

Abstract
Particle-based Brownian dynamics simulations offer the opportunity to not only simulate diffusion of particles but also the reactions between them. They therefore provide an opportunity to integrate varied biological data into spatially explicit models of biological processes, such as signal transduction or mitosis. However, particle based reaction-diffusion methods often are hampered by the relatively small time step needed for accurate description of the reaction-diffusion framework. Such small time steps often prevent simulation times that are relevant for biological processes. It is therefore of great importance to develop reaction-diffusion methods that tolerate larger time steps while maintaining relatively high accuracy. Here, we provide an algorithm, which detects potential particle collisions prior to a BD-based particle displacement and at the same time rigorously obeys the detailed balance rule of equilibrium reactions. We can show that for reaction-diffusion processes of particles mimicking proteins, the method can increase the typical BD time step by an order of magnitude while maintaining similar accuracy in the reaction diffusion modelling.
Key words: Brownian dynamics, protein-protein interactions, reaction-diffusion
1. Introduction
Biological processes, such as the selective nucleo-cytoplasmic protein transport or gene expression regulation typically involve the intricate relationship of hundreds of cellular components. Due to advances in experimental technology (including fluorescence imaging, quantitative mass spectroscopy and cryo electron tomography), quantitative information is increasingly available about the kinetics in reaction networks of cellular components and also about the molecular organization of living cells. There is a pressing need to integrate these varied data into spatially explicit, predictive models of biological processes such as signal transduction, genome separation, and mitosis.
Mathematical and computational modeling has been critical for predicting the systems level behavior of reaction networks. Typically, mathematical modeling treats the entire system or parts of it as well mixed solutions with a spatially homogeneous environment, which can be modeled by ordinary or stochastic differential equations. However, the cellular environment is highly inhomogeneous (Beck et al., 2011) due to spatial gradients in the distribution of biomolecules, crowding effects (Ando and Skolnick, 2010; Kim and Yethiraj, 2010, 2011; Minton, 2001, 2006), and cellular compartmentalization (Delon et al., 2006). Many cellular processes, such as cell division and nucleo-cytoplasmic transport, are either spatially constrained or segregated (Terry et al., 2007). Moreover, when biomolecules are present in relatively low copy numbers, their local concentrations can fluctuate widely, which can cause stochastic effects in reaction processes (Dobrzynski et al., 2007; Turner et al., 2004). The effective behavior of such molecules may be very different from their behavior under a constant distribution, which can significantly influence gene regulation (Cai et al., 2006; Raser and O'Shea, 2005), signal transduction (Kollmann et al., 2005), and many other processes.
Particle based simulations naturally incorporate the concepts of space, crowding, and stochasticity (Dobrzynski et al., 2007; Dugosz and Trylska, 2011). Those methods treat proteins or other reactants explicitly, and the time-evolution of particle positions is sampled at discrete time intervals by Brownian dynamics (BD) simulations (Ermak and McCammon, 1978; Northrup and Erickson, 1992). In BD, the net force experienced by a particle contains a random element in addition to contributions from interactions with other particles. The random element is an explicit approximation to the statistical properties of Brownian forces, due to the effects of collisions with solvent molecules, which are not explicitly modeled. More specifically, the particles are displaced from their position at each time interval by a random vector whose norm is chosen from a probability distribution function that is a solution to the Einstein diffusion equation.
To incorporate reactions within a BD framework, reactions occur upon collisions of particles according to specific probabilities, which are chosen to reproduce the correct reaction kinetics (Morelli and ten Wolde, 2008). A number of reaction-diffusion algorithms incorporating Brownian dynamics have been developed with various levels of detail. Some include atomic level details (Gabdoulline and Wade, 1997, 2002), while others have various degrees of coarseness (Andrews, 2009; Andrews and Bray, 2004; Barenbrug et al., 2002; Boulianne et al., 2008; Byrne et al., 2010; Ermak and McCammon, 1978; Morelli and ten Wolde, 2008; Northrup and Erickson, 1992; Ridgway et al., 2008; Strating, 1999; Sun and Weinstein, 2007; van Zon and ten Wolde, 2005). For instance, Morelli and ten Wolde (2008) introduced a coarse-grained Brownian dynamics algorithm for simulating reaction-diffusion systems that rigorously obeys detailed balance for equilibrium reactions to omit systematic errors in the simulation.
The disadvantage of particle methods is that often they require relatively small time steps in order to accurately simulate the dynamics of diffusion and reaction kinetics. The reason lies partly in the approximations used to derive reaction event probabilities and the incomplete detection of particle collisions, which prohibit the use of larger time steps. The use of small time steps often prevents reaching simulation times that are relevant for biological processes. Biological processes occur on a wide range of time scales. Some proteins may encounter each other in fractions of a millisecond, while others take hours. For instance, DNA transcription and translation are completed in a few minutes, but these processes are made up of thousands of small diffusion-limited reactions. It is an ongoing challenge to develop particle-based simulations that can cover a wide range of time scales while accurately reproducing the properties of diffusion and reaction networks.
Several methods have been developed to increase simulation time steps. Event-driven simulations, such as the Green's function reaction dynamics (GFRD) scheme (Takahashi et al., 2005; van Zon and ten Wolde, 2005), use Green's function exact expression for one and two particle reaction-diffusion systems to take large steps in time when the particles are far apart from each other (Takahashi et al., 2005; van Zon and ten Wolde, 2005). When particle concentrations are low, GFRD is significantly more efficient than traditional Brownian Dynamics (Takahashi et al., 2005). However, for reactions near surfaces or in crowded cellular environments, the benefits of GRFD vanish as these methods also require relatively short time steps to omit unphysical particle overlaps to accurately simulate particle diffusion (van Zon and ten Wolde, 2005).
Here, we describe a Brownian Dynamic algorithm that tolerates increased time steps in comparison to traditional Brownian dynamics algorithms while maintaining relatively high accuracy in the reaction-diffusing modeling. Our method builds upon a more accurate description of particle collisions in comparison to the traditional Brownian dynamics scheme. One of the causes of simulation errors when using increasingly larger time steps lies in the treatment of collisions during a BD time step. Typically particle collisions are considered when particle overlaps are detected after the displacement of the particles in a time step. Such a procedure underestimates the number of collisions per time steps, as not all potential collisions of the particles during a time step are considered. With increasing length of the time step this underestimation becomes more pronounced, which leads to inflated reaction probabilities when parameterizing the microscopic reaction rates. Previously, Barenbrug et al. (2002) proposed a method that corrected the traditional BD step by augmenting the collision detection with a correction step for non-overlapping particles based on an analytical term for missed collisions. However, the method did not satisfy the detailed balance rule for reversible reactions. In contrast, we provide a method, which not only detects potential collisions prior to a BD move but also determines reaction probabilities consistent with the detailed balance rule for reversible reactions. We can show that our Reaction Before Move (RBM) method allows an increase of BD time steps by an order of magnitude while maintaining relatively high accuracy with respect to analytical solutions of a reaction system.
2. Methods
To study processes at biologically relevant time scales (in the microsecond to second time range) it is required to simulate reaction-diffusion systems with relatively large time steps. Here, we describe a Brownian Dynamic algorithm that allows increased time steps in comparison to traditional Brownian dynamics algorithms while maintaining minimal impact on accuracy. We begin with an introduction of BD.
2.1. Brownian dynamics
The motion of a particle undergoing diffusion can be described by Einstein's diffusion equation (Kim and Shin, 1999),
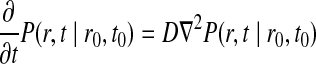 |
(1) |
where P(r, t|r0, t0) is the probability that the particle will be at position r at time t given the particle was initially at position r0 at time t0. The rate of diffusion is given by the parameter D. This problem can be solved with the addition of an initial condition (the particle starts at r0) and a boundary condition.
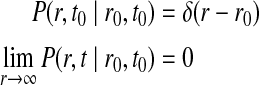 |
The solution that describes the position of the particle P(r, t) at a given time is known as Green's function, and for a single isolated particle is the Gaussian distribution associated with a continuous random walker (Agmon and Szabo, 1990; Rice, 1985).
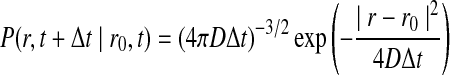 |
(2) |
When the diffusive motions of multiple particles are simulated, in a traditional Brownian dynamics algorithm the distribution of particle displacements P(r, t) is sampled for each particle every time step. To prevent unphysical overlaps between particles, typically individual moves are rejected that would result in overlaps of the excluded volume of the particles. Because collisions are only determined at the end of each time step, the length of the time step is naturally limited. The size must be small enough to prevent particles passing through each other without detecting a collision in a time step propagation.
2.2. Reaction-diffusion modeling
Brownian dynamics simulations cannot only be used to simulate particle diffusion but also the reactions between them (Morelli and ten Wolde, 2008). Within the Brownian dynamics framework, reactions can be simulated as follows. A second order reaction, where two species A and B form a product C (e.g. the formation of a protein complex), can be formulated as:
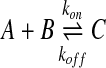 |
(3) |
where the kon and koff are the macroscopic reaction rates, which are related to the equilibrium(Keq) and the concentrations of the species by
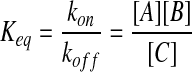 |
(4) |
where [A] is the concentration of the species A. This reaction can be decomposed into two events (Agmon and Szabo, 1990; Morelli and ten Wolde, 2008).
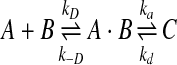 |
(5) |
The first event is the formation of the encounter complex (A·B), when the particles come into contact. In a typical Brownian dynamics framework, the formation of an encounter complex is naturally simulated when two particles are close to each other and a trial displacement of one of the two leads to an overlap. The formation of an encounter complex occurs at the diffusion limited rate kD. For spherical particles, this rate can be solved analytically (Agmon and Szabo, 1990; Rice, 1985) and is the diffusion limited Smoluchowski rate kD = 4πRD, where D is the sum of the diffusion rates, and R is the sum of their radii (Rice, 1985).
In the second step, the encounter complex (A·B) can either advance to form the reaction product C or the encounter complex can remain dissociated as the individual components A and B. The formation of the product from the encounter complex occurs by an intrinsic microscopic reaction rate ka. The correct calibration of this reaction probability accounts for the many forces and rotational motions involved in progressing from the encounter complex to the reaction products. This reaction rate reflects the time spent by the two reaction partners achieving an orientation that allows the reaction to progress. When dealing with proteins, the actual value depends on steering effects, due to electrostatic interactions from charged residues or dipole moment orientations, but also reflects the size and other physical properties of the binding sites (Gabdoulline and Wade, 1997, 2002). ka must be appropriately calibrated based on experiments so that physically meaningful reaction rates can be simulated.
The reverse first order reaction (e.g., the dissociation of a protein complex) can also be formalized in a two-step process. First, an interaction dissociates at an intrinsic microscopic dissociation rate of kd, resulting in the re-formation of the encounter complex of the two unbound proteins still positioned at contact distance R. In a second step, the individual proteins in the newly formed encounter complex can diffuse into the bulk solution.
The key point in accurate reaction-diffusion modeling is to correctly relate the microscopic rate constants to the probabilities that a particle collision leads to a reaction or a reaction product leads to particle dissociation during a time step. Following Morelli and ten Wolde (2008), the ratio of the microscopic reaction rates for forward and reverse reactions must be equal to the products of the corresponding reaction probabilities.
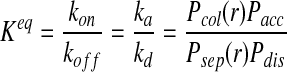 |
(6) |
where the individual event probabilities are defined as follows: Pcol(r) is the collision probability. It is the probability that particles initially separated by r will form an encounter complex during the time step of length Δt. Pacc is the acceptance probability. It is the probability that the reaction will occur to form C given that the reactants are in an encounter complex. Pdis is the probability that the reaction product C dissociates to form an encounter complex of particles A·B. The lifetime of a stable complex is modeled with a Poisson distribution of waiting times and is defined by a dissociation probability such that Pdis = 1 − exp(−kdΔt) ≈ kdΔt when kdΔt ≪ 1 (kd being the intrinsic dissociation rate) (Morelli and ten Wolde, 2008). Psep(r) is the distribution describing the separation distance between the particles A and B after the dissociation of the reaction product C. To omit systematic errors, the detailed balance in the reaction system must be satisfied, which states that a reverse move must be generated according to a probability distribution Psep(r) that is the same as that by which the forward move is generated (Pcol(r)), but properly normalized to a total probability of 1 (Morelli and ten Wolde, 2008). The normalization factor is the integral of Pcol(r) over all possible locations r.
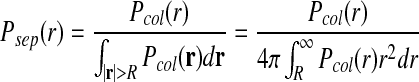 |
(7) |
Then the probability of accepting a reaction Pacc upon particle collision can be written as
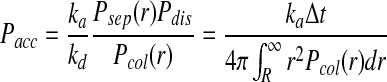 |
(8) |
where ka is the microscopic association rate, r is the distance between two particles A and B, and R is the sum of the radii of the two particles.
The key step for accurate reaction modeling is to analytically determine the Pcol(r). The analytical solution depends on the Brownian dynamics framework used. Morelli and ten Wolde (2008) derived the solution for the classic Brownian dynamics scheme. At each time step, a trial move for a particle is chosen based on the Einstein diffusion equation and a potential particle collision detected if the trial move leads to an overlap with another particle. In this scheme, the collision probability Pcol(r) is the analytically determined probability that two particles initially separated by a distance r will be placed after the time step Δt at positions that lead to a particle overlap.
However, a substantial number of actual collisions will not be detected by this criterion, as two particles can also experience collisions during the time step, even if their final positions do not overlap. The method therefore underestimates the number of collisions. This underestimation increases with the time step, and results in a simulation error proportional to the square root of the time step.
2.3. Increasing the time step by applying the RBM method
Here, we introduce an alternative Brownian dynamics scheme and determine all event probabilities in such a way that detailed balance in the reaction process is considered. Instead of checking for overlaps after a trial move of one of the two particles (i.e., at the end of the time step), we determine the probability of two particles colliding analytically prior to the particle displacement. Given the positions of the two proteins at the beginning of a time step, we derive an analytical solution for the probability that they collided during the time step without explicitly resolving their path. This probability distribution can be derived in analogy to the analytical solution of particle diffusion around an absorbing sphere (Barenbrug et al., 2002).
The probability of two particles coming into contact during a time step is given by the solution of the Green's function for the two-body problem (Kim and Shin, 1999). For two particles A and B diffusing with a combined diffusion rate of D = DA + DB, and separated by distance r, there exists a closed form expression for the probability of the particles coming into contact during a time step of length Δt (Agmon and Szabo, 1990; Rice, 1985). The probability of collision is
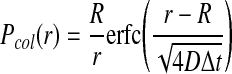 |
(9) |
where R is the sum of the two particle's radii and D the sum of the diffusion coefficients for both particles. The function erfc is the complementary error function which can be defined in terms of the error function erf:
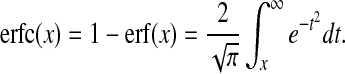 |
(10) |
With an expression for Pcol(r), we can now calculate the normalizing factor found in the denominator of Pacc, which we call N, in order to derive the remaining reaction probabilities:
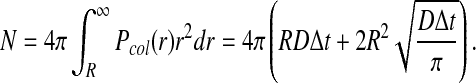 |
(11) |
The acceptance probability Pacc(r) is now determined:
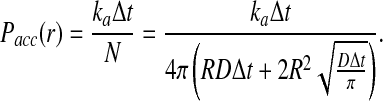 |
(12) |
Correspondingly the new distribution with which dissociated particles must be placed while satisfying the detailed balance condition is calculated as
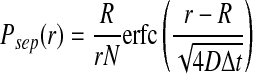 |
(13) |
The main outcome of this section is the formulation of new event probabilities (Pcol(r), Psep(r), and Pacc) that are consistent with the detailed balance rule but incorporate a more accurate description of the collision frequency in comparison to the traditional BD scheme. As we will demonstrate in the results section, this formulation will allow an increase in time step length by an order of magnitude while maintaining high accuracy in the reaction-diffusion kinetics.
2.4. The RBM algorithm
With the derivation of all event probabilities, we can now formulate the modified algorithm for reaction-diffusion simulations. Our algorithm can be divided into two main parts: First, the detection of particle collisions and reactions prior to the BD particle movement; and second, the actual particle displacement according to the diffusion equation post reaction detection (Fig. 1).
FIG. 1.

Flowchart of the RBM method.
2.4.1. Detection of potential reactions for a given configuration
First, all potential reactions between particles located within a certain cutoff distance are detected. For these pairs of particles the collision probability Pcol(r) and acceptance probability Pacc of the potential reaction is determined.
2.4.2. Acceptance of potential reactions for a given configuration
Next, reactions are accepted or rejected according to the reaction probabilities. For second-order reactions (A + B → C), the reaction probability is the product of the collision and acceptance probability (Pcol(r)Pacc) for the corresponding pairs of particles. For dissociation reactions and other first order reactions, the reaction is accepted with probability Pdis.
2.4.3. Resolving conflicting reactions
A particle can only be involved in at most one reaction per time step. If a particle participates in multiple accepted reactions, only one of these reactions is randomly chosen, while all other reactions are rejected.
2.4.4. Placement of reaction products
Finally, new positions of the reaction products are determined. For second-order reactions, (A + B → C), both reactant particles are removed and the product particle is placed at the location of one of the randomly chosen reactant particles. If the placement is rejected because of hard-sphere overlap, the reaction is not carried out, and both reactant particles are restored to their positions at the beginning of the time step. For dissociation reactions (A → B + C), the reactant particle is removed, and one of the product particles is placed at the reactant particles position. Then the second particle is placed at a distance from the first product particle according to the probability distribution Psep(r) so that the detailed balance relationship is fulfilled. If overlap with other particles are encountered this placement is repeated several times. If after several attempts no location for the second particle is found, the reaction is rejected, and the reactant is restored, while the product particles are removed.
2.4.5. Displacement of all non-reacting particles according to the BD scheme
After all reacting particles have been moved all other particles that did not participate in a reaction or whose reaction was rejected are displaced according to the Einstein's diffusion equation. Each particle move is considered in a random order. Several trial moves are performed if a move resulted in particle overlaps. If after several attempts, a new position has not been found, the particle is left at its current position.
3. Results
After establishing our approach, we now assess its accuracy and compare its performance with the traditional reaction-diffusion BD scheme, particularly in view of larger simulation time steps. Examples are shown for particle radii and diffusion constants typically observed for proteins.
3.1. Collision probability
We first focus on the differences in the collision probabilities of two particles observed in the traditional BD and the just discussed RBM approach. When particle collisions are detected during the time step, the collision probability is significantly increased in comparison to the traditional BD scheme. For instance, when two equally sized particles are initially placed close to their contact distance (i.e., the sum of their radii), the collision probability in our approach is more than twice as large as in traditional BD (Fig. 2). This observation is true also when the initial particle distance is larger then the collision distance. The increased collision probability has important consequences for calculating the acceptance probability Pacc and the expected error in simulations with larger time steps. First, an increased collision probability leads to an increase in the normalization factor N, which in turn influences the acceptance probability Pacc. With increasing time step Δt, the behavior of N differs dramatically between the traditional BD and the RBM approach. Whereas in the traditional method the function NBD(Δt) reaches a plateau at already relatively small time steps, in our approach NRBM(Δt) increases almost linearly with increasing time steps, leading to a significantly larger slope for the function (Fig. 3A). For example, for particle sizes and diffusion constants typically observed in proteins, NRBM is about 10 times larger than NBD when a time step of 1 ns is used (Fig. 3A). As a consequence the resulting acceptance probability 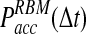 as a function of the time step Δt is significantly smaller in our approach (Fig. 3B). Indeed, the function Pacc(Δt) differs dramatically between the traditional BD approach and our RBM method (Fig. 3B). Most importantly, the values of
as a function of the time step Δt is significantly smaller in our approach (Fig. 3B). Indeed, the function Pacc(Δt) differs dramatically between the traditional BD approach and our RBM method (Fig. 3B). Most importantly, the values of 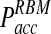 remain at relatively low values over a wide range of time steps, while
remain at relatively low values over a wide range of time steps, while 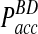 in the traditional BD scheme increases dramatically with increasing time step (Fig. 3B). For instance, for relatively fast reaction rates,
in the traditional BD scheme increases dramatically with increasing time step (Fig. 3B). For instance, for relatively fast reaction rates, 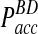 grows at significantly faster rates and reaches levels above
grows at significantly faster rates and reaches levels above 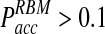 already at time steps of Δt ≈ 0.3. In contrast
already at time steps of Δt ≈ 0.3. In contrast 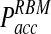 reaches a plateau and remains below
reaches a plateau and remains below 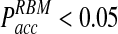 even at time steps as large as Δt = 2 ns (Fig. 3B).
even at time steps as large as Δt = 2 ns (Fig. 3B).
FIG. 2.

Comparison of the collision probabilities 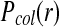 as a function of initial particle distance between our RBM and the traditional BD scheme.
as a function of initial particle distance between our RBM and the traditional BD scheme. 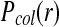 is the probability of observing a collision during a time step between two particles initially located at a distance r. (Green line)
is the probability of observing a collision during a time step between two particles initially located at a distance r. (Green line) 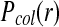 in the RBM scheme, where particle collisions are considered that occur during the time step and (blue line) the traditional BD scheme, where particle collisions are detected only at the end of each time step. The specific example is shown for two particles with typical parameters of proteins, each with a radius of 0.5 nm and diffusion rate D = 0.5 nm2/ns, and a time step of 0.1ns.
in the RBM scheme, where particle collisions are considered that occur during the time step and (blue line) the traditional BD scheme, where particle collisions are detected only at the end of each time step. The specific example is shown for two particles with typical parameters of proteins, each with a radius of 0.5 nm and diffusion rate D = 0.5 nm2/ns, and a time step of 0.1ns.
FIG. 3.

(A) The normalization factor 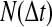 as a function of the simulation time step
as a function of the simulation time step 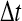 . (Green line)N as observed in the RBM method and (blue line) N as observed in the traditional BD scheme. (B) The probability of accepting a reaction upon the collision between two particles
. (Green line)N as observed in the RBM method and (blue line) N as observed in the traditional BD scheme. (B) The probability of accepting a reaction upon the collision between two particles 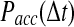 as a function of the time step
as a function of the time step 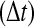 used in the simulation. The function
used in the simulation. The function 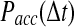 differs dramatically between the traditional BD approach and our RBM method. Most importantly, the values of
differs dramatically between the traditional BD approach and our RBM method. Most importantly, the values of 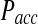 remain at relatively low values over a wide range of time steps, while
remain at relatively low values over a wide range of time steps, while 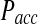 in the traditional BD scheme increases with the length of the time step. Simulation parameters are the same as Figure 2.
in the traditional BD scheme increases with the length of the time step. Simulation parameters are the same as Figure 2.
Importantly, due to approximations used in the derivation of the reaction-diffusion formulas, it is generally considered that the time step Δt must be selected so that Pacc < 0.1 (Morelli and ten Wolde, 2008). Because 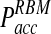 is sufficiently lower even at larger time steps in comparison to the traditional method, larger time steps can be tolerated while maintaining high accuracy in the reaction kinetic modeling (Fig. 3B). For instance, for reacting particles with radii of 5 nm and D = 1 nm2/ns, using Pacc < 0.1 as a limit, traditional BD schemes would require a time step Δt < 0.03 ns, while time steps can be as large as Δt = 2 ns in our approach with sufficiently small Pacc.
is sufficiently lower even at larger time steps in comparison to the traditional method, larger time steps can be tolerated while maintaining high accuracy in the reaction kinetic modeling (Fig. 3B). For instance, for reacting particles with radii of 5 nm and D = 1 nm2/ns, using Pacc < 0.1 as a limit, traditional BD schemes would require a time step Δt < 0.03 ns, while time steps can be as large as Δt = 2 ns in our approach with sufficiently small Pacc.
3.2. Assessment of the RBM
Next, we assess the accuracy of our method by comparing our simulations with known analytical solutions. In the following section, we first focus on the radial distribution functions of two reacting particles. The integral of the radial distribution function will provide the survival probability of the reaction partners for a given elapsed simulation time (i.e., the fraction of particles not reacted). Analytical solutions for both, the radial distribution function and survival probability can be determined for the case of two reacting particles (Kim and Shin, 1999).
Radial distribution function. The radial distribution function describes the probability of distances between two reacting particles after an elapsed simulation time. The two particles are initially separated by their contact distance R and diffuse for an elapsed time. If particles react during this time period, the simulation is terminated. If after the simulation time the particles have not reacted yet, their final separation is measured. The distribution of the final separation distances is measured from 30,000 independent simulations. Comparison of the radial distribution function with the analytical solution (Kim and Shin, 1999) therefore allows assessment of both, the reaction and diffusion of the particles.
We have calculated the radial distribution functions based on simulations with 6 different time steps, which differ over 6 orders of magnitude (Fig. 4A). We demonstrate that the determined radial distribution functions and the survival probabilities show excellent agreement with theory, even for the simulations with relatively very large time steps (see time step Δt = 10) (Fig. 4A). The reaction method we have described is exact for the two particle case, so all of the curves are very close to the theoretical curve. The sources of error are mainly due to the placement of particles and can be rationalized as follows. The RBM method places particles randomly, with equal probability also near other potentially reacting particles. This situation deviates from the theoretical distribution, which will have potentially reacting particles being located with a lower probability near other potentially reacting particles, because some fraction of these particles will have undergone the reaction. Although the reaction calculations are exact, this error in particle placement is more pronounced when using relatively small time steps, because more time steps are needed to reach the simulation time. With an increase in the number of time steps, the simulation error accumulates to larger values. For larger time steps, the error is lower, since a smaller number of time steps is needed.
FIG. 4.

(A) The radial distribution functions for a two particle system generated from simulations from analytical solution. Simulations are performed for 6 different time steps with length spaning 6 orders of magnitude. Particle radii and diffusion constant are chosen as they are observed for typical proteins. Particles have a radius 0.5nm, and diffusion rate of 0.5 nm2/ns, and react with the intrinsic rate, 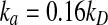 , where kD is the diffusion limited rate, and corresponds to reaction upon any contact. The reaction rate is chosen close to the diffusion-limited rate in order to accent any deviation from theory. At the beginning of each simulation the particles are placed at contact distance, with centers separated by 1 nm. The elapsed total simulation time is 20ns. (B) Survival probabilities with respect for the elapsed simulation time for simulations with 6 different time steps. (C) Relative error rate in the survival probabilities with respect to the analytical solution shown for the 6 different simulations and calculated at the elapsed simulation time of 20 ns.
, where kD is the diffusion limited rate, and corresponds to reaction upon any contact. The reaction rate is chosen close to the diffusion-limited rate in order to accent any deviation from theory. At the beginning of each simulation the particles are placed at contact distance, with centers separated by 1 nm. The elapsed total simulation time is 20ns. (B) Survival probabilities with respect for the elapsed simulation time for simulations with 6 different time steps. (C) Relative error rate in the survival probabilities with respect to the analytical solution shown for the 6 different simulations and calculated at the elapsed simulation time of 20 ns.
Survival probabilities. The integral of the radial distribution function is the survival probability of the reacting particles. We have plotted the survival probabilities for each time step length and compared it with the analytical solution (Fig. 4B). The errors in the survival probability are relatively small and range between <0.1% and <3% (Fig. 4B,C). Interestingly, in the RBM method the errors with respect to the analytical solution decreases significantly with increasing time steps Δt (Fig. 4C). This behavior is opposite to the one observed in the BD approach and demonstrate the good performance of the RBM method for larger time steps.
3.3. Annihilation experiment
Next, we perform a simulation of a diffusion-controlled second order reaction. Consider a reaction system A + A → ∅ where particles that come into contact instantly react and annihilate each other. Assuming the particles are distributed at a steady state, there is a closed form solution for the number of particles that have survived (Barenbrug et al., 2002). We chose 105 particles with radius 1 nm, which initially are randomly placed into a box with periodic boundary conditions and diffuse at a rate of D = 1 nm2/ns (Fig. 5). Any collision will lead to a reaction, resulting that both particles are removed from the simulation. At each time step in the simulation, the number of particles that have survived is calculated. We now compare the analytical solution of this annihilation reaction with the simulations from the traditional BD and RBM methods. More specifically, we perform the simulations at three different time steps of variable length.
FIG. 5.

Annihilation reaction of 10,000 particles that react upon collision. Shown are the results of simulations for three different time steps for the traditional BD method and our RBM method. The analytical solution for the annihilation reaction is also shown. The tests are performed with the following parameters: 105 particles with radius 1 nm, are placed into a cubic box and periodic boundary conditions are imposed. The particles diffuse at a rate of 1.0 nm2/ns.
Interestingly, for the RBM method the total error between simulation and theory remains almost constant with increasing time steps (Fig. 5). In contrast, the traditional BD scheme performs better at very small time steps, however with increasing time steps the error in the traditional BD scheme increases dramatically leading to errors that are an order of magnitude larger in comparison to the RBM method even at relatively modest time steps (Fig. 5).
3.4. Diffusion in crowded environments using large time steps
Finally, we investigate the errors in diffusion rates when relatively large time steps are used. The most challenging scenario is the diffusion of particles under crowded conditions (Dugosz and Trylska, 2011; Kim and Yethiraj, 2010, 2011; Minton, 2001, 2006). More specifically, we have placed crowding particles inside a box and measured the time for a central particle to escape a given volume defined by a sphere (Fig. 6). Simulations are performed for 4 different time steps, of varying length and also at different crowding levels, ranging from 0% to 20% of total amount of volume occupied by crowding particles (Fig. 6A,B). For each time step 5000 independent simulations are performed each time with a random configuration of the crowding particles. From the 5000 simulations, we measure the average time required for a central particle to escape the crowded environment from its starting location in the center of the crowding box. Results are then compared for different time steps and crowding levels (Fig. 6B).
FIG. 6.

The effect of time step on the diffusion rates of particles in crowded environments. Shown are the average elapsed times needed for a particle of radius 1 nm and a diffusion rate of 0.25 nm2/ns to reach a distance of 10 nm away from its initial starting location. The escape time is measured as the average over 5000 simulations, each time containing random configurations of crowding particles at the given crowding level. Simulations are performed at 6 different crowding levels, which is defined as the total volume fraction occupied by the crowding particles.
As expected, the escape times increase with the crowding level (Banks and Fradin, 2005; Kozer and Schreiber, 2004; Sun and Weinstein, 2007). Generally, with increasing time steps, the escape time increases. We observe that, for crowding levels between 0% and 15% volume occupancy, increasing the time step by two orders of magnitude does have only a modest effect on the escape times and hence the motion of the particles. However, at the highest crowding level of 20% volume occupancy, a more significant increase of escape times is observed, indicating that errors in particle diffusion must be considered.
4. Conclusion
We have introduced a particle-based reaction-diffusion method for the use of larger time steps in comparison to traditional BD while maintaining similar accuracy for reaction-diffusion events. Our method builds upon a more accurate description of particle collisions in comparison to traditional BD. Moreover, the method obeys the detailed balance rule for equilibrium reactions.
In our method, particle collisions are detected analytically prior to the trial displacements of particles in BD, which allows the detection of collisions during a time step, which are otherwise missed in the traditional BD scheme. In particular for longer time steps, this approach allows a more accurate detection of particle collisions. This procedure therefore increases the collision probability between particles in comparison to the traditional BD scheme, which in turn naturally reduces the acceptance probabilities for reaction events. Because for accurate simulations the time steps must be chosen so that the acceptance probabilities remain below a cutoff value, our approach naturally increases the range of allowable time steps for accurate reaction-diffusion modeling. The testing of our approach confirms its applicability. Our approach therefore provides a step towards the goal of increasing time scales in reaction-diffusion modeling of biological processes.
Acknowledgments
We would like to acknowledge Dr. Harianto Tjong and Dr. M.S. Madhusudhan for useful discussions and Dr. Harianto Tjong for help with numerical integration. This work is supported by the Human Frontier Science Program (grant RGY0079/2009-C to F.A.), Alfred P. Sloan Research Foundation (grant to F.A.), NIH (grants 1R01GM096089 and 2U54RR022220 to F.A.), and NSF CAREER grant 1150287 (to F.A.). F.A. is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts.
Disclosure Statement
No competing financial interests exist.
References
- Agmon N. Szabo A. Theory of reversible diffusion-influenced reactions. J. Chem. Phys. 1990;92:5270. [Google Scholar]
- Ando T. Skolnick J. Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc. Natl. Acad. Sci. USA. 2010;107:18457–18462. doi: 10.1073/pnas.1011354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S.S. Accurate particle-based simulation of adsorption, desorption and partial transmission. Phys. Biol. 2009;6:046015. doi: 10.1088/1478-3975/6/4/046015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S.S. Bray D. Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys. Biol. 2004;1:137. doi: 10.1088/1478-3967/1/3/001. [DOI] [PubMed] [Google Scholar]
- Banks D.S. Fradin C. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 2005;89:2960–2971. doi: 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenbrug T.M.A.O.M. Peters E.A.J.F.F. Schieber J.D. Accurate method for the brownian dynamics simulation of spherical particles with hard-body interactions. J. Chem. Phys. 2002;117:9202–9214. [Google Scholar]
- Beck M. Topf M. Frazier Z., et al. Exploring the spatial and temporal organization of a cells proteome. J. Struct. Biol. 2011;173:483–496. doi: 10.1016/j.jsb.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulianne L. Al Assaad S. Dumontier M., et al. Gridcell: a stochastic particle-based biological system simulator. BMC Syst. Biol. 2008;2:66. doi: 10.1186/1752-0509-2-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M.J. Waxham M.N. Kubota Y. Cellular dynamic simulator: an event driven molecular simulation environment for cellular physiology. Neuroinformatics. 2010;8:63–82. doi: 10.1007/s12021-010-9066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L. Friedman N. Xie X.S. Stochastic protein expression in individual cells at the single molecule level. Nature. 2006;440:358–362. doi: 10.1038/nature04599. [DOI] [PubMed] [Google Scholar]
- Delon A. Usson Y. Derouard J., et al. Continuous photobleaching in vesicles and living cells: a measure of diffusion and compartmentation. Biophys. J. 2006;90:2548–2562. doi: 10.1529/biophysj.105.069815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynski M. Rodríguez J.V. Kaandorp J.A., et al. Computational methods for diffusion-influenced biochemical reactions. Bioinformatics. 2007;23:1969–1977. doi: 10.1093/bioinformatics/btm278. [DOI] [PubMed] [Google Scholar]
- Dugosz M. Trylska J. Diffusion in crowded biological environments: applications of Brownian dynamics. BMC Biophys. 2011;4:3. doi: 10.1186/2046-1682-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermak D. L. McCammon J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978;69:1352–1360. [Google Scholar]
- Gabdoulline R.R. Wade R.C. Simulation of the diffusional association of Barnase and Barstar. Biophys. J. 1997;72:1917–1929. doi: 10.1016/S0006-3495(97)78838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabdoulline R.R. Wade R.C. Biomolecular diffusional association. Curr. Opin. Struct. Biol. 2002;12:204–213. doi: 10.1016/s0959-440x(02)00311-1. [DOI] [PubMed] [Google Scholar]
- Kim H. Shin K.J. Exact solution of the reversible diffusion-influenced reaction for an isolated pair in three dimensions. Phys. Rev. Lett. 1999;82:1578–1581. [Google Scholar]
- Kim J.S. Yethiraj A. Crowding effects on association reactions at membranes. Biophys. J. 2010;98:951–958. doi: 10.1016/j.bpj.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.S. Yethiraj A. Crowding effects on protein association: effect of interactions between crowding agents. J. Phys. Chem. B. 2011;115:347–353. doi: 10.1021/jp107123y. [DOI] [PubMed] [Google Scholar]
- Kollmann M. Lovdok L. Bartholome K., et al. Design principles of a bacterial signalling network. Nature. 2005;438:504–507. doi: 10.1038/nature04228. [DOI] [PubMed] [Google Scholar]
- Kozer N. Schreiber G. Effect of crowding on protein-protein association rates: fundamental differences between low and high mass crowding agents. J. Mol. Biol. 2004;336:763–74. doi: 10.1016/j.jmb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Minton A.P. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- Minton A.P. How can biochemical reactions within cells differ from those in test tubes? J. Cell Sci. 2006;119:2863–2869. doi: 10.1242/jcs.03063. [DOI] [PubMed] [Google Scholar]
- Morelli M.J. ten Wolde P.R. Reaction brownian dynamics and the effect of spatial fluctuations on the gain of a push-pull network. J. Chem. Phys. 2008;129:054112. doi: 10.1063/1.2958287. [DOI] [PubMed] [Google Scholar]
- Northrup S. Erickson H. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. USA. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser J.M. O'Shea E.K. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice S.A. Vol. 25. Elsevier; New York: 1985. Diffusion-Limited Reactions. [Google Scholar]
- Ridgway D. Broderick G. Lopez-Campistrous A., et al. Coarse-grained molecular simulation of diffusion and reaction kinetics in a crowded virtual cytoplasm. Biophys. J. biophysj. 2008;107:116053. doi: 10.1529/biophysj.107.116053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating P. Brownian dynamics simulation of a hard-sphere suspension. Phys. Rev. E. 1999;59:2175–2187. [Google Scholar]
- Sun J. Weinstein H. Toward realistic modeling of dynamic processes in cell signaling: quantification of macromolecular crowding effects. J. Chem. Phys. 2007;127:155105. doi: 10.1063/1.2789434. [DOI] [PubMed] [Google Scholar]
- Takahashi K. Arjunan S.N.V. Tomita M. Space in systems biology of signaling pathways—towards intracellular molecular crowding in silico. FEBS Lett. 2005;579:1783–1788. doi: 10.1016/j.febslet.2005.01.072. [DOI] [PubMed] [Google Scholar]
- Terry L.J. Shows E.B. Wente S.R. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318:1412–1416. doi: 10.1126/science.1142204. [DOI] [PubMed] [Google Scholar]
- Turner T.E. Schnell S. Burrage K. Stochastic approaches for modelling in vivo reactions. Comput. Biol. Chem. 2004;28:165–178. doi: 10.1016/j.compbiolchem.2004.05.001. [DOI] [PubMed] [Google Scholar]
- van Zon J.S. ten Wolde P.R. Green's-function reaction dynamics: a particle-based approach for simulating biochemical networks in time and space. J. Chem. Phys. 2005;123:234910. doi: 10.1063/1.2137716. [DOI] [PubMed] [Google Scholar]