

Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) a leading cause of cancer death. Smoking, diabetes and pancreatitis are risk factors. We have shown that the growth of PDAC and pancreatic duct epithelial cells is regulated by beta-adrenoreceptors (β-ARs). The activity of β-ARs in the central nervous system is counteracted by γ-aminobutyric acid (GABA) via GABAB receptor-mediated inhibition of adenylyl cyclase. The aim of this study was to investigate if the GABABR inhibits β-AR signaling in PDAC and pancreatic duct epithelial cells, thus blocking driving forces of cancer progression, such as cell proliferation and cell migration.
Methods
Intracellular cAMP was measured by immunoassays, DNA synthesis by BRDU incorporation assays, activation of ERK1/2 by ERK activation assays and Western blots and metastatic potential by cell migration assays in the human PDAC cell lines PANC-1 and BXPC-3 and immortalized human pancreatic duct epithelial cells HPDE6-C7. The expression of norepinephrine, PKARIIα and GABA in PDAC microarrays was assessed by immunohistochemistry.
Results
Stimulation of the GABABR by GABA or baclofen inhibited isoproterenol-induced cAMP signaling below base levels. ERK1/2 activity in response to isoproterenol was blocked by GABA, an effect enhanced by transient overexpression of the GABABR and abolished by GABABR knockdown. DNA synthesis and cell migration were stimulated by isoproterenol, responses blocked by GABA and baclofen. Norepinephrine and PKARIIα were overexpressed while GABA was underexpressed in human PDAC tissue arrays.
Conclusions
Our data suggest the stimulation ofGABABR signaling as a novel target for the treatment and prevention of pancreatic cancer.
Keywords: GABAB, signaling pancreatic cancer
Background
Pancreatic cancer is the fourth leading cause of cancer mortality in Western countries(1). About 95% of pancreatic cancers are ductal adenocarcinomas (PDAC). PDAC is among the most aggressive human cancers and is generally unresponsive to conventional therapy, resulting in a mortality near 100% within one year of diagnosis(1).
The regulation of PDAC and pancreatic duct epithelia is poorly understood. Most PDACs harbor activating point mutations in K-ras while also over-expressing the epidermal growth factor receptor (EGFR), leading to the hypothesis that EGFR signaling via ras and the extracellular signal regulated protein kinases (ERK1/2) pathway regulate PDAC(2). Moreover, the arachidonic acid-metabolizing enzyme cyclcooxygenase 2 (COX-2) is over-expressed in about 90% of PDACs(3). Studies in vitro and in human pancreatic cancer mouse xenografts have shown significant reductions in tumor growth by inhibitors of farnesyltransferase, EGFR tyrosine kinases, ERK1/2(1; 4). Likewise, inhibitors of COX-2 have yielded impressive antitumorigenic effects in vitro(4). Suppression of vascular endothelial cell growth factor (VEGF) has also shown promising responses in vitro and in an orthotopic mouse model of PDAC(5). However, clinical trials with inhibitors of tyrosine kinases, ra, COX-2, VEGF, or the combination of such agents have had disappointing results(6).
Smoking(7), diabetes mellitus and pancreatitis are risk factors for PDAC(7). However, the mechanisms by which these risk factors contribute to the development of PDAC are poorly understood.
We have shown that cell lines derived from human PDACs express β1-and β2-adrenoreceptors (ARs) and respond to their stimulation by agonists with the release of arachidonic acid and cell proliferation(8; 9). Our findings provided evidence, for the first time, that neurotransmitter receptors of the beta-adrenergic family participate in the growth regulation of PDAC.
Nicotine causes the release of the physiological agonists for β-ARs, the catecholamine stress hormones epinephrine and norepinephrine, from the adrenal medulla(10). Identification of beta-adrenergic signaling as a growth regulator of pancreatic cancer cells(8) suggests that the elevated systemic catecholamine levels in smokers may contribute to the development of pancreatic cancer. Our studies showed that the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) mimicked the actions of the catecholamines by binding as an agonist to β1- and β2-ARs(8). We recently showed that the beta-adrenergic agonist isoproterenol and NNK stimulated the proliferation of human pancreatic duct epithelial cells by signaling via cAMP/PKA/p-CREB while additionally transactivating the EGFR and ERK1/2 in a PKA-dependent manner(11). Beta-adrenergic activity of NNK resulting in mitogenic and/or anti-apoptotic signaling has additionally been reported in cell lines from human small airway-derived adenocarcinomas(12; 13), human small airway epithelia(12) and in colon cancer cells(14). It has been shown that the migration and invasiveness of adenocarcinomas of the colon, prostate and breast are also under beta-adrenergic control(15–17), and that ERK1/2-mediated proliferation of breast cancer cells is stimulated by beta-adreneregic agonists(18). Recent publications additionally demonstrated that the nicotine-induced increase in systemic catecholamines stimulated the growth of xenografts from human gastric carcinoma via activation of β-ARs(19) and that epinephrine increased the invasiveness of ovarian cancer cells via stimulation of β-ARs(20). A novel concept is thus emerging of β-AR hyperstimulation as an important etiologic factor for some of the most common human cancers, including PDAC.
Beta-adrenoreceptors consist of β1, β2, and β3-ARs and are G-protein coupled cell membrane receptors. Beta1 and β2-ARs are expressed in most mammalian cells. Binding of an agonist to β-ARs activates the adenylyl cyclase stimulating G-protein, Gαs, resulting in the formation of cAMP. In turn, cAMP activates PKA by binding to the PKA subunit complex, resulting in the release of inhibitory R subunits and activation of the transcription factor, cAMP response element binding protein (CREB)(21). It has been shown that β1- and β2-ARs can additionally transactivate the EGFR pathway in a PKA-dependent manner(22) and we have shown that such transactivation occurs in human pancreatic duct epithelial cells(11; 23),.
Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system, and counteracts the stimulatory actions of the physiological agonists for β-ARs, epinephrine and norepinephrine in the brain(24). Three types of GABA receptors have been identified: the ion channel receptor families GABAA and GABAC, and GABAB receptors which exist as two isoforms termed GABABR1 and GABABR2(25). Both GABABR isoforms are coupled to the adenylyl cyclase inhibiting G-protein, Gαi(25). GABAA and GABAC receptors mediate excitatory functions of GABA whereas the GABAB receptors mediate the inhibitory actions of GABA(25). In the pancreas, GABA, its synthesizing enzyme, glutamic acid decarboxylase, and its metabolizing enzyme, GABA-transaminase, are expressed in the beta cells of pancreatic islets at high concentrations similar to those in the central nervous system, and GABA is secreted from beta-cells into the extracellular space(24). However, the function of GABA in the pancreas is poorly understood. GABA is decreased in the pancreas of individuals with diabetes mellitus(24) due the reduction of functional islet beta cells. The number of functional pancreatic islets is also diminished in pancreatitis. The inhibitory role of GABA in the central nervous system and the fact that the GABABRs inhibits adenylyl cyclase via activation of Gαi, prompted us to explore a potential inhibitory action of GABA on the cAMP-mediated beta-adrenergic stimulation of human PDAC and pancreatic duct epithelial cells. Our data show that stimulation of the GABAB receptor potently inhibited isoproterenol-induced cAMP signaling, cell proliferation and cell migration. Moreover, human PDACs overexpressed norepinephrine and PKARIIα while underexpressing GABA. These data suggest that the reduction in pancreatic GABA may contribute to the development of PDAC. Therapeutic administration of GABA or a GABABR selective agonist may thus provide an effective novel tool for the treatment and prevention of pancreatic cancer.
Methods
Cell Lines
The human PDAC cell lines, PANC-1, which carries an activating point mutation in K-ras and BXPC-3, without ras mutation (American Type Culture Collection Rockville, MD) were cultured as suggested by the vendor: RPMI medium supplemented with 10% fetal bovine serum and L-glutamine (2 mM) for BXPC-3 and DMEM medium supplemented with 10% fetal bovine serum, L-glutamine (2 mM) for PANC-1. The immortalized human pancreatic duct epithelial cell line HPDE6-C7(26) (Dr. M.S. Tsao, Ontario Cancer Center, Ontario, Canada) was maintained in serum-free keratinocyte medium supplemented with bovine pituitary extract (25 mg/500 ml), EGF (2.5 mg/500 ml). Cells were washed to remove supplements prior to each assay, and all assays were conducted in basal media without supplements.
Analysis of intracellular cAMP by Immunoassay
Cells were plated at 4×105 cells/well in 6-well plates in their growth medium until 65–70 % confluence. The medium was replaced by basal medium without supplements following three washes with PBS and incubated for 24 hours. The cells were pre-incubated (30 minutes) with the phosphodiesterase inhibitor isobutylmethylxanthine (IBMX;1 mM) to prevent the enzymatic breakdown of cAMP formed in response to beta-adrenergic stimulation. The beta-adrenergic agonist isoproterenol (1 µM; Sigma, St Louis) was added to the culture medium with or without a 4-hour pre-incubation with GABA (30µM) or the selective GABAB receptor agonist, baclofen (30µM). After 3 washes with water, cells were treated with 0.1 M HCL for 20 min, then, lysed by sonication. Intracellular cAMP was measured with an enzyme immunoassay kit (Assay Designs Inc, Ann Arbor, MI) as previously described(27) Assays were conducted in triplicate, each with triplicate samples. Statistical analysis of data was by one-way ANOVA, Tukey-Kramer multiple comparison test and two-tailed unpaired t-test.
BRDU incorporation assays
BrdU incorporation assays were conducted with a kit (Roche Applied Science) as previously described Cells were cultured in 96 well plates (1 × 104/well), deprived of serum and supplements for 24 hours and then either treated with isoproterenol (10 nM) for 72 hours, pretreated for 4 hours with GABA (30µM) or baclofen (30µM), or they were exposed to each of the inhibitors alone for the duration of the assay. Cells were labeled with 10 ml/well BrdU and reincubated at 37°C for 4 hours. After removal of the labeling medium, cells were fixed and probed with anti-BrdU monoclonal antibody and its substrate, tetramethyl-benzidine for one hour. After removal of the antibody conjugate, the cells were rinsed three times with washing solution and substrate solution (100 µl/well) was added. After color development, the absorbance of each sample was measured in an ELISA reader at 370 nm. A blank was run in each experiment to provide information about BrdU and anti-BrdU nonspecific binding. The non-specific binding was subtracted from all other values. Each experiment was conducted twice with 5 replicates per data point. Statistical analysis of data was by one-way ANOVA and Tukey-Kramer multiple comparison test.
Cell migration assay
The metastasis of cancer cells is facilitated by their ability to migrate. Measurement of cell migration is therefore frequently used as a tool to assess the metastatic potential of cancer cells. We have used a colorimetric cell migration assay kit (Cell Biolabs) consisting of 24-well plates that contain polycarbonate membrane filter inserts (8µM pore size). Cells (0.5×106 cells per ml of basal medium) were seeded onto the top chamber above the filter insert and pretreated for 4 hours with GABA (30 µM) or baclofen (30 µM). Isoproterenol (10 nM) was then added. Following a 24-hour incubation period, non-migratory cells were removed from the top of the filters by cotton swab. The filter with cells that had migrated to its bottom surface were incubated with staining solution for 10 min, washed three times with tap water, air-dried and photographed. Each filter was extracted and 100 µL per sample of the extract was transferred to a 96-well microtiter plate. Optical density at 560nm was read with a plate reader. Each assay was conducted in triplicate. Statistical analysis of data was by ANOVA and Tukey-Kramer multiple comparison test.
Transient transfection with stealth select RNAi for GABAB-R1 or with GABAB-R1 cDNA
Cells (> 90% viable) were plated at 3×104 cells/well in 24-well plates in complete medium without antibiotics and allowed to reach 60% confluence. They were then transfected in triplicate for each treatment group with 100 ml of 100 nM of GABAB-R1 Stealth RNAI (Invitrogen) or GABAB-R1 cDNA complexed with 2mg/ml Lipofectamine (Invitrogen). Following a 24-hour incubation in a humidified incubator (5% CO2, 37°C), transfection efficiency, transfection toxicity and percent of transfected cells were determined (Block-iT Alexa Fluor Red Fluorescent Control, dead cell stain ethidium homodimer-1, Nuclear stain Hoechst 33342, Invitrogen). The growth medium was then replaced by basal medium without additives and responses to isoproterenol and GABA assessed in untransfected cells versus cells transiently over-expressing the GABABR or cells with GABABR knockdown, using the ERK1/2 activation assay desribed below. Negative Stealth RNAi provided by the vendor served as negative control.
ERK1/2 activation assays(12)
This assay takes advantage of the fact that ELK-1 is a downstream effector of activated ERK1/2. Equal amounts of protein were incubated overnight with 15 ml of agarose hydrazide beads immobilized p44/42 (Cell Signaling). Immune precipitates were washed three times in 100 mM tris (pH 7.5), 1% Nonidet P-40, 2 mM sodium orthovanadate; one time in 100 mM TRis (Ph 7.5), 0.5 M lithium chloride; and once in kinase buffer (12.5 mM MOPS, pH 7.5, 12.5 mM b-glycerophosphate, 7.5 mM MgCl2). Proteins were incubated for 20 minutes at 30°C in a 30 ml kinase reaction containing 2 mg ELK-1 fusion protein (GST-ELK-1 codons 307–428; Cell Signaling), and 10 mM ATP. Proteins were separated by electrophoresis on a 12% SDS-polyacrylamide gel, transferred to nitrocellulose membranes and probed with anti-phospho ELK (Ser383) antibody. After incubation with the secondary antibody, bands were visualized by enhanced chemiluminescence detection. The density of p-ELK-1 and ERK1/2 protein bands was quantitated by densitometry (four densitometric readings per band using NIH Scion image analysis software). The ratios of p-ELK-1/ERK were statistically analyzed by one-way analysis of variance and Tukey-Kramer multiple comparison test.
Immunohistochemistry
To provide in vivo evidence for a potential role of c-AMP-mediated and GABA signaling in the regulation of human pancreatic cancer, slides from human tissue arrays (US Biomax, Inc., Rockville, MD) were used. Microarray PA242 contained sections of 24 human PDACS and 4 normal unrelated pancreatic tissues. Microarray PA241 contained sections from 6 additional PDACs with matched normal tissues 1.5 cm away from tumor. The sections were de-paraffinized in xylene, washed with phosphate buffered saline (PBS, pH 7.4), and incubated with 3% hydrogen peroxide in 50% methanol for 20 min at room temperature. Incubations with protein block solution, primary and byotynilated secondary antibodies and diaminobenzydine substrate were conducted with reagents provided by the universal Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to instructions by the kit. Incubations with primary polyclonal antibodies (Chemicon Millipore, Billericam MA) to mammalian GABA (1:1000), human norepinephrine (1:1000), or human PKARIIα (1:1000; BD Biosciences, San Jose, CA) which recognizes the regulatory RIIα subunit when it has been released from the PKA subunit complex upon binding of cAMP(28), were conducted at 4°C overnight in a humid chamber. Tissue slides processed without exposure to primary antibodies served as negative controls and showed no detectable immunoreactivity. Hematoxylin was used as counterstain.
Results
In accordance with our earlier publications(8; 11; 23), the beta-adrenergic agonist isoproterenol caused a highly significant (p<0.001) increase in intracellular cAMP in the pancreatic duct epithelial cells and in both PDAC cell lines (Figure 1). This response was completely abrogated (p<0.001) by preincubation with GABA which is an agonist for all GABA receptors (Figure 1). Pre-incubation of the cells with the selective agonist for GABABRs baclofen similarly blocked the responses of all three cell lines to isoproterenol (p<0.001; Figure 1), suggesting involvement of the GABABR in the observed inhibition. Exposure of unstimulated pancreatic duct or PDAC cells to GABA or baclofen significantly (p<0.001) reduced base levels of cAMP below that of control cells (Figure 1).
Figure 1.
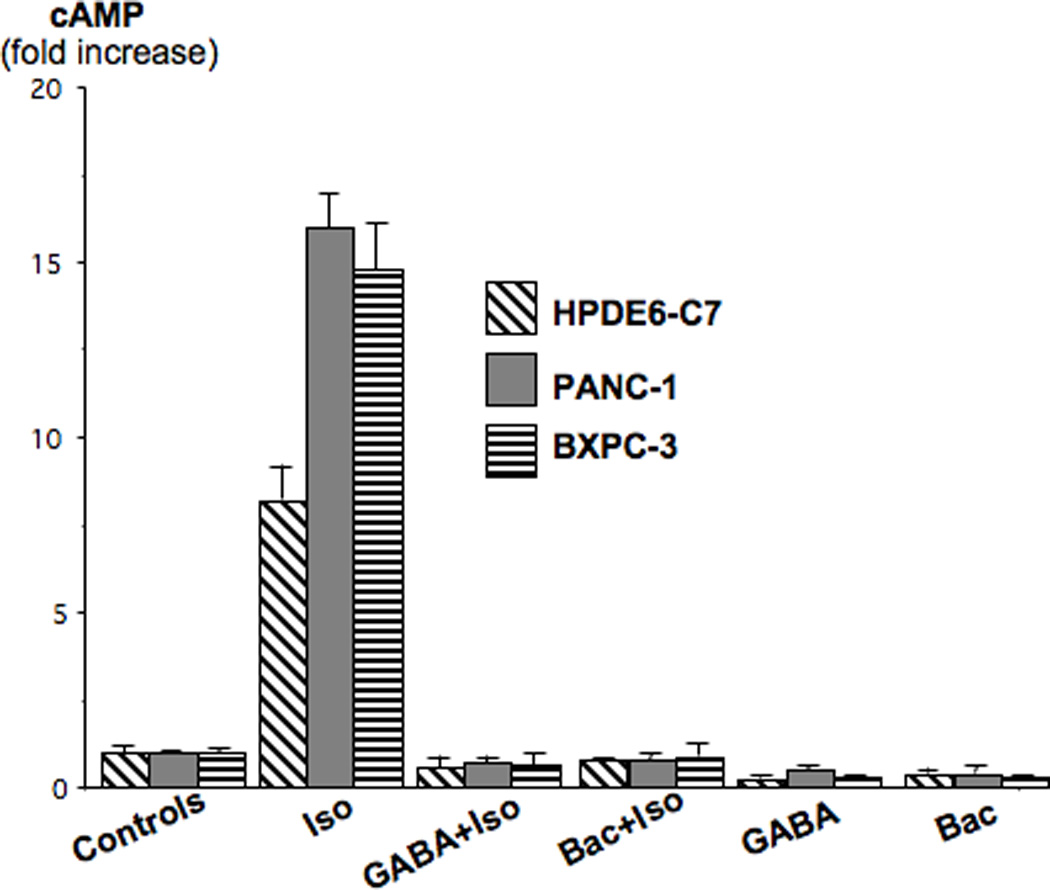
The beta-adrenergic agonist isoproterenol (1 µM) significantly (p<0.001) increased intracellular cAMP levels in the human PDAC cell lines PANC-1 and BXPC-3 in immortalized human pancreatic duct epithelial cell line HPDE6-C7. Pre-incubation with the physiological agonist for all GABA receptors, GABA (30µM), or with the selective agonists for GABABRs, baclofen (30µM) completely abrogated this response. Data are mean values and standard errors of triplicate samples per treatment group.
We have previously shown that β-AR-induced cAMP activates the ERK1/2 cascade via transactivation of the EGFR in a PKA dependent manner in human pancreatic duct epithelial cells and that the resulting activation of ERK1/2 serves as a major mitogenic stimulus(11). In the current study, we therefore used an ERK1/2 kinase reaction and Western blot in anti-p-ERK1/2 immunoprecipitates of p-ELK-1 protein phosphorylated by activated ERK1/2 to assess the modulating effects of the GABABR on ispoproterenol-evoked mitogenic signaling in these cells. Our data show that isoproterenol caused a significant (p<0.001) increase in p-ELK-1 protein (Figure 2, lane 2), a response significantly (p<0.001) reduced by pre-incubation with GABA (Figure 2, lane 3). Pre-incubation with GABA failed to reduce isoproterenol-induction of p-ELK-1 in cells with GABABR1 knockdown by transient transfection with GABABR1 stealth RNAi (p<0.001; Figure 2, lane 4). Conversely, over-expression of the GABABR1 by transient transfection of GABABR1 cDNA reduced (p<0.001) isoproterenol-induced p-ELK-1 below control levels in cells pre-incubated with GABA (Figure 2, lane 5). The inhibitory effects of GABA in cells over-expressing the GABABR1 were significantly (p<0.001) greater than in cells without GABABR1 over-expression (compare lane 5 with lane 3 in Figure 2). Collectively, these findings strongly suggest that activation of the GABABR may inhibit mitogenic signaling in response to β-AR stimulation in pancreatic duct epithelial cells and the cancers derived from them. This interpretation is supported by the results generated by BRDU incorporation assays that monitored modulation of DNA synthesis in pancreatic duct epithelial cells HPDE6-C7 and the two PDAC cell lines PANC-1 and BXPC-3. As Figure 3 shows, isoproterenol strongly (p<0.001) induced DNA synthesis in all three cell lines. Pre-incubation with GABA or baclofen each significantly (p<0.001) reduced this response below base levels observed in the controls. In addition, both agents significantly (p<0.001) reduced base level DNA synthesis in unstimulated cells from each of the three cell lines (Figure 3).
Figure 2.
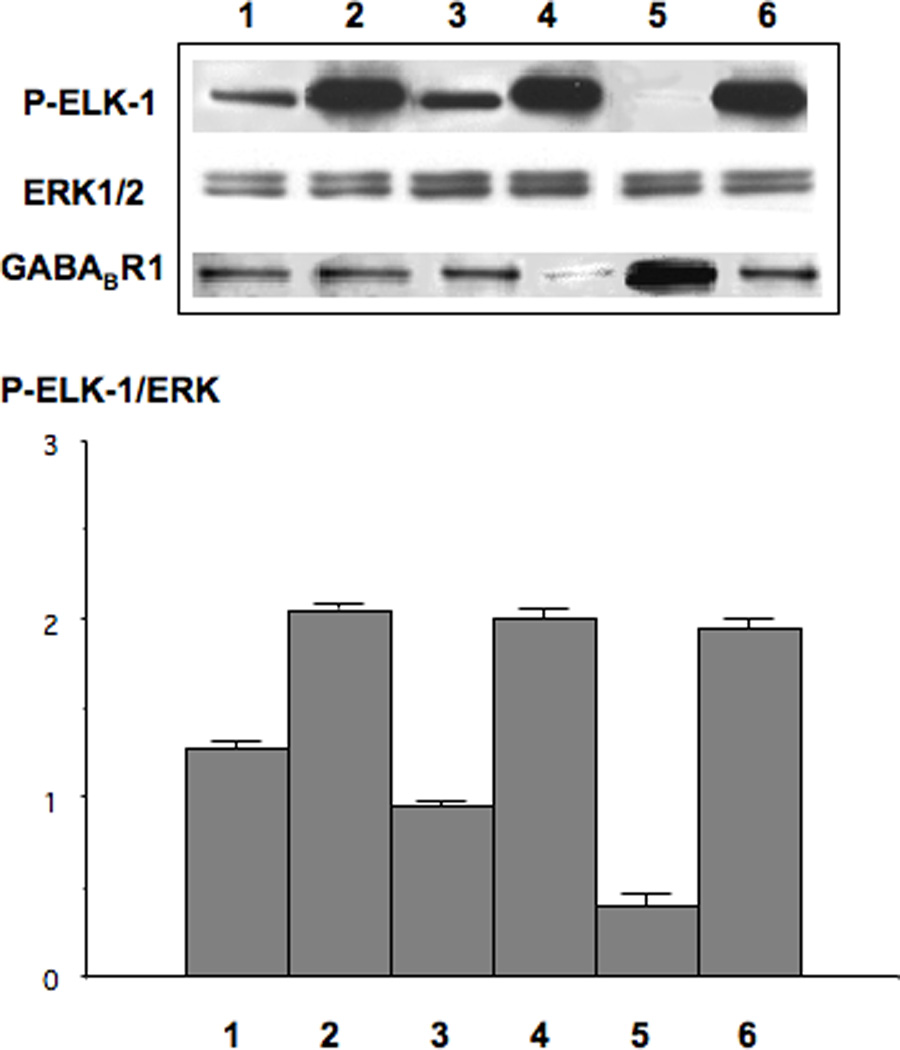
Modulation of p-ELK-1 protein in HPLDE6-C7 cells. Isoproterenol (1µM for 1 hour) significantly (p<0.001) increased p-ELK-1 expression (lane 2). This response was completely abolished by preincubation with GABA (30mM; lane 3). The inhibitory effects of GABA on p-ELK-1 induction by isoproterenol were completely blocked by transient GABABR knock-down (lane 4) whereas transient over-expression of GABABBR significantly (p<0.001) enhanced these inhibitory actions of GABA (lane 5). Lane 6: negative control of cells transiently transfected with negative Stealth RNAi and treated with isoproterenol. The columns in the graph represent the ration of p-ELK-1/ERK densitometry readings (mean values and standard errors of 5 readings per band).
Figure 3.
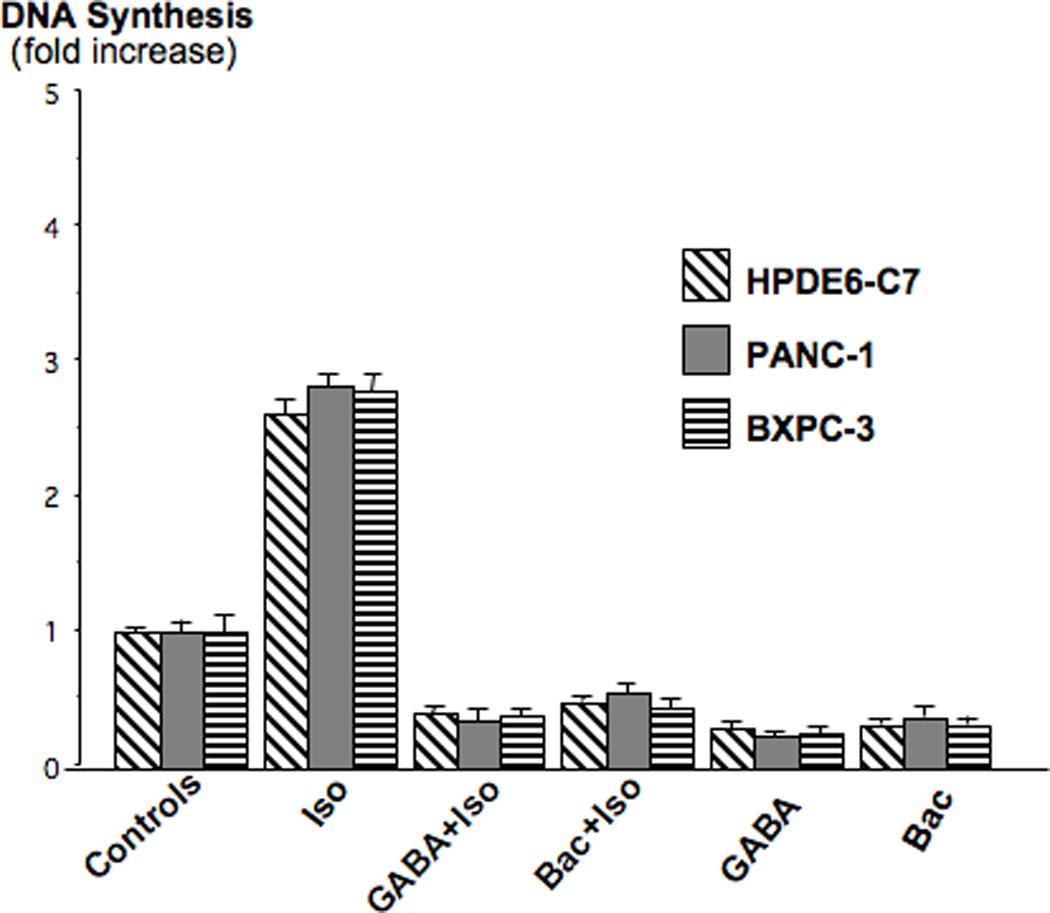
Isoproterenol (10 nM) significantly (p<0.001) induced DNA synthesis in HPDE6-C7, PANC-1 and BXPC-3 cells. These responses were completely inhibited by preexposure to GABA (30µM) or baclofen (30µM). Both agents also significantly (p<0.001) suppressed DNA synthesis below control levels. Data are mean values and standard errors of five samples per treatment group.
The high mortality rate of pancreatic cancer is caused by the aggressive behavior of this malignancy, resulting in extensive invasiveness and metastasis. We therefore assessed the effects of β-AR stimulation on the migration of PDAC cells and the potential modulation of this response by GABA and baclofen. As Figures 4 and 5 show, both PDAC cell lines demonstrated a highly significant (p<0.001) increase in cell migration when treated with isoproterenol. In both cell lines, this response was suppressed below control levels (p<0.001) by pre-incubation with GABA or baclofen. In addition, the migration of cells not stimulated by isoproterenol was significantly (p<0.00!) reduced by each of these GABA-ergic agents (Figures 4, 5). These data suggest, for the first time, that beta-adrenergic signaling increases the invasiveness and metastatic potential of PDAC and that activation of the GABABR by agonists may serve as a valuable tool to suppress this aggressive behavior.
Figure 4.
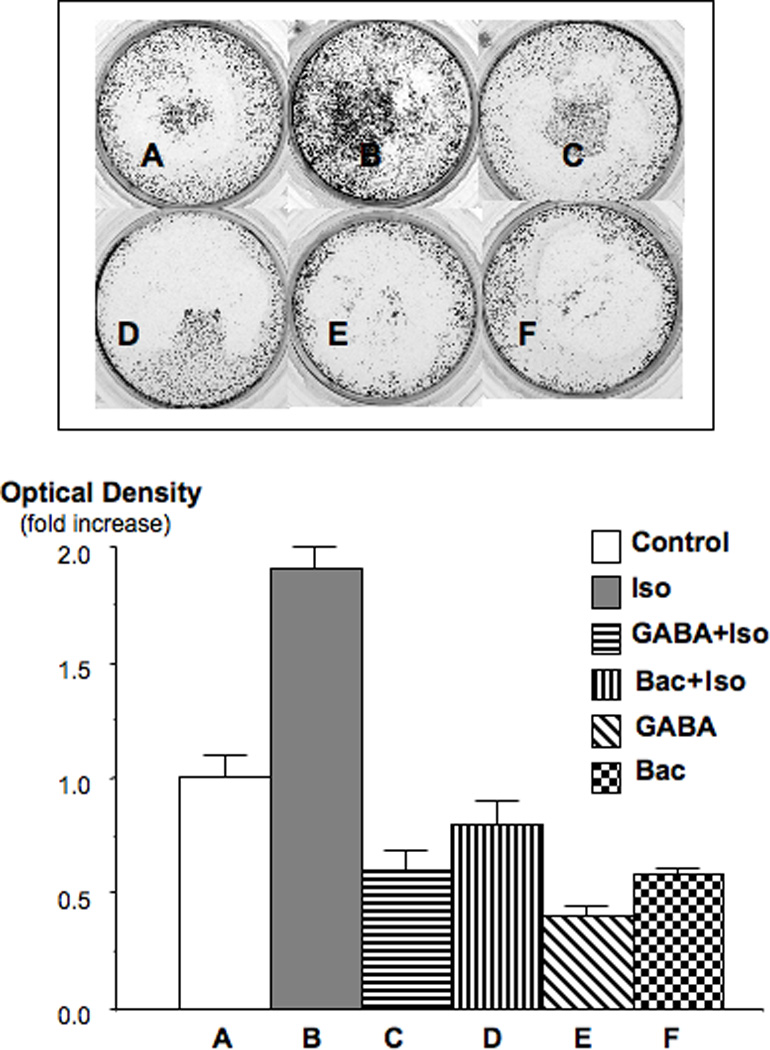
Cell migration assay in PDAC cell line PANC-1. Isoproterenol (10 nM) significantly (p<0.001) stimulated cell migration, an effect completely blocked by GABA (30 µM) and baclofen (30 µM). The photograph shows the bottom side of the filter inserts with cells that have migrated through the filter pores. Columns in the graph are mean values and standard errors of four optical density readings per treatment group and are expressed as fold increase over controls.
Figure 5.
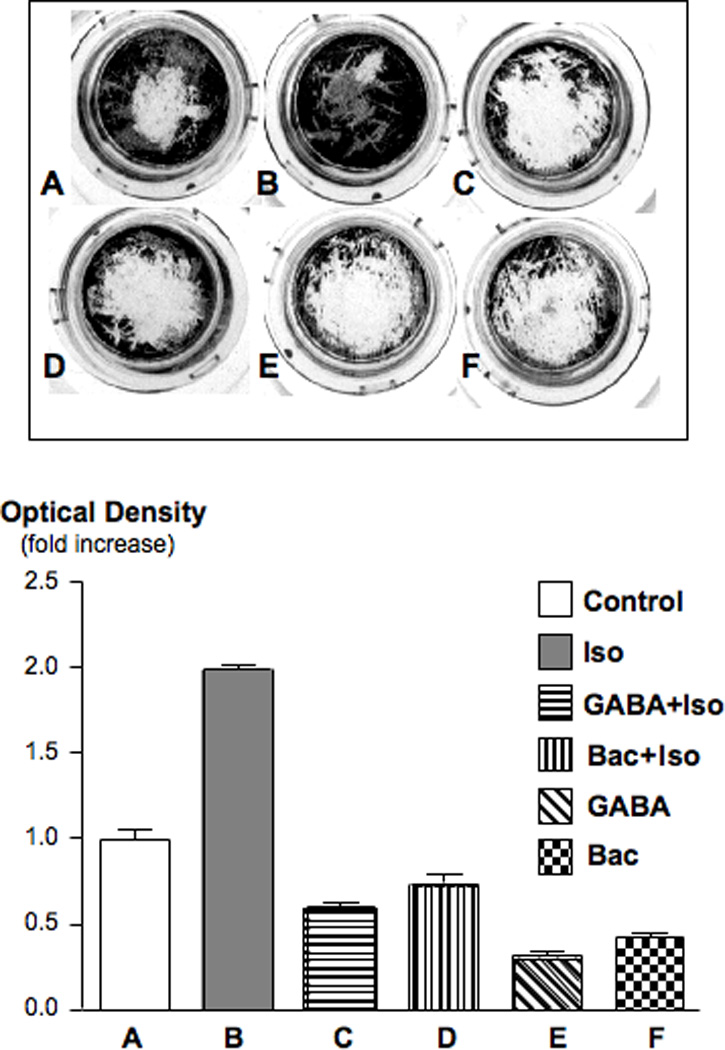
Cell migration assay in PDAC cell line BXPC-3. Isoproterenol (10 nM) significantly (p<0.001) stimulated cell migration, an effect completely blocked by GABA (30 µM) and baclofen (30 µM). The photograph shows the bottom side of the filter inserts with cells that have migrated through the filter pores. Columns in the graph are mean values and standard errors of four optical density readings per treatment group and are expressed as fold increase over controls.
Our in vitro data suggest an important role of cAMP/PKA-mediated signaling in the aggressive behavior of PDAC while indicating that activation of GABA signaling could be useful for the prevention and treatment of this malignancy. However, there is no published evidence to date that identifies adrenergic agonists in the tumor environment of human PDAC or indicates hyperactivity of cAMP-mediated signaling in these tumor cells. Similarly, there are no reports on GABA levels in human PDAC. Our immunohistochemical data generated in human PDAC tissue arrays show low but detectable levels of the physiological agonist for β-ARs, norepinephrine, in all normal tumor adjacent (Figure 6A) and unrelated tissues while norepinephrine was overexpressed in all tumor tissues (Figure 6B). In addition, we observed a strong overexpression of PKARIIα that is released from the PKA subunit complex upon binding of cAMP, in all PDAC samples (Figure 7B) as compared with normal tumor adjacent (Figure 7A) and unrelated pancreatic tissues. This finding suggests hyperactivity of cAMP/PKA signaling in PDAC. By contrast, positive immunoreactivity to GABA was below detectable levels under the conditions used in 29 of the 30 investigated PDAC samples (Figure 8B)) whereas each of the tumor adjacent (Figure 8A) and unrelated normal pancreatic tissues showed strong positive immunorecativity to GABA. These findings are suggestive of a pronounced underexpression of GABA in most human PDACs.
Figure 6.
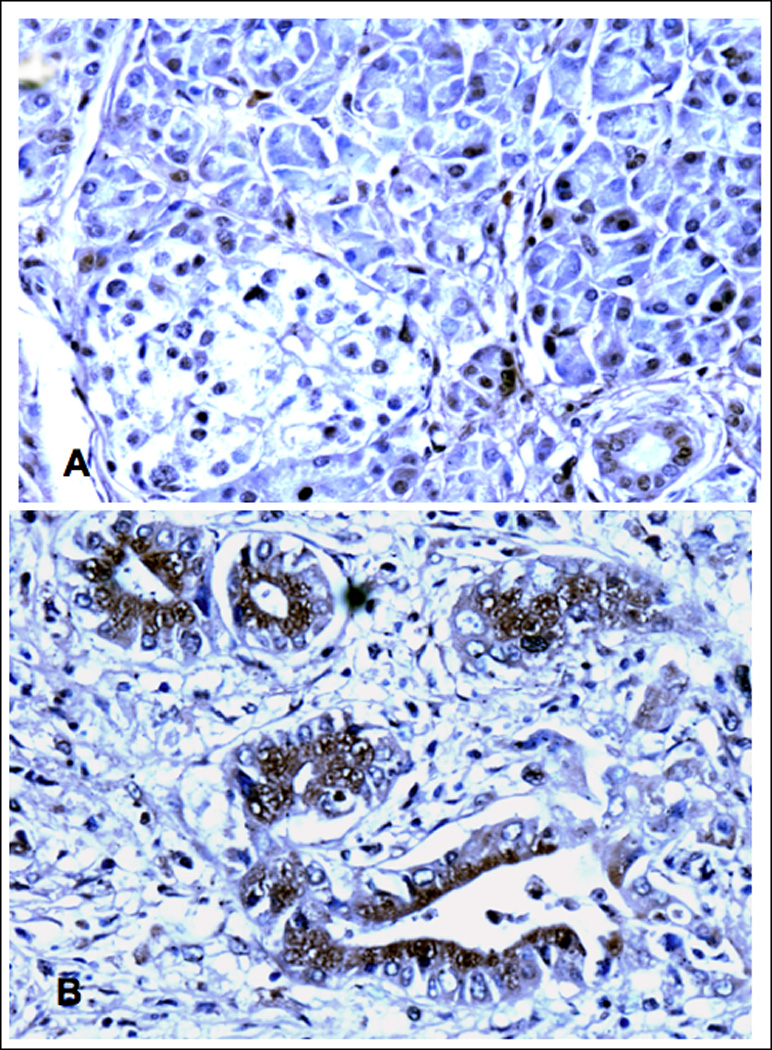
Photomicrographs of normal human pancreas (A,) 1.5 cm away from a PDAC (B). illustrating immunoreactivity to norepinephrine. In keeping with its role as a ubiquitous neurotransmitter of the sympathicus, normal pancreatic cells as well as tumor stroma cells expressed low but detectable levels. The epithelial cancer cells showed distinct overexpression of norepinephrine (B). X 200
Figure 7.
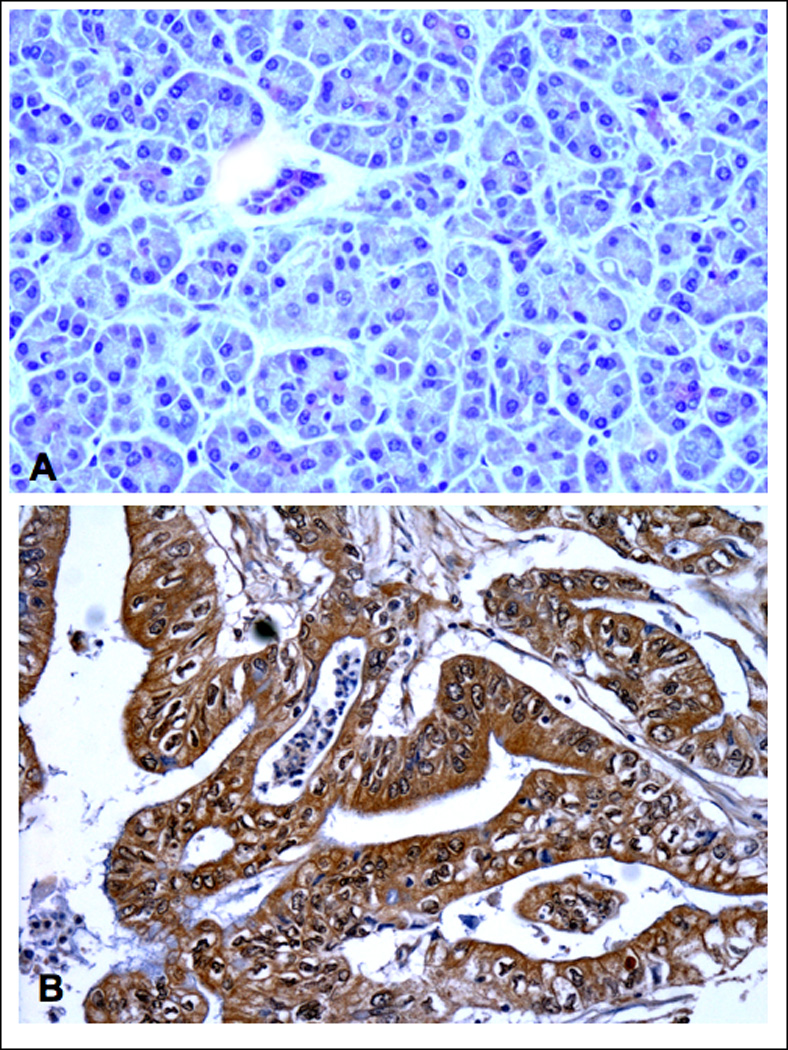
Photomicrographs of normal human pancreas (A,) 1.5 cm away from a PDAC (B). illustrating immunoreactivity to PKARIIα. In keeping with its role as a second messenger of the ubiquitous neurotransmitter of the sympathicus norepinephrine, normal pancreatic cells as well as tumor stroma cells expressed low but detectable levels. The epithelial cancer cells showed distinct overexpression of PKARIIα (B). X 200.
Figure 8.
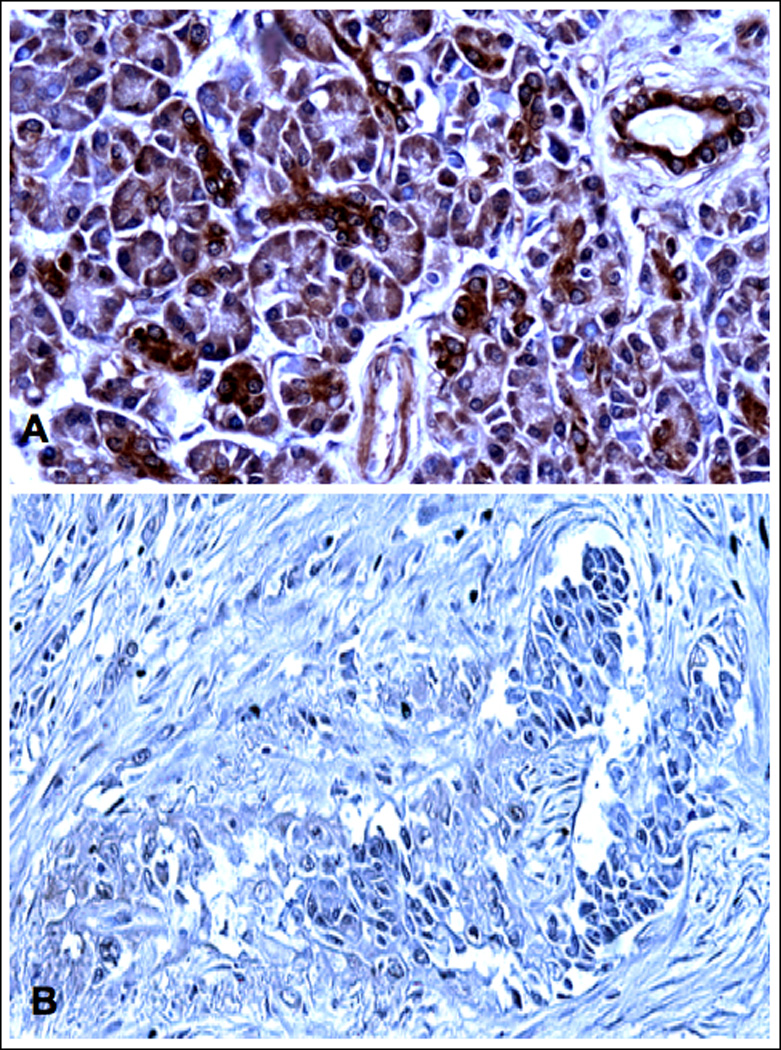
Photomicrographs of normal human pancreas (A,) 1.5 cm away from a PDAC (B). illustrating immunoreactivity to GABA. All cell types in normal pancreatic tissue areas cells expressed prominent levels of GABA. GABA was underexpressed in cancer cells and cancer stroma (B). X 200.
Discussion
This is the first report that implicates the GABABR as a target for the prevention and therapy of PDAC. Our data show that this receptor, which inhibits adenylyl cyclase by activating Gαii(24), acts as a powerful control for cAMP-dependent signaling, including the transactivated EGFR-mediated ERK1/2 cascade, that drive the proliferation and migration of PDAC. Unlike therapeutics that block only individual downstream components of this signaling network, agonists for the GABABR act as the "physiological brake" to counterbalance the entire cAMP-driven stimulatory signaling network activated by cell surface receptors coupled to the stimulatory G-protein Gαs. As our current and published(8; 9; 11; 23) data suggest, among the family of Gαs-coupled receptors that are counterbalanced by the GABABR, β-ARs play a prominent role in pancreatic carcinogenesis. Recent studies by other laboratories have also implicated β-ARs as important mediators of cancer growth and/or invasiveness in adenocarcinoma of the lungs(12; 29; 30), prostate(16), colon(15), stomach(19), breast(17) and ovary(20). In addition, it has been shown that GABA and baclofen inhibit β-AR-mediated migration of breast cancer cells(17) and colon cancer cells(31). Accordingly, the GABABR may be a promising target for the prevention and treatment of all of these cancers.
Intracellular cAMP can be increased by the activation of numerous Gαs-coupled receptors, by activators of adenylyl cyclase or cAMP, as well as phosphodiesterase inhibitors. All of these factors could contribute to pancreatic carcinogenesis. The more pronounced overexpression of PKARII than norepinephrine in PDACs detected by us suggest that cAMP stimulators in addition to β-AR agonists may contribute to this response.
The three known risk factors for PDAC, smoking, diabetes and pancreatitis, disturb the balance between stimulatory beta-adrenergic signaling and inhibitory GABABR signaling in the pancreas. A classic biological effect of nicotine is the enhanced release of the stress hormones epinephrine and norepinephrine(10; 32), resulting in increased stimulation of β-ARs. Beta-ARs in smokers are additionally stimulated by the nicotine-derived carcinogenic nitrosamine NNK that acts as a high affinity agonist for these receptors(8; 29). On the other hand, diabetes and pancreatitis are associated with the loss of pancreatic islet beta cells, the major source of inhibitory GABA in the pancreas(24). It stands to reason that a patient diagnosed with PDAC, a malignancy with an extremely poor prognosis, will suffer extreme anxiety. In turn, anxiety has been shown to increase the sensitivity of β-ARs to agonists(33). In addition, any kind of stress, including anxiety, causes the release of the physiological agonists for β-ARs, norepinephrine and epinephrine from the sympathetic nervous system and the adrenal medulla, increasing their systemic levels(34). Increased systemic levels of norepinephrine have also been reported as a response to gastrointestinal cancer surgery(35; 36). While these stress responses are not unique to pancreatic cancer, they have the potential to contribute to the poor prognosis of this malignancy. The overexpression of norepinephrine in the cancer cells suggest upregulation of the β-ARs that mediate its uptake.
GABA was below detectable levels in 29 of 30 human PDAC samples investigated whereas each of the tumor adjacent and unrelated normal pancreatic tissue samples demonstrated pronounced positive immunoreactivity to GABA. In accordance with its function in the regulation of secretion by pancreatic duct epithelia(37), acinar cells(38) and islet cells(39), norepinepohrine was expressed in all normal and cancerous pancreatic tissues. While positive immunoreactivity to norepinephrine was higher in cancer cells than in normal cells PKARIIα that is released from the PKA subunit complex upon activation of PKA by cAMP(28),, demonstrated a more pronounced overexpression in the cancer cells of each PDAC sample. These findings suggest that the balance between stimulatory and inhibitory signaling in the human pancreatic cancer samples was significantly shifted in favor of stimulatory cAMP/PKA signaling. In addition, the marked underexpression of GABA in the human PDACs in conjunction with our in vitro data suggest GABA as a novel type of tumor suppressor for PDAC.
GABA and baclofen are FDA approved for the treatment of anxiety. In addition, GABA is widely used as a dietary supplement to ease anxiety and promote sleep. Our data suggest that the use of these GABA-ergic agents as adjuvants to standard cancer therapy may have significant beneficial effects on the outcome of pancreatic cancer by inhibiting the proliferation and migration of cancer cells. In addition, the regular intake of GABA as a dietary supplement may significantly reduce the risk for the development of PDAC in individuals with diabetes and pancreatitis.
It is intriguing that GABA is a natural product of plants that mediates communication between plants and other organisms(40) and participates in the plants' defenses against stress, wounding and attack by parasites and microbes(41). Significant levels of GABA have been reported in grape berries and the Merlot wine made from them(42), in tomatoes(40), and in Soy(43). These natural products also have significant cancer preventive activities(44)(45). While research into the mechanisms of action of these and other plant-derived cancer preventive agents have focused on their antioxidant effects and inhibition of carcinogen metabolism(45), our data suggest that inhibition of Gαs-mediated signaling by GABA may be a novel and hitherto unexplored mechanism of action of some plants with cancer preventive activity.
Acknowledgments
Grant Support: RO1 CA42829, NIH.
References
- 1.Mancuso A, Calabro F, Sternberg CN. Current therapies and advances in the treatment of pancreatic cancer. Crit Rev Oncol Hematol. 2006;58:231–241. doi: 10.1016/j.critrevonc.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Sakorafas GH, Tsiotou AG, Tsiotos GG. Molecular biology of pancreatic cancer, oncogenes, tumour suppressor genes, growth factors, and their receptors from a clinical perspective. Cancer Treat Rev. 2000;26:29–52. doi: 10.1053/ctrv.1999.0144. [DOI] [PubMed] [Google Scholar]
- 3.Hong SH, Avis I, Vos MD, Martinez A, Treston AM, Mulshine JL. Relationship of arachidonic acid metabolizing enzyme expression in epithelial cancer cell lines to the growth effect of selective biochemical inhibitors. Cancer Res. 1999;59:2223–2228. [PubMed] [Google Scholar]
- 4.Mimeault M, Brand RE, Sasson AA, Batra SK. Recent advances on the molecular mechanisms involved in pancreatic cancer progression and therapies. Pancreas. 2005;31:301–316. doi: 10.1097/01.mpa.0000175893.04660.1b. [DOI] [PubMed] [Google Scholar]
- 5.Fukasawa M, Korc M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin Cancer Res. 2004;10:3327–3332. doi: 10.1158/1078-0432.CCR-03-0820. [DOI] [PubMed] [Google Scholar]
- 6.El-Rayes BF, Zalupski MM, Shields AF, Ferris AM, Vaishampayan U, Heilbrun LK, Venkatramanamoorthy R, Adsay V, Philip PA. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Invest New Drugs. 2005;23:583–590. doi: 10.1007/s10637-005-1028-z. [DOI] [PubMed] [Google Scholar]
- 7.Lowenfels AB, Maisonneuve P. Risk factors for pancreatic cancer. J Cell Biochem. 2005;95:649–656. doi: 10.1002/jcb.20461. [DOI] [PubMed] [Google Scholar]
- 8.Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis. 2001;22:473–479. doi: 10.1093/carcin/22.3.473. [DOI] [PubMed] [Google Scholar]
- 9.Schuller HM. Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat Rev Cancer. 2002;2:455–463. doi: 10.1038/nrc824. [DOI] [PubMed] [Google Scholar]
- 10.Li Q, Forsberg EJ. Catecholamine secretion induced by nicotine is due to Ca++ channel but not Na+ channel activation in porcine adrenal chromaffin cells. J Pharmacol Exp Ther. 1996;277:1209–1214. [PubMed] [Google Scholar]
- 11.Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol. 2005;131:639–648. doi: 10.1007/s00432-005-0002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laag E, Majidi M, Cekanova M, Masi T, Takahashi T, Schuller HM. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer. 2006;119:1547–1552. doi: 10.1002/ijc.21987. [DOI] [PubMed] [Google Scholar]
- 13.Jin Z, Gao F, Flagg T, Deng X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J Biol Chem. 2004;279:40209–40219. doi: 10.1074/jbc.M404056200. [DOI] [PubMed] [Google Scholar]
- 14.Wu WK, Wong HP, Luo SW, Chan K, Huang FY, Hui MK, Lam EK, Shin VY, Ye YN, Yang YH, Cho CH. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone from cigarette smoke stimulates colon cancer growth via beta-adrenoceptors. Cancer Res. 2005;65:5272–5277. doi: 10.1158/0008-5472.CAN-05-0205. [DOI] [PubMed] [Google Scholar]
- 15.Masur K, Niggemann B, Zanker KS, Entschladen F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. 2001;61:2866–2869. [PubMed] [Google Scholar]
- 16.Palm D, Lang K, Niggemann B, Drell TLt, Masur K, Zaenker KS, Entschladen F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118:2744–2749. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- 17.Drell TLt, Joseph J, Lang K, Niggemann B, Zaenker KS, Entschladen F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res Treat. 2003;80:63–70. doi: 10.1023/A:1024491219366. [DOI] [PubMed] [Google Scholar]
- 18.Cakir Y, Plummer HK, 3rd, Tithof PK, Schuller HM. Beta-adrenergic and arachidonic acid-mediated growth regulation of human breast cancer cell lines. Int J Oncol. 2002;21:153–157. [PubMed] [Google Scholar]
- 19.Shin VY, Wu WKK, Chu KM, Koo MWL, Wong HPS, Lam EKY, Tai EKK, Cho CH. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Tox Sci. 2007 doi: 10.1093/toxsci/kfl118. [DOI] [PubMed] [Google Scholar]
- 20.Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, Li Y, Gershenson DM, Lutgendorf S, Cole SW. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006;12:369–375. doi: 10.1158/1078-0432.CCR-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maki T, Kontula K, Harkonen M. The beta-adrenergic system in man: physiological and pathophysiological response. Regulation of receptor density and functioning. Scand J Clin Lab Invest Suppl. 1990;201:25–43. [PubMed] [Google Scholar]
- 22.Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- 23.Askari MD, Tsao MS, Cekanova M, Schuller HM. Ethanol and the tobacco-specific carcinogen, NNK, contribute to signaling in immortalized human pancreatic duct epithelial cells. Pancreas. 2006;33:53–62. doi: 10.1097/01.mpa.0000226883.55828.e9. [DOI] [PubMed] [Google Scholar]
- 24.Gladkevich A, Korf J, Hakobyan VP, Melkonyan KV. The peripheral GABAergic system as a target in endocrine disorders. Auton Neurosci. 2006;124:1–8. doi: 10.1016/j.autneu.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Franek M. History and the present of metabotropic GABAB receptor. Cesk Fysiol. 2004;53:117–124. [PubMed] [Google Scholar]
- 26.Ouyang H, Mou L, Luk C, Liu N, Karaskova J, Squire J, Tsao MS. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157:1623–1631. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Wadei HA, Takahashi T, Schuller HM. Growth stimulation of human pulmonary adenocarcinoma cells and small airway epithelial cells by beta-carotene via activation of cAMP, PKA, CREB and ERK1/2. Int J Cancer. 2006;118:1370–1380. doi: 10.1002/ijc.21537. [DOI] [PubMed] [Google Scholar]
- 28.Das R, Esposito V, Abu-Abed M, Anand GS, Tayloy SS, Melacini G. cAMP activation of PKA defines an ancient signaling mechanism. Proc Natl Acad Sci U S A. 2007;104:93–98. doi: 10.1073/pnas.0609033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuller HM, Tithof PK, Williams M, Plummer H., 3rd The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999;59:4510–4515. [PubMed] [Google Scholar]
- 30.Schuller HM, Porter B, Riechert A. Beta-adrenergic modulation of NNK-induced lung carcinogenesis in hamsters. J Cancer Res Clin Oncol. 2000;126:624–630. doi: 10.1007/PL00008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joseph J, Niggemann B, Zaenker KS, Entschladen F. The neurotransmitter gamma-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res. 2002;62:6467–6469. [PubMed] [Google Scholar]
- 32.Miele E. The nicotinic stimulation of the cat adrenal medulla. Arch Int Pharmacodyn Ther. 1969;179:343–351. [PubMed] [Google Scholar]
- 33.Kang EH, Yu BH. Anxiety and beta-adreneregic receptor function in a normal population. Progr Neuropsychopharmacol Biol Psychiatry. 2005;29:733–777. doi: 10.1016/j.pnpbp.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 34.Carrasco GA, Van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Pharmacol. 2003;463:235–272. doi: 10.1016/s0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- 35.Nishiyama T, Yamashita K, Yokoyama T. Stress hormone changes in general anesthesia of long duration: isoflurane-nitrous oxide vs sevoflurane-nitrous oxide anesthesia. J Clin Anesth. 2005;17:586–591. doi: 10.1016/j.jclinane.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Furuya K, Shimizu R, Hirabayashi Y, Ishii R, Fukuda H. Stress hormone responses to major intra-abdominal surgery during and immediately after sevoflurane-nitrous oxide anaesthesia in elderly patients. Can J Anaesth. 1993;40:435–439. doi: 10.1007/BF03009513. [DOI] [PubMed] [Google Scholar]
- 37.Novak I. beta-Adrenergic regulation of ion transport in pancreatic ducts: patch-clamp study of isolated rat pancreatic ducts. Gastroenterology. 1998;115:714–721. doi: 10.1016/s0016-5085(98)70151-9. [DOI] [PubMed] [Google Scholar]
- 38.Joachim S, Schwoch G. Localization of cAMP-dependent protein kinase subunits along the secretory pathway in pancreatic and parotid acinar cells and accumulation of the catalytic subunit in parotid secretory granules following beta-adrenergic stimulation. Eur J Cell Biol. 1990;51:76–84. [PubMed] [Google Scholar]
- 39.Zijlstra FJ, Vincent JE, Mol WM, Hoogsteden HC, Van Hal PT, Jongejan RC. Eicosanoid levels in bronchoalveolar lavage fluid of young female smokers and nonsmokers. Eur J Clin Invest. 1992;22:301–306. doi: 10.1111/j.1365-2362.1992.tb01466.x. [DOI] [PubMed] [Google Scholar]
- 40.Shelp BJ, Bown AW, Faure D. Extracellular gamma-aminobutyrate mediates communication between plants and other organisms. Plant Physiol. 2006;142:1350–1352. doi: 10.1104/pp.106.088955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bown AW, Macgregor KB, Shelp BJ. Gamma-aminobutyrate: defense against invertebrate pests? Trends Plant Sci. 2006;11:424–427. doi: 10.1016/j.tplants.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Pereira GE, Gaudillere JP, Pieri P, Hilbert G, Maucourt M, Deborde C, Moing A, Rolin D. Microclimate influence on mineral and metabolic profiles of grape berries. J Agric Food Chem. 2006;54:6765–6775. doi: 10.1021/jf061013k. [DOI] [PubMed] [Google Scholar]
- 43.Shizuka F, Kido Y, Nakazawa T, Kitajima H, Aizawa C, Kayamura H, Ichijo N. Antihypertensive effect of gamma-amino butyric acid enriched soy products in spontaneously hypertensive rats. Biofactors. 2004;22:165–167. doi: 10.1002/biof.5520220133. [DOI] [PubMed] [Google Scholar]
- 44.Kim H, Hall P, Smith M, Kirk M, Prasain JK, Barnes S, Grubbs C. Chemoprevention by grape seed extract and genistein in carcinogen-induced mammary cancer in rats is diet dependent. J Nutr. 2004;134:3445S–3452S. doi: 10.1093/jn/134.12.3445S. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;14:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]