regression analysis using SigmaPlot version 8.02 (SPSS, Chicago, IL).
where A is A360, t is time, A0 is initial A360, Af is final A360, mf is the gradient of the stationary phase, T50 is time to 50% maximal absorbance, and n is a constant (n < 1). In Equation 1, the growth rate is described by Afn/4tm; in Equations 2 and 3, the growth rate is described by 1/τ, lag time is described by T50 − 2τ, and the baseline after growth phase is described by Af + mft. Where required, A0 and mf were set at 0 to obtain a representative fit. The results are presented as change relative to the control, set as Af = 1.
Expression and Purification of Transthyretin
Plasmid pMMHa encoding the L55P mutant of transthyretin (TTR) was a kind gift from Dr. J. W. Kelly (Scripps Research Institute, La Jolla, CA). Protein expression and purification was performed as described by Wilkinson-White and Easterbrook-Smith (33).
TTR concentration was calculated from A280 (ϵ280 = 18,450, Prot-Param tool of SwissProt). Purified TTR was precipitated with ammonium sulfate (90% (w/v)) and stored as pellets at 4 °C. When required, the TTR was resuspended in appropriate buffer, desalted using PD-10 desalting columns (GE Healthcare), concentrated using a Amicon Ultrafree-MC centrifugal filter device (Millipore), and filtered through a Ultrafree MC 0.22-μm centrifugal filter device (Millipore).
TTR Amyloid Formation in Presence of BSA
TTR samples (200 μg/ml) were induced to form amyloid in 100 mm sodium acetate with 100 mm NaCl at pH 4.2 (WT) or pH 5.2 (L55P) in the presence and absence of various concentrations of BSA at 37 °C for 168 h. Turbidity measurements and thioflavin T (ThT) assays (as described by Wilkinson-White and Easterbrook-Smith (33)) were performed at regular intervals. In the latter, the samples were diluted 3:2 in 0.5 m Tris (pH 8.2) to obtain a protein concentration of 0.1 mg/ml. The modified exponential decay equation was fitted to the fluorescence data,
where F0 is initial fluorescence intensity, Ff is final fluorescence intensity, t is time, and k is growth rate. T50 was calculated using ln2/k. To derive kinetic parameters from the A350 and ThT assay, Equations 3 and 4 were fitted to L55P-TTR and WT-TTR data, respectively.
Binding between BSA and Target Proteins: ELISA
Between all steps in the procedure, all of the wells were washed 10 times with water, and all of the incubations were performed at 37 °C for 1 h unless otherwise specified. Solutions of ADH or insulin (100 μg/ml) were prepared in 0.1 m NaHCO3 (pH 9.5), and the proteins were allowed to adsorb to the well surface of the ELISA plate. ADH plates were stressed at 70 °C (1 h). Insulin plates were stressed with 15 mm DTT in PBS (30 min), and exposed cysteines were blocked by incubating with 10 mm iodoacetamide in PBS. The plates were blocked with 0.025% (v/v) Tween in PBS (30 min). BSA (1 mg/ml in PBS) was serially diluted (1:3) down the plate and incubated. Primary antibody (monoclonal anti-BSA) and then secondary antibody (sheep anti-mouse conjugated to horseradish peroxidase) were incubated on the plate, both diluted 1:1000 in 1% (w/v) casein, 2.7 mm thymol in PBS (pH 7.4). Substrate solution was then applied (2.5 mg/ml o-phenylenediamine hydrochloride, 30% (w/v) hydrogen peroxide, 50 mm citrate, 100 mm phosphate, pH 5.0), and color was allowed to develop. The reaction was stopped with 0.5 volume of 2 m H2SO4. A492 was measured (Labsystems Multiskan RC microplate photometer and Labsystems Genesis v3.03 software), and Equation 5 was fitted to derive kinetic parameters,
where Af is final A492, [c] is [BSA], and Kd is the dissociation constant.
High Molecular Weight (HMW) Complexes
Target proteins were left in their native form or stressed, in the presence or absence of 10 mg/ml BSA, with or without FFAs also being present (mixture of palmitic, stearic, oleic, and linoleic acids 27:10:32:15 molar ratio to a total of 683 μm). ADH (800 μg/ml) was stressed at 45 °C for 60 min, and insulin (500 μg/ml) was stressed at 37 °C with 15 mm DTT for 10 min. The samples were then placed on ice (5 min), filtered using an Ultra-MC 0.22-μm centrifuge filter device (1 min), and returned to ice. The samples were applied to a Superose 6 HR 10/30 or a Superdex 75 10/30 gel filtration column (GE Healthcare) using a BioLogic DuoFlow chromatography system (Bio-Rad), then loaded onto the column equilibrated in PBS, and eluted (0.5 ml/min). Protein elution was monitored at A280, 0.5-ml fractions were collected, and the quantity of protein in the peaks was calculated by finding the area under the curve using the trapezoidal rule. Peaks at the excluded volume of the column were analyzed by SDS-PAGE and compared with pure, untreated samples of BSA, insulin, and ADH.
Depletion of Albumin from Plasma
Human blood samples were anticoagulated with 1.5 mg/ml EDTA, and plasma was obtained by taking the supernatant after blood was centrifuged at 3,000 rpm for 10 min. NaN3 (0.02%) added as a preservative. Albumin was depleted from plasma using a CM Affi-Gel® Blue Gel (Bio-Rad) according to the manufacturer's instructions, using PBS with 0.02% NaN3 as the running buffer. Where specified, plasma was diluted with PBS containing 0.02% NaN3. Heat or shear stress were applied; the latter was achieved by orbital shaking at 241 and 316 rpm with a orbit diameter 25 mm (Innova 4300), calculated to correspond to ∼40 and 60 dynes/cm2, respectively (34).
RESULTS
In the experiments performed here, the bovine homologue of human serum albumin, BSA, was used. It is likely that these two proteins possess similar chaperone-like properties, given that they share high sequence similarity with 76% homology (35, 36), their ligand binding properties are almost indistinguishable (37, 38), and chaperone-like activity has been demonstrated in both (2, 17, 19).
Chaperone-like Behavior of BSA toward ADH and Insulin
Chaperone-like activity was assessed by observing the influence of BSA on the aggregation of two test client proteins: alcohol dehydrogenase and insulin. These proteins were denatured either by heating (55 °C, ADH) or by treatment with a reducing agent (15 mm DTT at 37 °C, insulin), and the extent of aggregation was measured as turbidity (A360). BSA was tested for aggregation under the assay conditions, and no detectable aggregation was observed over the time course analyzed (results not shown).
First, BSA was screened for chaperone-like activity at physiological concentrations (Fig. 1A). In the presence of 45 mg/ml BSA, the aggregation of both ADH and insulin was drastically reduced. It can be observed that although physiological concentrations of BSA reduced total ADH aggregation, the presence of BSA also reduced the lag time to aggregation (Fig. 1A, panel i). This behavior is predicted by macromolecular crowding theory: under crowding conditions, association constants increase significantly, and equilibrium is reached earlier (21). In subsequent assays using BSA with ADH, the high BSA concentrations required to inhibit ADH aggregation ensured that such macromolecular crowding effects remained significant. This behavior was not observed for assays using insulin (Fig. 1A, panel ii), for which there may be a number of explanations: stabilization of the native fold may be the predominant crowding effect of BSA on insulin, the lower temperature at which insulin was aggregated may reduce the crowding effect of BSA, or the inherently idiosyncratic nature of crowding dynamics may be responsible.
FIGURE 1.

BSA displays chaperone-like activity at physiological concentrations, and its protection of ADH and insulin is titratable. Aggregation assays contained ADH (350 μg/ml) at 55 °C (panels i) or insulin (350 μg/ml) at 37 °C with 15 mm DTT (panels ii) in the absence (triangles) or presence (circles) of BSA (A and B), transthyretin (C), or lysozyme (D) at concentrations as indicated (mg/ml). Aggregation was monitored as A360, with a representative data set from two or more independent experiments shown in the upper panel, and the mean final aggregation (Af) from kinetic analysis of the pooled data (± 95% confidence interval) shown in the lower panel. A, chaperone-like activity of physiological concentrations of BSA (45 mg/ml): protection of ADH (panel i) and insulin (panel ii). B, chaperone-like activity of BSA lends titratable protection to ADH (panel i) and insulin (panel ii) against aggregation. C, transthyretin (control), when present at physiological levels does not protect either ADH (panel i) or insulin (panel ii) against aggregation. D, lysozyme (control) at concentrations matched to BSA does not protect either ADH (panel i) or insulin (panel ii) against aggregation.
To explore whether the chaperone-like effect was titratable, client proteins were aggregated in the presence of varying concentrations of BSA (Fig. 1B). A dose-dependent relationship was observed between BSA concentration and protein aggregation for both ADH and insulin: increasing BSA concentration led to a reduction in net aggregation of both proteins. Very little ADH aggregation was observed at concentrations of 40 mg/ml BSA, with a subunit molar ratio of ∼31:1 (Fig. 1B, panel i). A plot of final aggregation (Af) versus BSA concentration (Fig. 1B, panel i, lower panel) confirmed that increasing BSA concentration reduced aggregation. A linear relationship was observed at concentrations greater than 5 mg/ml, with a goodness of fit of the relationship (R2) of >0.98 (R2 was also >0.98 for each data set individually analyzed).
BSA was a more potent inhibitor of insulin aggregation than it was for ADH: minimal insulin aggregation was achieved at 1 mg/ml BSA, with a subunit molar ratio of ∼0.16:1 (Fig. 1B, panel ii). Increasing BSA concentration led to an exponential decrease in insulin aggregation, with an R2 of 0.96 (R2 = 0.96 and 0.93 in each data set individually analyzed).
At low concentrations, BSA had an unusual effect on ADH aggregation: BSA concentrations of 1 mg/ml increased the aggregation of ADH (Fig. 1B, panel i), an effect that was consistently reproducible. It is possible that low level crowding is affecting the folding equilibrium, that is, because crowding favors more compact species (pushing unfolded proteins either toward the native fold or to aggregate), at subphysiological concentrations the proaggregation crowding effects may dominate the chaperone-like activity of BSA. Other experimenters have observed equally complex crowding effects on aggregation kinetics (30, 39).
Wild type TTR and lysozyme were employed as controls. Both are thermally stable, and although transthyretin is an abundant (0.2 mg/ml) circulating protein that forms tetramers of similar molecular weight to BSA, lysozyme is highly soluble (allowing it to be concentration-matched to BSA), and, like BSA, contains disulfide bonds which stabilize its structure. Neither of these proteins detectably aggregated over the time course analyzed (results not shown).
Physiological concentrations of TTR did not inhibit ADH aggregation (Fig. 1C, panel i), nor did they alter insulin aggregation (Fig. 1C, panel ii). Conversely, lysozyme actually enhanced aggregation of both proteins (Fig. 1D).
Physiological Ligands
Of the numerous ligands of BSA, some interact with one another in a complex manner: binding cooperatively (40), competitively (41), or independently (37). Therefore, it was investigated whether three important BSA ligands: Ca2+, Cu2+, and FFAs, had any effect on the chaperone-like activity of BSA.
Target proteins were aggregated as previously described, with [BSA] kept constant, whereas [Ca2+] was varied in a range corresponding to ∼0.5–4 times the physiological level relative to [BSA]. Samples with BSA and Ca2+ at absolute physiological levels (45 mg/ml and 1.2–2.4 mm, respectively) were also included. In a similar manner, the effect of Cu2+ was varied in a range corresponding to 1–10 times the physiological level relative to [BSA]. Again absolute physiological concentrations were also included ([Cu] = 15.5 μm (42)).
Neither Ca2+ nor Cu2+ caused any measurable interference with the chaperone-like activity of BSA toward either ADH or insulin under the titration conditions presented, nor when the cations and BSA were present at absolute physiological levels (data not shown). These observations suggest that at concentrations from 0 to well beyond physiological levels, neither Ca2+ nor Cu2+ significantly inhibits the chaperone-like activity of BSA.
Because hydrophobic interactions are known to be important in the binding between chaperones and their client proteins (17, 43), the effect of changing FFA concentrations on BSA chaperone-like activity was investigated. Physiological FFA concentration was taken as 512 μm (44), and total FFA levels were varied in a range corresponding to 1–10 times those present in the serum relative to BSA, alongside samples containing BSA and FFAs at absolute physiological concentrations. Palmitic, stearic, oleic, and linoleic acids together make up 90% of circulating FFAs (44–46) and 80% of the FFAs bound to serum albumin (47); therefore to simulate physiological conditions, a mixture of these acids was prepared in proportion to their relative quantities in human serum.
Titrating FFAs into the ADH aggregation reaction resulted in a progressive reduction in the protective effect of BSA (Fig. 2A). Net aggregation (Fig. 2A, inset) was seen to be directly proportional to FFA concentration, with an R2 of >0.98 (R2 of 0.98 and 0.94 in two separate experiments). Using this model, the addition of a physiological level of FFAs relative to BSA alone resulted in a 9–10% increase in total aggregation. This proagreggatory effect was not merely an anomaly of increasing amounts of unbound FFA in solution: controls in which ADH or insulin were aggregated in the presence of FFAs alone showed that unbound FFAs exhibited an antiaggregation influence on the precipitation of these proteins (results not shown).
FIGURE 2.
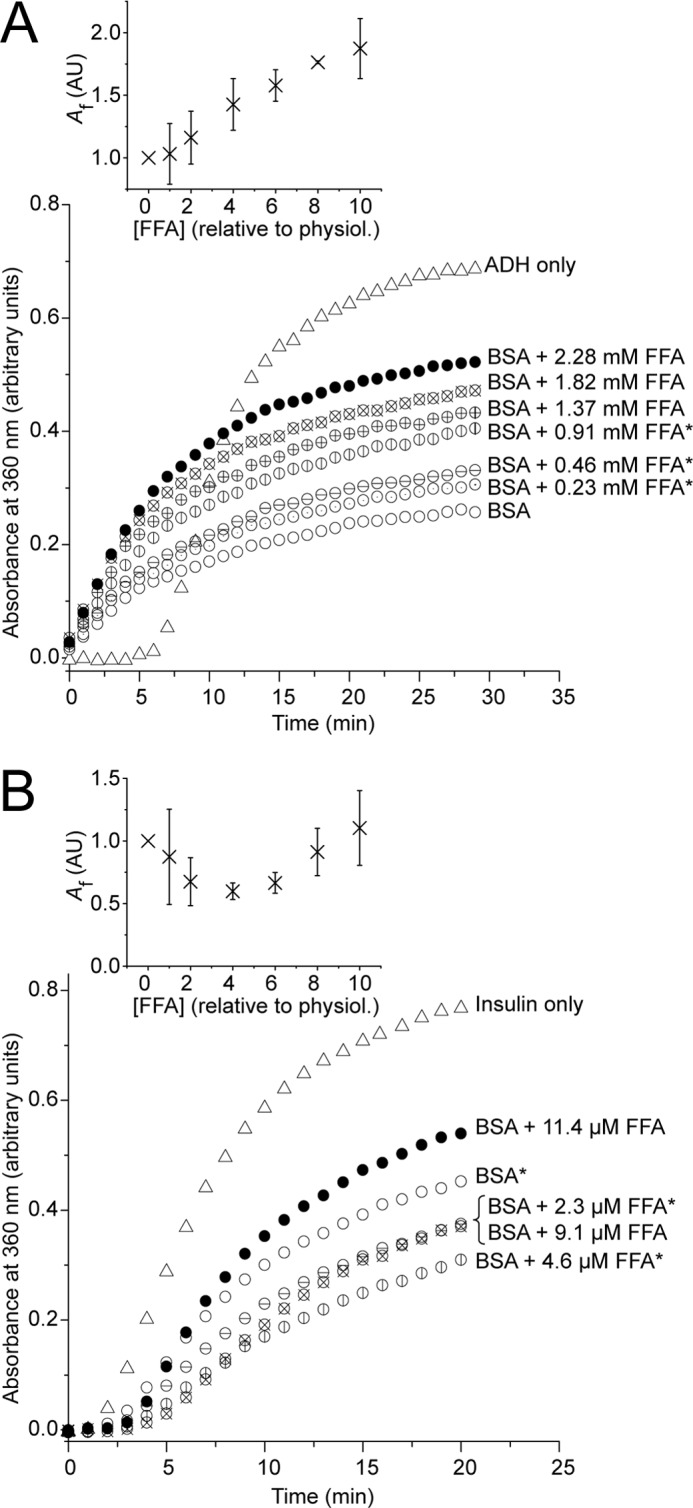
The effect of free fatty acids on BSA chaperone-like activity. Aggregation assays contained ADH (350 μg/ml) at 55 °C (A) or insulin (350 μg/ml) at 37 °C with 15 mm DTT (B) in the absence (triangles) or presence (circles) of BSA at either 20 mg/ml (A) or 0.1 mg/ml (B), with FFAs added at the concentrations indicated. Aggregation was monitored as A360. FFAs were present as a mixture of palmitic, stearic, oleic, and linoleic acids (27:10:32:15 molar ratio) according to their relative proportions in serum, and, on a mole to mole ratio compared with BSA, the concentrations used correspond to a range from 0 (○) to 10 (●) times the physiological concentration of FFAs in normal human serum. Those ≤4 times normal levels (which are known to occur in vivo) are marked with an asterisk. The main panels show a representative data set from two independent experiments, with the insets displaying mean final aggregation (Af) from kinetic analysis of the pooled data (± 95% confidence interval).
The effect of BSA on insulin aggregation showed a more complex relationship with varying FFA concentration. In repeat experiments, low levels of FFAs were seen to marginally increase the chaperone-like properties of BSA (Fig. 2B), however, as the concentration of FFAs increased protection progressively declined. Kinetic analysis (Fig. 2B, inset) displayed a parabolic relationship over the relative range of 1–8 times physiological FFA concentration, with an R2 of 0.93 (R2 = 0.98 and 0.99 in each data set individually analyzed), and using this model, the inflection point was determined to be ∼3–5 times physiological FFA concentration.
The parabolic relationship between insulin aggregation and FFA concentration is not unexpected. Although BSA itself does not aggregate over the assay time course, it is destabilized and over a longer time course will aggregate in the presence of 15 mm DTT. It was observed by others (48) and also here that FFA-bound BSA is more resistant to DTT denaturation. Thus, FFAs at low concentrations may stabilize BSA and increase its chaperone-like efficiency, whereas higher concentrations result in a trend closer to that seen for ADH.
Aggregation and Amyloid Formation by TTR
To assess whether BSA exhibited anti-amyloid activity, its effect on TTR aggregation and amyloid formation was assessed. WT-TTR and L55P-TTR (a highly aggregation-prone mutant) at physiological levels (200 μg/ml (49)) were induced to form amyloid by incubation at 37 °C in acetate buffer at pH 4.2 and 5.2, respectively, in the absence or presence of BSA. Formation of amyloid was determined by induction of ThT fluorescence (50).
For WT TTR, a dose-dependent decrease in both aggregation (Fig. 3A, panel i) and amyloid formation (panel ii) was observed with increasing concentrations of BSA. The effect of BSA on L55P-TTR was similar (Fig. 3B), although this effect was achieved at lower concentrations of BSA.
FIGURE 3.

BSA inhibits aggregation and amyloid formation by WT-TTR and L55P TTR. WT-TTR (A) and L55P-TTR (B) at 200 μg/ml were incubated at 37 °C for 168 h in acetate buffer pH 4.2 and 5.2, respectively. Amyloid formation was carried out in the absence (crosses) and presence (circles) of BSA at concentrations as indicated (mg/ml). The aliquots were removed at the specified times and were assessed for A350 (panels i, net aggregation) and ThT fluorescence (panels ii, amyloid formation). Equations 4 and 3 were fitted to the WT-TTR and L55P-TTR data, respectively, to give kinetic values for the time taken for 50% of the maximum turbidity and ThT fluorescence to be achieved (T50) (panels iii; solid marker and solid trace for turbidity, and open marker and dashed trace for ThT fluorescence). Each data set is representative of three independent experiments.
Equations 3 and 4 were fitted to the data to derive values for total aggregation, total amyloid formation, and time taken for 50% of the maximum turbidity/ThT fluorescence to be achieved (T50). Total aggregation and total amyloid formation confirmed what was seen in panels i and ii of Fig. 3: aggregate formation occurred at the same rate as amyloid formation, with an inverse relationship between total aggregate/amyloid formed and [BSA]. These changes were significant (p < 0.05). Analysis of the T50 emphasized the differences between the WT and L55P-TTR (Fig. 3, panels iii); whereas BSA showed no significant effect on the T50 for WT-TTR, it significantly lengthened the T50 for L55P-TTR for both aggregate and amyloid formation (p < 0.005). Thus, BSA not only reduces aggregation and amyloid formation by WT and L55P-TTR in a dose-dependent manner but also titratably slows the formation of aggregates and amyloid by the L55P mutant.
Binding between BSA and Target Proteins
Small heat shock proteins, a class of chaperone-like proteins, reduce aggregation of client proteins by preferentially binding stressed proteins over those that are in their native fold and form relatively stable high molecular weight complexes with their client (3). ELISAs were performed to assess whether BSA shows similar preferential binding to denatured proteins. Client proteins were adsorbed to wells and either left in their native form or exposed to stress (ADH: 70 °C for 60 min; insulin: 15 mm DTT for 30 min), followed by the addition of BSA.
BSA displayed preferential binding to stressed ADH and insulin over the native form (Fig. 4). Although BSA showed moderated dose-dependent binding to the unstressed proteins immobilized on the ELISA plate, kinetic analysis revealed no significant difference between binding to the two unstressed target proteins. The dose-dependent binding to stressed proteins, however, was significantly higher for both ADH and insulin compared with unstressed clients (p < 0.001), confirmed by kinetic analysis: final absorbances (Af) were higher and dissociation constants (Kd) were lower for stressed versus unstressed protein, and these differences were significant.
FIGURE 4.
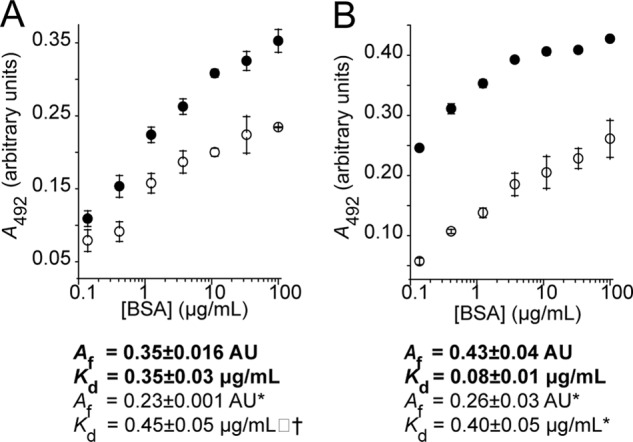
ELISA: BSA preferentially binds to adsorbed stressed target proteins compared with the native form. ADH (A) was stressed by incubation at 45 °C, whereas insulin (B) was denatured using DTT at 37 °C. Bound BSA was detected using a monoclonal BSA antibody followed by a secondary HRP-conjugated antibody. Equation 5 was fitted to the data to obtain a maximum absorbance (Af) and a dissociation constant (Kd). Stressed proteins are shown in filled circles with kinetic parameters in bold type, whereas native target proteins are shown in open circles with kinetic parameters in normal type. Significance was tested using a Student's t test. Differences between binding of BSA to stressed and unstressed proteins was significant for both ADH and insulin (p < 0.001), and differences in kinetic parameters between stressed and unstressed series had significance as indicated. *, p < 0.005; †, p < 0.05. The data shown are the means ± standard deviation of triplicate measurements; each data set provided is representative of three independent experiments.
Formation of High Molecular Weight Complexes
Clusterin and small heat shock protein are known to form HMW complexes with partially unfolded proteins under stressed conditions (3, 51, 52). Therefore, size exclusion chromatography was used to determine whether BSA also preferentially binds stressed proteins to form HMW complexes and also whether FFAs inhibit the formation of such complexes. Target proteins were stressed in the presence or absence of BSA with or without FFAs, and soluble fractions were analyzed by size exclusion chromatography (Fig. 5A).
FIGURE 5.
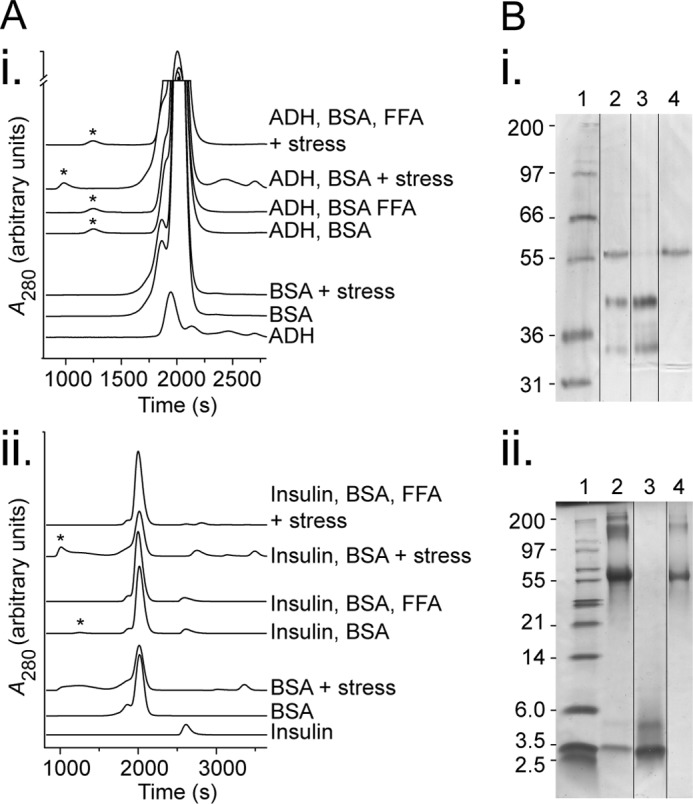
BSA forms stable high molecular weight species with stressed target proteins. A, size exclusion analysis of BSA and target proteins. Solutions of individual proteins or mixtures of BSA, FFAs (683 μm of physiological mixture), and target protein (ADH at 800 μg/ml (panel i) or insulin at 500 μg/ml (panel ii)) were left in their native state or stressed (45 °C for 60 min, or 37 °C with 15 mm DTT for 10 min, respectively). The product was then filtered to remove precipitated proteins and analyzed by size exclusion chromatography, and asterisks indicate the excluded volume from the column (different gel filtration matrices gave rise to slightly shifted elution profiles for the first, third, and fourth traces (top down in each)). B, SDS-PAGE of excluded volume peaks from stressed target protein with BSA conditions in A. The samples were run in Tris-Tricine gels and stained with silver stain (panel i) or reduced with DTT prior to loading and stained with Coomassie Blue (panel ii) with lanes as follows: standard (lane 1, band sizes indicated in kDa), excluded volume from condition stressed target protein with BSA (lane 2), target protein (lane 3), and BSA (lane 4). Lane order was rearranged as demarcated for ease of comparison. The data are representative of three independent experiments.
When unstressed ADH was incubated with BSA, a HMW peak appeared at the excluded volume of the column (Fig. 5A, asterisk) in addition to the background from the individual native proteins. This peak represented ∼0.8% of the total protein (from the area under curve; Fig. 5A, panel i). When ADH was stressed in the presence of BSA, the size of this peak increased to 1.0% of total protein, and the introduction of FFAs reduced the HMW peak to 0.9% of total protein, although neither of these differences was statistically significant.
With insulin as the target protein, BSA with the unstressed client led to formation of HMW species representing 2.0% of total protein. This increased to 10.1% of total protein under stress conditions. Moreover, this effect was reversed with the addition of FFAs to 0.2% total protein, levels significantly lower than even the unstressed state (p < 0.05).
The above results are in line with both the greater ability of BSA to protect insulin from aggregation (Fig. 1B) and the differing affinities of BSA for stressed/unstressed AHD and insulin (Fig. 4). Not only does BSA bind insulin more strongly than ADH, it also shows greater preference for the stressed form of insulin over the native fold.
In an attempt to determine the compositions of these high molecular weight species, SDS-PAGE was performed. Only when BSA was in the presence of stressed target protein were the components of the complexes detectable, and it is clear that these high molecular weight species contain mixtures of BSA and client proteins (Fig. 5B). The composition of HMW peaks derived from other conditions could not be determined despite 10-fold concentration and SYPRO® Ruby staining; however, it is likely that these HMW species are also mixtures of BSA and client proteins.
Aggregation of Plasma
To determine whether serum albumin maintains chaperone-like properties in vivo, human plasma was induced to aggregate using physiologically relevant stresses. Samples were stressed either with heat (41 °C for 42 h) or shear stress (40 dynes/cm2 for 5 days followed by 60 dynes/cm2 for 2.5 days), and aggregation was measured using A360.
Plasma depleted of albumin was compared with plasma controls that were either diluted to a comparable level of non-albumin proteins (10% plasma; Fig. 6A, panel i) or diluted to the same A280 (i.e., approximately the same total protein concentration). Under both the heat and shear stress, serum depleted of albumin aggregated significantly more than either of the samples containing serum albumin (Fig. 6A, panel ii).
FIGURE 6.

Aggregation of human serum under physiological shear and heat stress. The samples were exposed to either shear (40 dynes/cm2 for 5 days followed by 60 dynes/cm2 for 2.5 days) or thermal stress (41 °C for 42 h for A, panel ii, and B; 41 °C for 5 days for A, panel iii). Aggregation was measured by turbidity (A360). A, depletion of albumin from plasma leads to increased aggregation under physiologically relevant stresses. Panel i, serum albumin was depleted from plasma, and successful depletion was confirmed by SDS-PAGE, with lanes as follows: molecular mass standard (lane 1); 10% plasma (lane 2); and plasma depleted of serum albumin (lane 3). Panel ii, depletion of serum albumin results in a significant increase in aggregation of plasma. Whole plasma was diluted to concentrations comparable with the depleted plasma, either as 10% plasma or to the same A280, and exposed to stresses as described (n = 3 for shear; n = 4 for thermal). Panel iii, reintroduction of serum albumin to albumin-depleted plasma results in a significant reduction in aggregation under heat stress. Three conditions were run: whole plasma (diluted to the same A280 as albumin-depleted plasma), albumin-depleted plasma, and albumin-depleted plasma with serum albumin (BSA) added to a concentration proportionate to its normal concentration in blood (3.75 mg/ml) (n = 24 for each condition). B, addition of FFAs did not significantly alter aggregation of plasma. Native plasma was compared with plasma with an additional 3-fold physiological level (1.5 μm) of FFA mixture and exposed to thermal and shear stress as described (n = 3 for shear; n = 4 for thermal). Black columns, whole plasma or diluted to 10%; crossed columns, plasma to the same A280 as depleted plasma; white columns, albumin-depleted plasma; gray columns, albumin-depleted plasma with BSA added. The data shown are the means ± 95% confidence interval, with significance as indicated. *, p < 0.05; **, p ≤ 0.005; ***, p < 0.0005.
A number of other proteins, including known chaperones, also bind the albumin depletion column. To ensure that the changed aggregation behavior was a result of the removal of albumin, BSA was reintroduced to albumin-depleted plasma. This led to a significant reduction in aggregation (p = 0.005) to a level comparable with that seen in whole plasma diluted to the same A280 (Fig. 6A, panel iii). Because the protein concentration in the depleted plasma was roughly half that used in Fig. 6A (panel ii), a longer time course (5 days) was used to ensure a representative aggregation end point was met.
To elucidate whether FFA elevations present in normal physiology may alter the chaperone-like activity of serum albumin, additional FFA at a concentration three times that of physiological levels was added to undiluted plasma, and the aggregation behavior was compared with native plasma (Fig. 6B). There was no significant difference in aggregation between the native samples and those where additional FFAs had been added under either of the stress conditions.
DISCUSSION
This study demonstrates that serum albumin preferentially binds stressed over unstressed client proteins, forms soluble high molecular weight complexes with stressed client proteins, and, when present at physiological levels, reduces aggregation or amyloid formation of client proteins under stress conditions. Moreover, it is shown that significant chaperone-like activity is maintained in the presence of Ca2+, Cu2+, and FFAs and that plasma depleted of serum albumin is more susceptible to aggregation when exposed to physiological stresses, a susceptibility reversible with the reintroduction of serum albumin.
The difference in the protection afforded ADH by serum albumin compared with its effect on insulin was stark: insulin was protected far more effectively than ADH. This is unlikely to be due to an association between molecular weight of the client protein and the efficacy of serum albumin chaperone-like activity, as has been reported in clusterin (3), given that Marini et al. (2) investigated the effect of serum albumin on a larger sample of proteins and found no correlation between protein size and serum albumin efficacy. An alternative explanation for the differing efficacy of BSA could be the nature of the stressors used: whereas ADH was heat-stressed, insulin was DTT-stressed. Hence, because BSA contains multiple disulfide bonds subject to reducing stress, the ability of BSA to minimize aggregation may be related to the reduction of these bonds. Torricelli et al. (53) implied such a role for disulfide bonds in the interaction between atrial natriuretic peptide (the amyloid-forming species in isolated atrial amyloid) and serum albumin; the apparently protective complexes formed between these two proteins were only broken down with exposure to a reducing agent.
Serum albumin binds ∼50% of the Ca2+ present in the blood; therefore any influence of Ca2+ on serum albumin chaperone-like activity is highly relevant. It has been reported that Ca2+ abolishes the protective action of serum albumin toward the thermally stressed sorbitol dehydrogenase (2). Here, at physiologically relevant concentrations, the presence of Ca2+ did not observably alter the chaperone-like properties of serum albumin. A likely cause of these differing results is the ratios of Ca2+/serum albumin, because Marini et al. (2) used a ratio ∼100-fold above that present in human serum. It was observed here that free Ca2+ increased the propensity of proteins to aggregate, and therefore Marini et al. may have observed the direct effects of Ca2+ on aggregation rather than the influence of Ca2+ on serum albumin chaperone-like activity.
Approximately 10% of circulating Cu2+ is bound to serum albumin, and free Cu2+ has a known proaggregatory influence (11, 54, 55). In this study, the effect of a wide range of Cu2+ concentrations was assessed, yet no significant effect on serum albumin activity was observed.
Of the identified roles of serum albumin, FFA transport is arguably the most important, because 99% of the ∼0.5 mm FFAs present in human serum are bound to serum albumin (44, 47), and hyperlipidemia is one of the surprisingly few clinical symptoms of analbuminemia (56, 57). Serum albumin has six high affinity and numerous low affinity binding sites for this group of ligands (58, 59), and although under physiological conditions each serum albumin molecule has only approximately one FFA bound to it, this may rise to four following exercise or to as many as six during heparin treatment (60). Here it is reported that FFAs significantly alter the chaperone-like activity of serum albumin toward both client proteins (Fig. 2). Despite some stabilizing effect on albumin itself, higher FFA concentrations increased the extent of aggregation. Although normal physiological levels of FFA had little effect on serum albumin activity, very high FFA concentrations significantly altered serum albumin-mediated protection (Fig. 2) and were able to almost completely block the formation of high molecular weight complexes between stressed insulin and serum albumin (Fig. 5).
Significantly, although occupation of the FFA-binding sites reduces the chaperone-like activity, activity does not cease completely. Even with the six high affinity FFA-binding sites saturated, BSA still significantly protects both client proteins (Fig. 2), and when both BSA and FFAs were present at absolute physiological concentrations, the target proteins displayed aggregation behavior akin to that seen in Fig. 1A. Similarly, the addition of FFAs to stressed plasma did not result in any significant difference in aggregation (Fig. 6B). This is likely because BSA vastly outpopulates its client proteins when present at physiological concentrations: although FFAs may reduce the chaperone-like activity of serum albumin, with BSA at such high concentrations in serum, there is sufficient residual chaperone-like activity to prevent experimentally observable aggregation.
The above results suggest that FFAs and denatured proteins compete for the same binding sites on serum albumin. Hydrophobic interactions are known to be important in the binding between chaperones and their client proteins (43), and in BSA it has been previously demonstrated that these interactions play a critical role in the protection of β-galactosidase from thermal inactivation (61). The fatty acid-binding sites on serum albumin are long hydrophobic pockets capped by polar side chains in which each methylene tail is buried in a deep hydrophobic cavity (59), and thus occupation of this site by a fatty acid may be at the exclusion of a misfolded protein. It is possible that the observed result is not due to competition for the same site per se but rather due to the fact that FFAs and denatured proteins require different serum albumin conformers to bind (the case for the binding of bilirubin and palmitic acid (41)) because FFA binding leads to significant domain rotations in the serum albumin structure (62). Regardless of the cause, serum albumin chaperone-like activity is likely to be inhibited to some degree if FFA concentrations are elevated to very high levels.
Although there have been previous reports of anti-amyloid properties of serum albumin (16–19, 53), here it is directly demonstrated that the presence of serum albumin at physiologically relevant concentrations reduces the total amount of aggregate and amyloid formation by both WT and L55P-TTR (Fig. 3). TTR is the amyloid-forming species in the transthyretin amyloidoses, a group of systemic amyloidoses that includes familial amyloid polyneuropathy, senile systemic amyloidosis, and senile cardiac amyloidosis. Recently a group studying familial amyloid polyneuropathy observed that low serum albumin not only correlated with clinical disease progression in human patients, but that analbuminaemia led to more severe disease at a younger age in a rat model (63). Thus, TTR is a potential client protein of physiological and medical relevance.
In addition, serum albumin was seen to induce a lag phase in the aggregation and amyloid formation of L55P-TTR that was not observed for the WT, an observation that may suggest a mechanism of action of serum albumin. It is known that TTR amyloid formation involves the dissociation of tetramers to monomers, followed by conversion of the native monomer to the amyloidogenic monomer, the former being the rate-limiting step (64–66). The fact that serum albumin does not induce a lag phase for WT-TTR whereas it does for L55P-TTR (the tetramer-destabilized mutant) may suggest that serum albumin inhibits conversion from the native monomer to the amyloidogenic monomer.
Despite the fact that serum albumin is observed to be inherently “sticky,” forming complexes with both stressed and unstressed proteins, serum albumin binds stressed proteins more avidly than their native counterparts (Figs. 4 and 5). This finding is consistent with the observations of others: serum albumin is a major component of the soluble HMW species formed when human plasma is exposed to shear stress (5) and preferentially binds transthyretin amyloid over the native fold (63). Experimenters investigating atrial natriuretic peptide recognized that whole serum is an effective inhibitor of atrial natriuretic peptide amyloid formation, that atrial natriuretic peptide is incorporated into a number of stable complexes in this process, and that serum albumin is the most abundant counterpart in these complexes (53).
Here it is shown that the removal of serum albumin from plasma leaves plasma more susceptible to aggregation under two physiological stress conditions (Fig. 6A, panel ii). Heat stresses used here (41 °C) occur in vivo: during exercise, core temperatures can exceed 40 °C (67) and muscle temperatures may reach 41 °C (68), heat stroke is defined as temperatures exceeding 40.6 °C (69), and during drug-induced fevers core temperatures may reach 42 °C (70). Similarly, shear stress of 40–60 dynes/cm2 is physiologically relevant and simulates a degree of the variability present in human circulation: blood is constantly exposed to a variable arterial shear stress ∼10–70 dynes/cm2 in health, although this ranges up to levels greater than 100 dynes/cm2 in pathological states (71). Similar stresses achieved using similar techniques have been used to demonstrate physiologically relevant chaperone activity in clusterin (5, 72).
The results presented here show that plasma exposed to these physiological stresses aggregates to a greater extent when depleted of serum albumin than when serum albumin is present. It is important to recognize that the affinity gel used to deplete albumin from plasma also binds clusterin (73), haptoglobin, and α2-macroglobulin (74), among other proteins. Nonetheless, the observation that aggregation reverts to normal levels with the reintroduction of serum albumin (Fig. 6A, panel iii) suggests that serum albumin is responsible for much of the antiaggregatory activity in the original plasma sample.
Although the experiments on human plasma used physiological stresses, and these experiments aimed to simulate physiological conditions, the stresses required to destabilize ADH, insulin, and TTR within an experimentally appropriate time frame were nonphysiological. Similar practical concerns dictated that the physiological heat stress of 41 °C was sustained for a nonphysiological time frame to achieve experimental outcomes: although whole undiluted plasma aggregated within hours, the dilution inherent in the depletion process meant that a longer time course was required to produce aggregation in the depleted plasma. Furthermore, although the measurement of turbidity is a commonly employed method for quantifying protein aggregation, care must be taken in extrapolating turbidity to all aggregation: particles below a certain size are not detected by turbidity, and thus potentially significant smaller aggregates are not accounted for in this study.
The loss of serum albumin chaperone-like activity may be pertinent to our understanding of a number of pathological states. Despite possessing a lower intrinsic activity than the other extracellular chaperones, serum albumin is present at levels over 18 times that of haptoglobin (75), and 250 times that of clusterin (76). Because it makes up more than 50% of plasma protein, serum albumin vastly outpopulates any of its potential clients. Thus, serum albumin may exert a significant antiaggregatory effect in the extracellular compartment.
Footnotes
- Aβ
- amyloid β peptide
- ADH
- alcohol dehydrogenase
- HMW
- high molecular weight
- ThT
- thioflavin T
- TTR
- transthyretin
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Kragh-Hansen U. (1990) Structure and ligand binding properties of human serum albumin. Dan. Med. Bull. 37, 57–84 [PubMed] [Google Scholar]
- 2. Marini I., Moschini R., Del Corso A., Mura U. (2005) Chaperone-like features of bovine serum albumin. A comparison with α-crystallin. Cell Mol. Life Sci. 62, 3092–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Humphreys D. T., Carver J. A., Easterbrook-Smith S. B., Wilson M. R. (1999) Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 274, 6875–6881 [DOI] [PubMed] [Google Scholar]
- 4. Yerbury J. J., Poon S., Meehan S., Thompson B., Kumita J. R., Dobson C. M., Wilson M. R. (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322 [DOI] [PubMed] [Google Scholar]
- 5. Wyatt A. R., Wilson M. R. (2010) Identification of human plasma proteins as major clients for the extracellular chaperone clusterin. J. Biol. Chem. 285, 3532–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poon S., Easterbrook-Smith S. B., Rybchyn M. S., Carver J. A., Wilson M. R. (2000) Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 39, 15953–15960 [DOI] [PubMed] [Google Scholar]
- 7. Poon S., Rybchyn M. S., Easterbrook-Smith S. B., Carver J. A., Pankhurst G. J., Wilson M. R. (2002) Mildly acidic pH activates the extracellular molecular chaperone clusterin. J. Biol. Chem. 277, 39532–39540 [DOI] [PubMed] [Google Scholar]
- 8. Yerbury J. J., Rybchyn M. S., Easterbrook-Smith S. B., Henriques C., Wilson M. R. (2005) The acute phase protein haptoglobin is a mammalian extracellular chaperone with an action similar to clusterin. Biochemistry 44, 10914–10925 [DOI] [PubMed] [Google Scholar]
- 9. Yerbury J. J., Kumita J. R., Meehan S., Dobson C. M., Wilson M. R. (2009) α2-Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J. Biol. Chem. 284, 4246–4254 [DOI] [PubMed] [Google Scholar]
- 10. French K., Yerbury J. J., Wilson M. R. (2008) Protease activation of α2-macroglobulin modulates a chaperone-like action with broad specificity. Biochemistry 47, 1176–1185 [DOI] [PubMed] [Google Scholar]
- 11. Davis D. P., Gallo G., Vogen S. M., Dul J. L., Sciarretta K. L., Kumar A., Raffen R., Stevens F. J., Argon Y. (2001) Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J. Mol. Biol. 313, 1021–1034 [DOI] [PubMed] [Google Scholar]
- 12. Hollander J. M., Martin J. L., Belke D. D., Scott B. T., Swanson E., Krishnamoorthy V., Dillmann W. H. (2004) Overexpression of wild-type heat shock protein 27 and a nonphosphorylatable heat shock protein 27 mutant protects against ischemia/reperfusion injury in a transgenic mouse model. Circulation 110, 3544–3552 [DOI] [PubMed] [Google Scholar]
- 13. Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 14. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 15. Aguzzi A., O'Connor T. (2010) Protein aggregation diseases. Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 9, 237–248 [DOI] [PubMed] [Google Scholar]
- 16. Kuo Y. M., Kokjohn T. A., Kalback W., Luehrs D., Galasko D. R., Chevallier N., Koo E. H., Emmerling M. R., Roher A. E. (2000) Amyloid-β peptides interact with plasma proteins and erythrocytes. Implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 268, 750–756 [DOI] [PubMed] [Google Scholar]
- 17. Bohrmann B., Tjernberg L., Kuner P., Poli S., Levet-Trafit B., Näslund J., Richards G., Huber W., Döbeli H., Nordstedt C. (1999) Endogenous proteins controlling amyloid β-peptide polymerization. Possible implications for β-amyloid formation in the central nervous system and in peripheral tissues. J. Biol. Chem. 274, 15990–15995 [DOI] [PubMed] [Google Scholar]
- 18. Galeazzi L., Galeazzi R., Valli M. B., Corder E. H., Giunta S. (2002) Albumin protects human red blood cells against Aβ25–35-induced lysis more effectively than ApoE. Neuroreport 13, 2149–2154 [DOI] [PubMed] [Google Scholar]
- 19. Rasmussen T., Tantipolphan R., van de Weert M., Jiskoot W. (2010) The molecular chaperone α-crystallin as an excipient in an insulin formulation. Pharm. Res. 27, 1337–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minton K. W., Karmin P., Hahn G. M., Minton A. P. (1982) Nonspecific stabilization of stress-susceptible proteins by stress-resistant proteins. A model for the biological role of heat shock proteins. Proc. Natl. Acad. Sci. U.S.A. 79, 7107–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zimmerman S. B., Trach S. O. (1991) Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 222, 599–620 [DOI] [PubMed] [Google Scholar]
- 22. Cheung M. S., Klimov D., Thirumalai D. (2005) Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 4753–4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buczek O., Green B. R., Bulaj G. (2007) Albumin is a redox-active crowding agent that promotes oxidative folding of cysteine-rich peptides. Biopolymers 88, 8–19 [DOI] [PubMed] [Google Scholar]
- 24. Snoussi K., Halle B. (2005) Protein self-association induced by macromolecular crowding. A quantitative analysis by magnetic relaxation dispersion. Biophys. J. 88, 2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rivas G., Fernandez J. A., Minton A. P. (1999) Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium. Theory, experiment, and biological significance. Biochemistry 38, 9379–9388 [DOI] [PubMed] [Google Scholar]
- 26. Ren G., Lin Z., Tsou C. L., Wang C. C. (2003) Effects of macromolecular crowding on the unfolding and the refolding of d-glyceraldehyde-3-phosophospate dehydrogenase. J. Protein Chem. 22, 431–439 [DOI] [PubMed] [Google Scholar]
- 27. Munishkina L. A., Cooper E. M., Uversky V. N., Fink A. L. (2004) The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464 [DOI] [PubMed] [Google Scholar]
- 28. Hatters D. M., Minton A. P., Howlett G. J. (2002) Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 277, 7824–7830 [DOI] [PubMed] [Google Scholar]
- 29. van den Berg B., Ellis R. J., Dobson C. M. (1999) Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18, 6927–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou B. R., Liang Y., Du F., Zhou Z., Chen J. (2004) Mixed macromolecular crowding accelerates the oxidative refolding of reduced, denatured lysozyme. Implications for protein folding in intracellular environments. J. Biol. Chem. 279, 55109–55116 [DOI] [PubMed] [Google Scholar]
- 31. Chen R. F. (1967) Removal of fatty acids from serum albumin by charcoal treatment. J. Biol. Chem. 242, 173–181 [PubMed] [Google Scholar]
- 32. Cohlberg J. A., Li J., Uversky V. N., Fink A. L. (2002) Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from α-synuclein in vitro. Biochemistry 41, 1502–1511 [DOI] [PubMed] [Google Scholar]
- 33. Wilkinson-White L. E., Easterbrook-Smith S. B. (2007) Characterization of the binding of Cu(II) and Zn(II) to transthyretin. Effects on amyloid formation. Biochemistry 46, 9123–9132 [DOI] [PubMed] [Google Scholar]
- 34. Dardik A., Chen L., Frattini J., Asada H., Aziz F., Kudo F. A., Sumpio B. E. (2005) Differential effects of orbital and laminar shear stress on endothelial cells. J. Vasc. Surg. 41, 869–880 [DOI] [PubMed] [Google Scholar]
- 35. McLachlan A. D., Walker J. E. (1977) Evolution of serum albumin. J. Mol. Biol. 112, 543–558 [DOI] [PubMed] [Google Scholar]
- 36. Huang B. X., Kim H. Y., Dass C. (2004) Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1237–1247 [DOI] [PubMed] [Google Scholar]
- 37. Masuoka J., Saltman P. (1994) Zinc(II) and copper(II) binding to serum albumin. A comparative study of dog, bovine, and human albumin. J. Biol. Chem. 269, 25557–25561 [PubMed] [Google Scholar]
- 38. Richieri G. V., Anel A., Kleinfeld A. M. (1993) Interactions of long-chain fatty acids and albumin. Determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry 32, 7574–7580 [DOI] [PubMed] [Google Scholar]
- 39. van den Berg B., Wain R., Dobson C. M., Ellis R. J. (2000) Macromolecular crowding perturbs protein refolding kinetics. Implications for folding inside the cell. EMBO J. 19, 3870–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soltys B. J., Hsia J. C. (1977) Fatty acid enhancement of human serum albumin binding properties. A spin label study. J. Biol. Chem. 252, 4043–4048 [PubMed] [Google Scholar]
- 41. Reed R. G. (1977) Kinetics of bilirubin binding to bovine serum albumin and the effects of palmitate. J. Biol. Chem. 252, 7483–7487 [PubMed] [Google Scholar]
- 42. Rahil-Khazen R., Bolann B. J., Ulvik R. J. (2000) Trace element reference values in serum determined by inductively coupled plasma atomic emission spectrometry. Clin. Chem. Lab. Med. 38, 765–772 [DOI] [PubMed] [Google Scholar]
- 43. Sheluho D., Ackerman S. H. (2001) An accessible hydrophobic surface is a key element of the molecular chaperone action of Atp11p. J. Biol. Chem. 276, 39945–39949 [DOI] [PubMed] [Google Scholar]
- 44. Püttmann M., Krug H., von Ochsenstein E., Kattermann R. (1993) Fast HPLC determination of serum free fatty acids in the picomole range. Clin. Chem. 39, 825–832 [PubMed] [Google Scholar]
- 45. Meng Z., Wen D., Sun D., Gao F., Li W., Liao Y., Liu H. (2007) Rapid determination of C12-C26 non-derivatized fatty acids in human serum by fast gas chromatography. J. Sep. Sci. 30, 1537–1543 [DOI] [PubMed] [Google Scholar]
- 46. Sztefko K., Panek J. (2001) Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology 1, 230–236 [DOI] [PubMed] [Google Scholar]
- 47. Saifer A., Goldman L. (1961) The free fatty acids bound to human serum albumin. J. Lipid Res. 2, 268–270 [Google Scholar]
- 48. Ross P. D., Shrake A. (1988) Decrease in stability of human albumin with increase in protein concentration. J. Biol. Chem. 263, 11196–11202 [PubMed] [Google Scholar]
- 49. Benvenga S., Bartalena L., Antonelli A., Li Calzi L., Di Pasquale G., Trimarchi F., Pinchera A. (1986) Radioimmunoassay for human thyroxine-binding prealbumin. Ann. Clin. Lab. Sci. 16, 231–240 [PubMed] [Google Scholar]
- 50. LeVine H., 3rd (1999) Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 [DOI] [PubMed] [Google Scholar]
- 51. Rao P. V., Horwitz J., Zigler J. S., Jr. (1993) α-Crystallin, a molecular chaperone, forms a stable complex with carbonic anhydrase upon heat denaturation. Biochem. Biophys. Res. Commun. 190, 786–793 [DOI] [PubMed] [Google Scholar]
- 52. Carver J. A., Aquilina J. A., Cooper P. G., Williams G. A., Truscott R. J. (1994) α-Crystallin. Molecular chaperone and protein surfactant. Biochim. Biophys. Acta 1204, 195–206 [DOI] [PubMed] [Google Scholar]
- 53. Torricelli C., Capurro E., Santucci A., Paffetti A., D'Ambrosio C., Scaloni A., Maioli E., Pacini A. (2004) Multiple plasma proteins control atrial natriuretic peptide (ANP) aggregation. J. Mol. Endocrinol. 33, 335–341 [DOI] [PubMed] [Google Scholar]
- 54. Atwood C. S., Moir R. D., Huang X., Scarpa R. C., Bacarra N. M., Romano D. M., Hartshorn M. A., Tanzi R. E., Bush A. I. (1998) Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 273, 12817–12826 [DOI] [PubMed] [Google Scholar]
- 55. Uversky V. N., Li J., Fink A. L. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 56. Nagase S., Shimamune K., Shumiya S. (1980) Albumin-deficient rat mutant. An animal model for analbuminemia. Jikken Dobutsu 29, 33–38 [DOI] [PubMed] [Google Scholar]
- 57. Russi E., Weigand K. (1983) Analbuminemia. Klin. Wochenschr. 61, 541–545 [DOI] [PubMed] [Google Scholar]
- 58. Spector A. A., John K., Fletcher J. E. (1969) Binding of long-chain fatty acids to bovine serum albumin. J. Lipid Res. 10, 56–67 [PubMed] [Google Scholar]
- 59. Choi J. K., Ho J., Curry S., Qin D., Bittman R., Hamilton J. A. (2002) Interactions of very long-chain saturated fatty acids with serum albumin. J. Lipid Res. 43, 1000–1010 [DOI] [PubMed] [Google Scholar]
- 60. Peters T., Jr. (1985) Serum albumin. Adv. Protein Chem. 37, 161–245 [DOI] [PubMed] [Google Scholar]
- 61. Chang B. S., Mahoney R. R. (1995) Enzyme thermostabilization by bovine serum albumin and other proteins. Evidence for hydrophobic interactions. Biotechnol. Appl. Biochem. 22, 203–214 [PubMed] [Google Scholar]
- 62. Curry S., Mandelkow H., Brick P., Franks N. (1998) Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 5, 827–835 [DOI] [PubMed] [Google Scholar]
- 63. Kugimiya T., Jono H., Saito S., Maruyama T., Kadowaki D., Misumi Y., Hoshii Y., Tasaki M., Su Y., Ueda M., Obayashi K., Shono M., Otagiri M., Ando Y. (2011) Loss of functional albumin triggers acceleration of transthyretin amyloid fibril formation in familial amyloidotic polyneuropathy. Lab. Invest. 91, 1219–1228 [DOI] [PubMed] [Google Scholar]
- 64. Quintas A., Saraiva M. J., Brito R. M. (1999) The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J. Biol. Chem. 274, 32943–32949 [DOI] [PubMed] [Google Scholar]
- 65. Quintas A., Vaz D. C., Cardoso I., Saraiva M. J., Brito R. M. (2001) Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem. 276, 27207–27213 [DOI] [PubMed] [Google Scholar]
- 66. Hurshman A. R., White J. T., Powers E. T., Kelly J. W. (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43, 7365–7381 [DOI] [PubMed] [Google Scholar]
- 67. Ely B. R., Ely M. R., Cheuvront S. N., Kenefick R. W., Degroot D. W., Montain S. J. (2009) Evidence against a 40 degrees C core temperature threshold for fatigue in humans. J. Appl. Physiol. 107, 1519–1525 [DOI] [PubMed] [Google Scholar]
- 68. González-Alonso J., Teller C., Andersen S. L., Jensen F. B., Hyldig T., Nielsen B. (1999) Influence of body temperature on the development of fatigue during prolonged exercise in the heat. J. Appl. Physiol. 86, 1032–1039 [DOI] [PubMed] [Google Scholar]
- 69. McGugan E. A. (2001) Hyperpyrexia in the emergency department. Emerg. Med. 13, 116–120 [DOI] [PubMed] [Google Scholar]
- 70. Kumar K. L., Reuler J. B. (1986) Drug fever. West J. Med. 144, 753–755 [PMC free article] [PubMed] [Google Scholar]
- 71. Malek A. M., Alper S. L., Izumo S. (1999) Hemodynamic shear stress and its role in atherosclerosis. J. Am. Med. Assoc. 282, 2035–2042 [DOI] [PubMed] [Google Scholar]
- 72. Wyatt A. R., Yerbury J. J., Wilson M. R. (2009) Structural characterization of clusterin-chaperone client protein complexes. J. Biol. Chem. 284, 21920–21927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Blaschuk O., Burdzy K., Fritz I. B. (1983) Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 258, 7714–7720 [PubMed] [Google Scholar]
- 74. Gianazza E., Arnaud P. (1982) Chromatography of plasma proteins on immobilized Cibacron Blue F3-GA. Mechanism of the molecular interaction. Biochem. J. 203, 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bowman B. H., Kurosky A. (1982) Haptoglobin. The evolutionary product of duplication, unequal crossing over, and point mutation. Adv. Hum. Genet. 12, 189–261, 453–454 [DOI] [PubMed] [Google Scholar]
- 76. Morrissey C., Lakins J., Moquin A., Hussain M., Tenniswood M. (2001) An antigen capture assay for the measurement of serum clusterin concentrations. J. Biochem. Biophys. Methods 48, 13–21 [DOI] [PubMed] [Google Scholar]