

Abstract
Objectives
To perform fine mapping of the autoimmunity susceptibility gene BLK and identify functional variants involved in systemic lupus erythematosus (SLE).
Methods
Genotyping of 1163 European SLE patients and 1482 controls and imputation were performed covering the BLK gene with 158 single-nucleotide polymorphisms. Logistic regression analysis was done using PLINK and conditional analyses using GENABEL's test score. Transfections of BLK constructs on HEK293 cells containing the novel mutation or the wild type form were analysed for their effect on protein half-life using a protein stability assay, cycloheximide and western blot. CHiP-qPCR for detection of nuclear factor κ B (NFkB) binding.
Results
Fine mapping of BLK identified two independent genetic effects with functional consequences: one represented by two tightly linked associated haplotype blocks significantly enriched for NFκB-binding sites and numerous putative regulatory variants whose risk alleles correlated with low BLK mRNA levels. Binding of NFkBp50 and p65 to an associated 1.2 Kb haplotype segment was confirmed. A second independent genetic effect was represented by an Ala71Thr, low-frequency missense substitution with an OR=2.31 (95% CI 1.38 to 3.86). The 71Thr decreased BLK protein half-life.
Conclusions
These results show that rare and common regulatory variants in BLK are involved in disease susceptibility and both, albeit independently, lead to reduced levels of BLK protein.
Introduction
The BLK gene located on chromosome 8p23-p22 encodes a tyrosine kinase of the SRC family of proto-oncogenes. A genome-wide association study identified a single-nucleotide polymorphism (SNP) rs13277113 in the 5′ upstream region associated with systemic lupus erythematosus (SLE),1 a chronic autoimmune disease with complex inheritance. To date, multiple studies have confirmed the association of BLK with SLE in European and Asian populations as well as with other autoimmune disorders.2–10 BLK protein has 505 amino acids with SH3 (58–118), SH2 (124–220) and protein kinase domains (241–494). Although BLK is expressed in various tissues such as pancreatic β cells, thymus, hair follicles and salivary ducts,11 12 it is preferentially expressed in B lymphoid cells and has a role in B cell receptor signalling and B cell development.13
The BLK-rs13277113 SNP is part of a group of common variants across several genes that have been associated with SLE through genome-wide association study.14 Together, they account for 15% of the disease's familial aggregation and few have been demonstrated to be functional.14 The SLE-risk allele rs13277113-A correlated with decreased expression of BLK mRNA1 but the mechanism by which this or another SNP in linkage disequilibrium (LD) with it affects gene expression has not been characterised yet.
To address which and if common and/or low frequency functional variants are involved in SLE susceptibility at the BLK locus, we performed fine mapping by genotyping tag SNPs aided by imputation.
Methods
Patients and controls
After quality control of the data, the study sample consisted of 1163 patients with SLE and 1482 ethnicity-matched healthy control subjects from the European multicentre collaboration network BIOLUPUS (supported by the European Science Foundation), comprising individuals from Argentina (60 patients and 109 controls), Belgium (70 patients and 59 controls), Germany (284 patients and 176 controls), Hungary (38 patients and 35 controls), Italy (264 patients and 320 controls), Portugal (163 patients and 165 controls), Spain (206 patients and 227 controls) and Sweden (78 patients and 391 controls). A completely new and independent set of 1103 healthy Spanish controls was also used. All patients fulfilled at least four of the American College of Rheumatology 1982 criteria for the classification of SLE.15 Subjects with an individual genotyping rate lower than 90% or duplicated and/or related samples were excluded. We removed individuals with <90% of European ancestry, as estimated with STRUCTURE v2.3.3 and 350 ancestry informative markers.
Tag SNP selection
From the LD structure of the BLK locus in the HapMap CEU population (data release 27; available at http://hapmap.ncbi.nlm.nih.gov/), tag SNPs selection capturing more than 90% of common variation at the gene locus (minor allele frequency of greater than or equal to 10%) including 20 kb upstream and downstream of the gene, at a r2 threshold of greater than or equal to 0.8, were selected using the tagger algorithm implemented in Haploview, V.4.1.16 After quality control of the data, 27 tag SNPs were used for genotyping and to guide the imputation.
Genotyping
All samples were genotyped at the Feinstein Institute for Medical Research (Manhasset, New York, USA) using a GoldenGate Custom Genotyping Assay and a BeadXpress Reader from Illumina (San Diego, California, USA). Two SNPs (rs1478895 and rs2248932) were genotyped by TaqMan 5′-exonuclease assay (ABI, Foster City, California, USA). SNP rs55758736 was genotyped by RFLP and sequencing for verification in 178 individuals. An Illumina custom BeadArray was used for genotyping this SNP in 1103 healthy controls.
Imputation
We used IMPUTE217 across a 250 kb genomic region (chr8:11,288,930-11,559,517) of the BLK locus. As reference panels we used haplotypes from all HapMap Consortium18 19 populations together (phase 3, release 2) and haplotypes of the CEU population from the pilot phase of 1000 Genomes Project20 (downloaded from http://mathgen.stats.ox.ac.uk/impute/impute_v2.html#filtered_1kg_hm3_haps on May 13th, 2011). HapMap haplotypes duplicated with 1000 Genomes data were removed. Default parameters were used and the Ne argument set to 15 000. Only imputed genotypes with a probability equal to or greater than 0.9, a minor allelic frequency (MAF) equal to or greater than 0.005, following Hardy–Weinberg equilibrium (p>0.001), and a genotyping rate equal to or greater than 0.95 were retained for association.
Statistical analysis
Genetic association analysis was done using PLINK v1.0721 and GenABEL v1.6-5.22 All SNPs with a genotype call rate <95% or not in Hardy–Weinberg equilibrium (p<0.001) were excluded. For single-marker tests, we performed a Cochran-Mantel-Haenszel (CMH) meta-analysis, using the country of origin as the stratification variable, and a Breslow-Day test to assess homogeneity of the ORs. p Values were adjusted using false discovery rate. We also performed an omnibus haplotype test for all windows of 2, 3 and 4 SNPs across the gene. In this test, the alternate hypothesis of each haplotype having a unique effect is compared with the null hypothesis (df=number of haplotypes-1).21 Omnibus as well as haplotype-specific tests for haplotype blocks were conducted by testing each haplotype against all others pooled together (1 df). A Bonferroni correction using the number of windows tested was applied (p<0.05/323 windows tested).
Conditional analysis was done by logistic regression using GenABEL's score test. By specifying the country of origin as the strata argument, we performed a structured test of association for all SNPs. In a stepwise manner, the SNP with higher p value was identified in each step and then used as covariate for the next association step until no SNP remained associated. GenABEL package was run using R version 2.9.2.
Genome sequence track analysis for epigenomic marks and transcription factor binding sites
Associated and non-associated SNPs within haplotype blocks were analysed for enrichment with eight epigenomic marks using GenomeRunner.23 For SNPs within each haploblock the same number of randomly selected nucleotides from the whole range of haploblocks within chr8:11374997-11475653 coordinates (NCBI36/hg18 genome assembly) was sampled over the course of 100 Monte-Carlo simulations. The average expected frequency of their association with a given genomic feature (eg, nuclear factor κ B (NFkB) binding site, epigenomic marks) was compared with the observed frequency using a χ2 test. Specifically, SNPs within haploblocks were tested for enrichment against experimentally validated NFκBp65, IRF4 and PAX5 transcription factor binding sites annotated in the wgencoderegtfbs clustered UCSC table of the genome browser.24 Other tables included histone acetylation and methylation marks (Regulation/Broad histone tables for Gm12878 lymphoblastoid cell line). p Values were –Log10 converted and a – was included for under-represented p values. The values are shown as a heatmap plotted using Matlab (Natick, Massachusetts, USA) (online figure S2). p Values are presented in the online table S3.
We used SIFT25 to predict the status of rs55758736. SIFT predicts whether an amino acid substitution affects protein function based on similarity to related sequences and physical properties of amino acids (http://sift.jcvi.org/).
CHiP-qPCR for NFkB p50 and p65 on the 1.2 Kb associated haplotype genomic segment
CHiP experiments were performed according to the manufacturer's recommendations (EZ-Magna CHiP A, Millipore, Billerica, California, USA). In brief, 4×10−6 Daudi cells were incubated with or without 10 mg/ml anti-immunoglobulin M (IgM) F(ab′)2 fragments (Jackson Immunoresearch, West Grove, Pennsylvania, USA) for the indicated time. Cells were fixed with 1% formaldehyde and nuclei were isolated and sonicated within 300 ml of lysis buffer. An amount of 50 ml of chromatin–protein complexes were immunoprecipitated overnight at 4°C by mild agitation with 5 mg of pAb specific for p65 (Santa Cruz, California, USA) or p50 (Abcam, Cambridge, Massachusetts, USA) or normal rabbit IgG (Millipore) as negative control. After elution from the immunoprecipitated chromatin complexes followed by reverse-crosslink and cleanup, DNA samples were subjected to real-time PCR analysis using primers as follows: rs2248932 neighbouring region within Blk intron 1 primer set: forward – TGCATCAGCATCACTGGG; reverse – GGCACAATAGTTCTGAAACCTC. IkBa was used as control for NFkB binding with the following primer set: forward – CACTTGCAGAGGGACAGGAT; reverse – GAGAAACTCCCTGCGATGAG.
Cloning and expression of BLK constructs with the Ala71Thr substitution
BLK sequence was PCR amplified using cDNA from a BJAB cell line and primers f-BLK: 5′-CACCatggggctggtaagtagc-3 and r-BLK 5′-gggctgcagctcgtactgcc-3. The start codon in the forward primer is underlined. The open reading frame was cloned in pcDNA3.1D/V5-His (Invitrogen, Life Technologies, Grand Island, New York, USA) and confirmed by sequencing. The protein tagged with the V5 epitope at the C terminal was produced by deletion of the stop codons. The BLK 71Thr isoform was produced by mutagenesis PCR with pfu-Ultra II fusion HS polymerase (Stratagene, Agilent Technologies, Santa Barbara, California, USA) and the primers BLKm71f: 5′-TATGACTACACCaCTATGAATGATCGGGACCT-3 and BLKm71r: 5′-ATCATTCATAGtGGTGTAGTCATACAGAG-3. Expression of the constructs was assayed by western blot with anti-V5 antibody (online figure S4).
Protein stability assay
Embryonic kidney HEK293T cells grown in RPMI 1640 medium supplemented with 10% fetal calf serumwere seeded on six-well plates and transfected with 4 µg of expression plasmids V5-tagged BLK isoforms using Lipofectamine 2000. At 22 h cells were harvested and distributed in six wells of 12-well plates. After 2 h cells were treated with 20 µg/ml cycloheximide in dimethyl sulphoxide (DMSO) or DMSO alone (time 0) for various times. Cellular extracts were prepared in lysis buffer: 1% NP40, 150 mM NaCl, 10 mM Tris (pH 7.5), 2 mM sodium orthovanadate, 1 mM EDTA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride and protease and phosphatases inhibitor cocktails (Roche). A fifth of the extract was resolved on SDS-PAGE and immmunoblotting carried out using standard protocols with anti-V5 antibodies (Invitrogen), b-tubulin (Sigma-Aldrich) and antimouse IgG-HRP (Zymed, San Francisco, California, USA). Quantification of the bands was done using ImageJ.
Results
Fine mapping and conditional analysis of BLK reveals two independent effects
A total of 158 SNPs with MAF>0.005 were tested for association in 1163 European patients with SLE and 1482 age- and country-of-origin-matched controls. A meta-analysis demonstrated that the strongest association signal was in the BLK promoter region (rs998683, PCMH-corrected=1.05×10−4 and OR 1.38 (1.21 to 1.58)) (online table S1). However, even after correction for multiple testing, multiple association signals across the gene remained significant. To refine the BLK association, we tested sliding windows, which narrowed it down to three blocks (denominated B1, B3 and B5 in figure 1). The best model of association was observed for a 1.2 kb haplotype window between SNPs rs2248932 and rs9329246 located at the end of BLK's first intron (p=1.58×10−10, omnibus haplotype test).
Figure 1.

Systemic lupus erythematosus-associated single-nucleotide polymorphisms (SNPs) and haplotype blocks and genomic features of the BLK locus. From top to bottom: –log10 of the p value corrected by false discovery rate for 158 SNPs. Below, the association for haplotype windows of two, three and four SNPs is displayed; the horizontal black line marks the threshold for Bonferroni correction (p=0.05/323 windows tested). Only SNP windows contained within block B1 (5′ upstream, exon 1 – UTR – and beginning of intron 1), B3 (end of intron 1) and B5 (exons 4, 5 and 6) were associated after correction. The exon/intron structure of BLK is displayed next and then NFkBp65 (blue), IRF4 (yellow) and PAX5 (green) Chip-Seq identified binding sites and promoter-associated histone marks (H3K4Me3) according to ENCODE project. Recombination rate (cM/Mb) is displayed at the bottom.
The first block (B1) covered BLK 5′ upstream region, exon 1 (untranslated-UTR) and beginning of intron 1, which correspond to the promoter. This block comprised a set of highly correlated SNPs including all variants previously associated with SLE (rs2409780, rs2618444, rs2736336, rs2736337, rs2736338, rs2736340, rs13277113) and our strongest hit (rs998683) (figure 2A and online figure S1). Haplotype-specific analysis demonstrated a common, risk haplotype tagged by the minor alleles of these SNPs, including the rs13277113-A variant (omnibus test p value=1.61×10−5, OR=1.32 (1.16 to 1.49)) (table 1).
Figure 2.

Structure of associated BLK haplotypes. (A) Haplotypes formed by single-nucleotide polymorphisms (SNPs) in each associated block B1, B3 and B5 as estimated with expectation maximisation algorithm (Haploview v.4.1). Blue and red squares represent major and minor alleles, respectively. To the right of each haplotype, its population frequency (cases and controls) and connections between blocks (thin lines if >1%, thick lines if >10%) are displayed. In crossing areas are the values of the multiallelic D′ which represent the level of recombination between two blocks. See table 1 for haplotype-specific ORs and p values. Haplotypes associated with increased risk of developing systemic lupus erythematosus are enclosed within rectangles (solid line if p<1×10−3; dashed line if p<0.05). B1 contains a risk haplotype tagged by rs998683-A(b) (strongest hit in this study) and its proxies including the rs13277113-A allele(a) previously described.1 Blocks B1 and B3 are highly correlated. B3 contains SNP rs9329246(c), which is shared by the two strongest four-SNP windows of the whole BLK locus. B5 contains the independent risk haplotype tagged by the mutant allele of the rs55758736/Ala71Thr non-synonymous SNP(d) and a set of highly correlated allelic variants (grey circles). The independence of B5 risk haplotype is further supported by (B) the linkage disequilibrium plot displaying r2 values between the representative variants of each block and (C) the relationship between their alleles.
Table 1.
Haplotype analysis of the BLK-associated blocks
| Block (SNPs)/Start-End (Size) | No. SNPs | Omnibus test | Haplotype | Frequency Cases | Frequency Controls | OR (95% CI) | P-value |
|---|---|---|---|---|---|---|---|
| B1 (rs2409780-rs2618478) | 21 | 1.75E-04 | |||||
| 11374997-11393411 (18.4 kb) | TACGGTAGCTGTAGGGGCCGA | 0.079 | 0.073 | ns | 0.4517 | ||
| CCCGTCCGTTATAGGAGCCGA | 0.299 | 0.245 | 1.32 (1.16-1.49) | 1.61E-05 | |||
| TACGGTAGCTGTAGGGGCGGA | 0.504 | 0.559 | 0.81 (0.73-0.91) | 7.56E-05 | |||
| TATAGTAACCGCCCAGAGCAA | 0.011 | 0.014 | ns | 0.3503 | |||
| TATAGTAACCGCCCAGAGCAC | 0.108 | 0.109 | ns | 0.9182 | |||
| B3 (rs58373594-8-11433666) | 17 | 3.85E-07 | |||||
| 11428036-11433666 (5.6 kb) | ATCGCATAGAGTAAAAG | 0.489 | 0.541 | 0.80 (0.72-0.89) | 2.70E-04 | ||
| TTGGCATCCAATAGAAG | 0.113 | 0.113 | ns | 0.9928 | |||
| AACACACAGGACAGTAG | 0.045 | 0.045 | ns | 0.9493 | |||
| AACACACAGGACGGTAG | 0.323 | 0.290 | 1.13 (1.01-1.27) | 0.013 | |||
| AACACATAGGACAGTAG | 0.030 | 0.011 | 2.75 (1.80-4.19) | 5.16E-07 | |||
| Strongest window (rs2248932-rs9329246) / 11429059-11430289 (1.2 kb) | 4 | 1.58E-10 | |||||
| B5 (rs2244931-rs17744726) | 15 | 2.56E-04 | |||||
| 11441178-11449232 (8.1 kb) | CAAGAGTACCGTTTA | 0.291 | 0.328 | 0.84 (0.75-0.95) | 3.94E-03 | ||
| CGAGGGCACCGTATG | 0.134 | 0.110 | 1.26 (1.06-1.49) | 0.011 | |||
| GAAGAGTACCGTATG | 0.014 | 0.010 | ns | 0.2331 | |||
| CAAGAGTACCGTATG | 0.061 | 0.067 | ns | 0.3859 | |||
| GGAGGGCACCGTATG | 0.370 | 0.362 | ns | 0.5478 | |||
| GAAGGGTACCGTATG | 0.077 | 0.078 | ns | 0.8688 | |||
| CAAGGGTACCGTATG | 0.012 | 0.010 | ns | 0.6186 | |||
| GGAGGGTACCGTATG | 0.019 | 0.024 | ns | 0.236 | |||
| CAAAAATATTTCATG | 0.023 | 0.010 | 2.48 (1.54-4.00) | 1.57E-04 |
The first column indicates the block for which the haplotype analysis corresponds to. For each block, the first and the last SNP, the starting and ending positions, as well as the size in kilobases (kb) are also presented. The Omnibus P value indicates the overall significance of the association of the block. Only haplotypes with frequencies equal to or greater than 0.01 were tested. For the haplotype-specific analyses, the odds ratio (OR) with 95% confidence interval (95% CI) and P-values were determined for each haplotype tested against all of the others pooled together. In block B3, the SNPs comprising the strongest associated window are underlined and the omnibus test for this window is presented. All analyses were performed using PLINK software, version 1.07. ns: non-significant
The second block (B3) was at the end of intron 1 (figure 1) and displayed moderate LD with B1 (figure 2A). Consistent with this observation, a risk haplotype correlated with the promoter risk haplotype and with similar haplotype frequency was over-represented in cases, although only with nominal association (p=0.013) (table 1).
In addition, an independent and low-frequency-haplotype within B3 (1.1% in controls) was strongly associated and had a large effect size (p=5.16×10−7, OR=2.75 (1.80 to 4.19)) (table 1). This haplotype correlated completely with a risk haplotype observed on the third associated block B5 (p=1.57×10−4, OR=2.48 (1.54 to 4.00)), which covered the protein-coding exons 4, 5 and 6 (figure 1). The minor alleles of six low-frequency variants occurred in this haplotype: rs55758736, a non-synonymous SNP in exon 4 resulting in an alanine to threonine substitution at amino acid position 71 (Ala71Thr; GCT>aCT); chr8:11443817 (intron 4); chr8:11444706 (intron 5), chr8:11446229 (intron 5); and chr8: 8-11446791 and rs2255227 (intron 6) (figure 2A).
Conditional analysis using multivariate logistic regression demonstrated that the association of SNPs in B3 disappeared after adjusting by the strongest hit located in B1 (rs998683) suggesting that the B3 and the B1 association was not independent (online table S2).
In contrast, the low-frequency associated SNPs located in B5 remained associated after adjustment to rs998683 supporting two independent effects in BLK. Further, we observed that SNP rs55758736 had an r2≈0 with SNPs tagging B1 and B3 (figure 2B,C and online table S2), although this may be an underestimation due to the low frequency of the SNP.
Analysis of epigenetic marks and transcription factor binding sites shows an over-representation of NFkB and IRF4 along the promoter region associated blocks
We first attempted the identification of functional regulatory variants across the promoter blocks B1 and B3. Bioinformatic analysis revealed that B1 was significantly enriched for H3K4me3 sites, a hallmark of active promoters (figure 1, online figure S2 and online table S3). Furthermore, SNPs in B1 and B3, when compared with randomly chosen nucleotides, were enriched for binding sites for the transcription factor NFκBp65 and IRF4 but not PAX5 (using data from ENCODE project Chip-Seq for lymphoblastoid cell lines), while SNPs in the non-associated blocks B2 and B4 were not (figure 1, online figure S2 and online table S3). However, we were unable to single out a unique variant to explain the correlation between low BLK gene expression and risk alleles of SNPs in B1 and B3 due to the strong LD. We hypothesised that multiple sites would therefore explain a coordinated regulatory effect on the expression of BLK. Nevertheless, the 1.2 Kb haplotype window with the strongest genetic association and within B3 contained SNP rs2248932 that overlapped with a DNAse hypersensitive cluster and with a strong NFκB binding site (online figure S3 and online table S4). To confirm if NFκB binds to this segment of BLK we performed a CHiP-qtPCR experiment in a human Daudi cell line (figure 3A–C). We observed that NFκBp50 binds somewhat earlier than NFκBp65 (30 min post-BcR stimulation). The IkBa promoter served as control (figure 3C). Simultaneously, expression of BLK is reduced after activation (figure 3A).
Figure 3.

CHiP-qtPCR showing binding of NFκB p50 as well as NFκBp65 within the genomic segment contained in the 1.2 Kb haplotype window in Daudi cells. (A) mRNA expression of the BLK gene after BcR stimulation. (B) qtPCR of the portion containing SNP rs2248932 within the 1.2 kb genomic segment in intron 1 of human BLK. (C) qtPCR of the IkBa gene used as positive control.
The 71Thr mutation leads to reduced half-life of BLK protein
The independent Ala71Thr substitution is located on the SH3 (Src homology 3) domain of the BLK protein, which is of importance in protein–protein interactions. The SIFT algorithm25 26 predicted this substitution to be potentially damaging. As rs55758736 was identified through imputation, we verified the presence of the mutation in 178 selected samples of cases and controls. Of these, we genotyped all of those individuals predicted by imputation to have the mutation and a group of individuals predicted not to have it. Verification was done using PCR, RFLP and sequencing (figure 4) reaching 92% concordance. The mutation was observed in 2.17% of SLE patients and 0.92% of controls, all in the heterozygous state (OR=2.31; 95% CI 1.38 to 3.86 with the Pearson's goodness-of-fitχ2 (df=1)). Adding 1103 new controls, the OR was adjusted to 1.79 (95% CI 1.15 to 2.51).
Figure 4.

Genotyping of the Ala71Thr substitution in exon 4 of BLK. (A) Schematic diagrams showing the procedure for genotyping the single-nucleotide polymorphism (SNP) rs55758736 in exon 4 of BLK. Genomic DNA was amplified with primers flanking exon 4 (In3-F: 5′AGAAGCCTGTCCTCCTTGGTAGC 3′ and In4-R: 5′GGAAAGATTTTGGAGAGGAAGACA 3′) and the PCR products were digested with the restriction enzyme AciI. The A allele disrupts the restriction site generating only two fragments of 48 and 322 bp while the G allele produces three fragments of 48, 80 and 242 bp. (B) PCR products of four subjects showing a unique band of 370 bp and (C) the subsequent AciI digestion of the amplification products. Subjects 2 and 3 are heterozygotes (G/A) and subjects 1 and 4 are homozygotes for the wild type allele (G/G). (D) Sequence chromatographs showing one subject heterozygous for the BLK mutation (above) and one subject homozygous for the wild type allele (below).
At the translational level, the substitution of alanine for threonine implies the formation of a putative phosphorylation site that could lead to the reduction of the half-life of the protein. We investigated if this was the case. Using constructs containing each allele of rs55758736 in transfection experiments we observed that, indeed, 71Thr had an accelerated degradation after treatment with the protein synthesis inhibitor cycloheximide as compared with the Ala71 allele (figure 5 A,B and figure S4).
Figure 5.
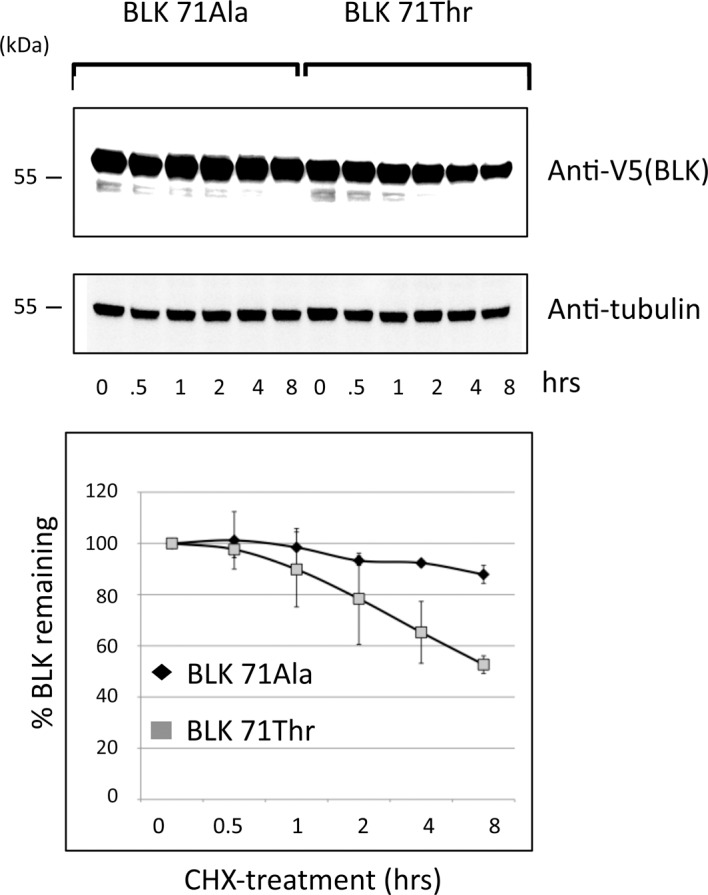
Reduced half-life of BLK protein in the presence of the 71Thr mutation. (A) Plasmids expressing each of the two BLK isoforms fused with V5 were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with 20 µg/ml cycloheximide (CHX) for various times as indicated. The recombinant proteins were detected by western blot using anti-V5 antibody. Tubulin was used as loading control. (B) Quantification of the blots from three independent experiments; the SD at each time point is represented by bars. After 8 h of inhibition of protein synthesis, the level of BLK 71Thr was reduced to half while the wild type isoform Ala71 remained highly stable.
Discussion
We have herein shown that blocks of associated SNPs within the promoter regions of the BLK gene were significantly enriched for NFκB and IRF4 binding sites, potentially involved in regulating levels of BLK mRNA and the related reduction in BLK gene expression levels correlating with the risk haplotypes. Also, through conditional analysis we have identified a potentially deleterious, low-frequency mutation leading to reduced BLK protein half-life. Unlike the wild type allele, the threonine residue of the mutated variant is capable of being phosphorylated, and there is evidence that while phosphorylation sites are important for activation of proteins in signal transduction pathways phosphorylation sites can act at the same time as signals for degradation by ubiquitin ligases.27 28
Protein stability has been extensively studied in the cycling protein family. The abundance of cycling E is controlled by phosphorylation-triggered ubiquitin-dependent proteolysis.29 30 Phosphorylation of the Thr380 allows cycling E to be recognised by Fbw7, an F box protein substrate of related E3 ubiquitin ligases.31 Mutation of this Thr380 to Ala prevents degradation of cyclin E.32 We are currently investigating if the reduction of the half-life of the BLK 71Thr protein is due to an active process of degradation mediated by phosphorylation or just the result of a general decrease in protein stability.
As the LD within the promoter blocks of BLK was very tight, it was not possible for us to define a single variant that could explain the correlation between low BLK levels and a polymorphism. Instead, we observed a clear correlation between associated blocks and transcription factor binding sites for NFκB as experimentally determined by ChIP-Seq and contained in the ENCODE site. However, the database contains only NFkBp65 experiments. NFkBp65 is known to have a after BcR activation. Alternatively, NFkBp50 homodimers that lack a transactivating domain bind to the human BLK promoter, as has been shown in mice.33 Here, homodimers of NFκB/p50 were shown to compete with Pax5 and reduce Blk gene expression upon mature B cell activation and development to antibody-producing plasma cells,33 opposite to what occurs during early B cell differentiation, when Blk expression plays an important role.33 We also do not know the role of IRF4 in this context and a much more detailed analysis of the regulatory landscape of BLK is required to understand the correlation between the promoter risk haplotypes and lowered BLK gene expression. Also, due to the strong LD within the promoter haplotypes, it has also not been possible to discard the region proximal to the C8orf13 or FAM167A region lying 5′ of BLK. In this context, the same associated SNPs correlate with increased levels of FAM167A gene expression.1 The meaning of this is currently unknown and there is no knowledge that this gene would have any role in immunity in general. In this context, we chose not to include the FAM146A gene in our fine mapping, as ours and the actual evidence in other autoimmune diseases support BLK as the susceptibility gene in this locus.
We propose that subtle quantitative differences between risk and protective haplotypes occur during mature B cell stimulation leading to lower BLK levels, and that epigenetically several sites show a variegated effect of NFκBp50 homodimers on the promoter region of BLK. Nevertheless, our genetic analysis did show a potentially unique location within the highly associated 1.2 Kb haplotype window where a single binding site for NFκBp65 was shown to be particularly strong in the ENCODE data and confirmed here for NFκBp50 as well.
In summary, we identified two independent effects within the BLK locus one of which covers the promoter region of the gene where the associated haplotype blocks are enriched for transcription factor binding sites for NFκB and IRF4, providing a potential mechanism for the correlation of the risk alleles and reduced levels of BLK expression. We also identified a novel mutation where the SLE-associated 71Thr leads to the reduced half-life of BLK protein.
What effect reduced BLK expression has on the function of human B lymphocytes is at present under investigation.
Supplementary Material
Acknowledgments
The authors would like to thank all SLE patients who consented to their recruitment in this project. Funding for the project was provided by The Swedish Research Council, the Gustaf Ve:80-Års Fond, the Swedish Association against Rheumatism, the Instituto de Salud Carlos III (PS09/00129) that cofinanced partly through FEDER funds of the European Union, the Research Network BIOLUPUS with support from the European Science Foundation, the Consejería de Salud de Andalucía (PI0012) and grants from the NIH RR020143, the Alliance for Lupus Research and OCAST to MEAR.
Footnotes
Contributors AMDV performed all the genetic analyses and wrote the initial draft of the manuscript; MGD and JDW performed the bioinformatic analyses; YYW performed the qPCR-CHIP; BMG did the genotyping for the Ala71Thr; CCL designed the protein degradation experiments on the Ala71Thr and the RFLP; MBQ performed the degradation experiments; JF, LT, EdR, MFGE, BAPE, SD'A, GDS, TW, BRL, EE, LK, CV, BMdaS and JM provided samples; MEAR directed the work; MEAR, ADG and CCL wrote the manuscript. AMDV, MEAR, CCL and MGD contributed equally to this work.
Funding Funding provided by FCT (Fundação Ciencia e Tecnologia, Portugal), DFG WI 1031/6-1 (Germany), Grupo Italiano LES and the CVDIMMUNE Project supported by the European Commission.
Competing interests None
Ethics approval Approval provided by the IRB from each participating institution.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We will share data related to this article.
References
- 1.Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 2008;358:900–9 [DOI] [PubMed] [Google Scholar]
- 2.Tsuchiya N, Ito I, Kawasaki A. Association of IRF5, STAT4 and BLK with systemic lupus erythematosus and other rheumatic diseases. Nihon Rinsho Meneki Gakkai Kaishi 2010;33:57–65 [DOI] [PubMed] [Google Scholar]
- 3.Ito I, Kawaguchi Y, Kawasaki A, et al. Association of the FAM167A-BLK region with systemic sclerosis. Arthritis Rheum 2010;62:890–5 [DOI] [PubMed] [Google Scholar]
- 4.Torres O, Palomino-Morales R, Vazquez-Rodriguez TR, et al. Role of the C8orf13-BLK region in biopsy-proven giant cell arteritis. Hum Immunol 2010;71:525–9 [DOI] [PubMed] [Google Scholar]
- 5.Yin H, Borghi MO, Delgado-Vega AM, et al. Association of STAT4 and BLK, but not BANK1 or IRF5, with primary antiphospholipid syndrome. Arthritis Rheum 2009;60:2468–71 [DOI] [PubMed] [Google Scholar]
- 6.Fan Y, Tao JH, Zhang LP, et al. Association of BLK (rs13277113, rs2248932) polymorphism with systemic lupus erythematosus: a meta-analysis. Mol Biol Rep 2011;38:4445–53 [DOI] [PubMed] [Google Scholar]
- 7.Nordmark G, Kristjansdottir G, Theander E, et al. Association of EBF1, FAM167A(C8orf13)-BLK and TNFSF4 gene variants with primary Sjögren's syndrome. Genes Immun 2011;12:100–9 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Zhu KJ, Xu Q, et al. The association of the BLK gene with SLE was replicated in Chinese Han. Arch Dermatol Res 2010;302:619–24 [DOI] [PubMed] [Google Scholar]
- 9.Gourh P, Agarwal SK, Martin E, et al. Association of the C8orf13-BLK region with systemic sclerosis in North-American and European populations. J Autoimmun 2010;34:155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orozco G, Eyre S, Hinks A, et al. Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 2011;70:463–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appel S, Filter M, Reis A, et al. Physical and transcriptional map of the critical region for keratolytic winter erythema (KWE) on chromosome 8p22-p23 between D8S550 and D8S1759. Eur J Hum Genet 2002;10:17–25 [DOI] [PubMed] [Google Scholar]
- 12.Borowiec M, Liew CW, Thompson R, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci USA 2009;106:14460–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dymecki SM, Niederhuber JE, Desiderio SV. Specific expression of a tyrosine kinase gene, blk, in B lymphoid cells. Science 1990;247:332–6 [DOI] [PubMed] [Google Scholar]
- 14.Delgado-Vega A, Sánchez E, Löfgren S, et al. Recent findings on genetics of systemic autoimmune diseases. Curr Opin Immunol 2010;22:698–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7 [DOI] [PubMed] [Google Scholar]
- 16.Barrett JC. Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb Protoc 2009;2009:pdb.ip71 [DOI] [PubMed] [Google Scholar]
- 17.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010;467:52–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.International HapMap Consortium A haplotype map of the human genome. Nature 2005;437:1299–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.1000 Genomes Consortium A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007;23:1294–6 [DOI] [PubMed] [Google Scholar]
- 23.Dozmorov MG, Cara LR, Giles CB, et al. GenomeRunner: automating genome exploration. Bioinformatics 2012;28:419–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004;32(Database issue):D493–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–81 [DOI] [PubMed] [Google Scholar]
- 27.Deckert M, Elly C, Altman A, et al. Coordinated regulation of the tyrosine phosphorylation of Cbl by Fyn and Syk tyrosine kinases. J Biol Chem 1998;273:8867–74 [DOI] [PubMed] [Google Scholar]
- 28.Rao N, Ghosh AK, Ota S, et al. The non-receptor tyrosine kinase Syk is a target of Cbl-mediated ubiquitylation upon B-cell receptor stimulation. EMBO J 2001;20:7085–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welcker M, Singer J, Loeb KR, et al. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell 2003;12:381–92 [DOI] [PubMed] [Google Scholar]
- 30.Hao B, Oehlmann S, Sowa ME, et al. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 2007;26:131–43 [DOI] [PubMed] [Google Scholar]
- 31.Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001;294:173–7 [DOI] [PubMed] [Google Scholar]
- 32.Strohmaier H, Spruck CH, Kaiser P, et al. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001;413:316–22 [DOI] [PubMed] [Google Scholar]
- 33.Zwollo P, Rao S, Wallin JJ, et al. The transcription factor NF-kappaB/p50 interacts with the blk gene during B cell activation. J Biol Chem 1998;273:18647–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.