

Abstract
Identification of splicing regulatory elements (SREs) deserves special attention because these cis-acting short sequences are vital parts of splicing code. The fact that a variety of other biological signals cooperatively govern the splicing pattern indicates the necessity of developing novel tools to incorporate information from multiple sources to improve splicing factor binding sites prediction. Under this context, we proposed a Varying Effect Regression for Splicing Elements (VERSE) to discover intronic SREs in the proximity of exon junctions by integrating other biological features. As a result, 1562 intronic SREs were identified in 16 human tissues, many of which overlapped with experimentally verified binding motifs for several well-known splicing factors, including FOX-1, PTB, hnRNP A/B, hnRNP F/H, and so on. The discovered tissue, region, and conservation preferences of the putative motifs demonstrate that splice site selection is a complicated process that needs subtle and delicate regulation. VERSE may serve as a powerful tool to not only discover SREs by incorporating additional informative signals but also precisely quantify their varying contribution under different biological contexts.
Key words: computational molecular biology, next generation sequencing, regulatory regions, RNA, statistical models
1. Introduction
Alternative splicing is a key biological process in higher eukaryotes to generate multiple transcript isoforms from a single gene, and thus it promotes protein diversity with functional or structural differences (Nilsen and Graveley, 2010; Wang et al., 2008a). With the boost of genomic data due to the recent development in deep sequencing techniques, new splicing events and novel transcript isoforms greatly extended our appreciation of its popularity. For instance, it has been reported that in humans up to 95% of the protein coding genes experienced alternative splicing (Pan et al., 2008; Wang et al., 2008a). However, its regulation mechanism, the so-called splicing code, still remains elusive and deserves additional effort (Barash et al., 2010; Wang and Burge, 2008b). In eukaryotes, despite core signal landmarks such as the junction consensus sequences and branch points, other auxiliary splicing regulatory elements (SREs) also play a vital role through the recruitment of splicing factors and thus facilitate accurate splicing site recognition. The identification of such SREs would greatly enhance our capability to understand the regulation rules, and even to predict splicing patterns under specific biological environment.
Numerous experimental methods were proposed to identify these cis-acting elements, which have been excellently summarized in a review article (Chasin, 2007). Experimental efforts mainly include Systematic Evolution of Ligans by Exponential Enrichment (SELEX) (Kim et al., 2003; Liu et al., 1998, 2000; Tacke and Manley, 1995), ultraviolet (UV) crosslinking and immunoprecipitation (CLIP) (Hafner et al., 2010; Konig et al., 2010; Ule et al., 2003, 2005; Yeo et al., 2009), and splicing reporter systems (Wang et al., 2004). Due to experimental constraints, SREs were reported for only a few splicing factors, but the vast remaining SREs are still unknown, especially those for the unidentified splicing factors.
Meanwhile, several computational efforts were also made to explore the existing genomic data to detect putative splicing factor binding motifs. Word count related enrichment analysis is the most widely used approach, which discovers the statistically over- or under-represented short oligonucleotides through the comparison of foreground and background sequences. For example, Fairbrother et al. (2002) compared exons with strong and weak splice sites and reported a list of 238 exonic splicing enhancers (ESE) with a high validation rate, known as RESCUE ESE. Zhang and Chasin (2004) compared non-coding exons with negative controls, such as the pseudo genes or the 5’ untranslated regions (UTR) of intronless genes, and discovered 2069 8-mer SREs. Brudno et al. (2001) identified 25 brain-specific alternative exons and compared the exonic and flanking intronic sequences therein with the background sequences, for instance, those in the proximity of other cassette exons, and provided several putative mRNA protein binding sites specific to the brain tissue. More recently, Wen et al. (2010) adopted a similar approach to uncover the tissue specific SREs from mouse RNA-seq data in brain, liver, and muscle tissues by comparing the regions near the included and excluded cassette exons. However, this type of analysis heavily relies on the accurate selection of background sequences. A second type of approach is the linear regression between motif counts and the expression or splicing levels of genes or exons. This methodology was first introduced by Bussemaker et al. (2001), and its several adapted versions have received great success in the prediction of transcript factor binding motifs (Zhong et al., 2005). In 2007, Das et al. first applied this method on a small group of muscle-specific cassette exons and discovered several potential SREs. However, the linear assumption of these models might oversimplify the complex relationship between motif counts and exon recognition.
Here, we developed a Varying Effect Regression model on Splicing Elements (VERSE) to predict the genome-wide intronic SREs from RNA-seq data (Fig. 1). Non-motif based biological features such as the phyloP conservation scores (Pollard et al., 2010) are treated as the baseline binding preference of splicing factors, and then our VERSE model utilizes this baseline preference to predict the contribution from each hexamer to the exon inclusion rate accordingly. Besides the capability to integrate additional information, another advantage of VERSE is that the estimated coefficient function is allowed to change with the non-motif based features nonlinearly. We applied VERSE to 16 human tissues and predicted the intronic SREs, among which many overlapped with experimentally verified SREs.
FIG. 1.

Work flow for the genome-wide tissue-specific SRE prediction through VERSE.
2. Results
2.1. VERSE identifies a list of novel motifs from 16 human tissue RNA-seq data
In the search of SREs, we focused on the upstream and downstream 200-bp regions flanking the tissue-specific alternative exons, which were repeatedly reported as enriched with splicing factor binding sites. The 10-bp regions in the direct vicinity of exon-intron boundaries were removed to avoid the effect of consensus sequences. Regression p-values were calculated from our VERSE model against the no effect model, and accordingly to distinguish random occurrences from functional SREs.
Considering the 6-mer SREs, as an example, ordered p-values for all the 4096 hexamers in the brain tissue were plotted and the top ten ranked hexamers are listed in Figure 2A,B. A total of 87 and 79 hexamers have a p-value less than 0.0005 for up- and downstream regions, respectively (FDR < 0.03). To demonstrate the varying contribution of the predicted SREs, we also plotted the estimated coefficient function of the well-known FOX-1 family binding motif TGCATG in brain and skeleton muscle tissues in Figure 2C,D. Our VERSE model not only reported that TGCATG can explain the variation of the exon inclusion rates well (p-value = 2.4 × 10−7 and 1.1 × 10−8 respectively), but also demonstrated the varying contribution with respect to the baseline preference. As indicated, the conserved TGCATG motifs would considerably promote brain or muscle specific alternative splicing, consistent with the conclusion in lines of references that FOX-1 family is enriched in the intronic regions following up-regulated alternative exons (Das et al., 2007; Yeo et al., 2009). Similar conclusions can be drawn from other tissues and the 5-mer SREs prediction results. The predicted SREs and their related p-values are provided in Supplementary Table 1 (for online Supplementary Material, see www.liebertonline.com/cmb).
FIG. 2.

Ordered p-values in brains and examples of the coefficient functions. (A, B) Ordered p-values from VERSE for the upstream and downstream intronic regions in brains. The y-axis is the absolute log transform of regression p-values, and the x-axis is the ranks of the hexamers. The top 10 ranked motifs in both regions are listed. (C, D) Coefficient functions of TGCATG for the downstream intronic regions in brains and muscles. The x-axis is the baseline preference from the phyloP conservation score. The larger values near 1 indicate the conserved TGCATG sites, while smaller values near −1 are the newly evolved ones. The p-values for TGCATG are as low as 2.4 × 10−7 and 1.1 × 10−8, respectively, indicating significant associations with exon inclusion rates.
Setting the p-value cutoff at 0.0005, we have predicted a total of 1562 6-mer SREs across all 16 human tissues or cell lines, many of which were verified through experiments in other literatures. For example, the FOX family protein binding sites (T)GCATG was ranked second and third in brains and muscles, respectively. Interestingly, this motif was proved to be phylogenetically conserved since ancient times (Minovitsky et al., 2005), and our estimated varying coefficient function also indicates that conservation should be an important factor in the functionality of the motif (Fig. 2C,D). Specifically, the conserved TGCATG sites make a much stronger contribution to promote alternative splicing as compared to the newly evolved ones. Similarly, the polypyrimidine-like motif CTCTCT and TCTCTC for PTB were ranked 4th and 47th in upstream intron flanks in brain, consistent with previous conclusions (Chan and Black, 1997; Gooding et al., 1998; Singh et al., 1995). The (GGG)n runs, which has been frequently mentioned as a SRE (Han et al., 2005; Martinez-Contreras et al. 2006), was also found to profoundly affect splice site selection. Detailed lists of previously discovered motifs are provided in Supplementary Table 2. The discovery of these well-known SREs provided convincing evidence of the statistical power of our proposed VERSE model.
Additionally, gene ontology (GO) analysis was also performed to validate the SRE functions. To use TGCATG and the coefficient function predicted in brains as an example, the foreground genes (i.e., real functional SREs) were selected as those with TGCATG and coefficients greater than 0.4, while the background genes were selected as those with coefficients between −0.05 and 0.05 to represent the random occurrences. Using the GO analysis tool GORILLA (http://cbl-gorilla.cs.technion.ac.il/), the most significant enrichment term in the foreground genes is the neuron-specific term “synapse” (p-value = 2.5 × 10−12). Besides, a list of other neuron-specific terms, such as synaptic membrane, presynaptic membrane, axon, and synapse part (p-value = 3.6 × 10−8, 2.2 × 10−7, 2.3 × 10−7, and 1.7 × 10−6), are also ranked 2nd, 3rd, 4th, and 8th. This does not only verify the role of FOX-1 family in regulating brain related alternative splicing events, but also offers convincing evidence of VERSE to make detailed inference of splicing regulation rules.
With the predicted SRE list from VERSE, it is also worthwhile to group these tissues according to their SRE similarities. Tissues similar to each other would probably under co-regulation to a larger extent. Hence we quantitatively measured the regulation similarity by dividing the number of shared SREs in each tissue/cell line pair to the number of total claimed ones. Heat maps of these matrices were provided in Figure 3. In the upstream intron flanks, the colon tissue demonstrates the lowest average regulation similarity with other 15 tissues/cell lines while the testes tissue displays the highest similarity (1.62% and 12.72%, respectively). Two clusters can be divided by hierarchical clustering (Fig. 3A): one is the more similar groups represented by muscle, heart, adipose, testes, breast, and breast cancer cell lines; and the other is composed of brain, liver, colon, and maqhc (Ambion's human brain reference RNA) with distinct motifs from other tissues. We made similar conclusions in the downstream regions.
FIG. 3.

Heat maps of regulation similarity among 16 human tissues. Similarity between each tissue pair is calculated as the percentage of shared motifs. Hierarchical clustering is used to group tissues with higher level of co-regulation.
2.2. Comparison with linear regression demonstrates the advantage of VERSE
As a comparison with existing methods, we performed SRE predictions using simple linear regressions. Because VERSE can detect both motifs with constant coefficients and varying coefficients, many motifs were uniquely discovered by VERSE but missed by the linear regression, some of which were well documented in previous literatures. For example, without considering the non-motif based feature, the FOX-1 family motif TGCATG failed to explain the inclusion rate variation in neither brains nor muscles by the linear regression (p-value = 0.42 and 0.11 for downstream regions). Similarly, in the upstream region, the well-known motif CTCTCT also showed no significance in brains (p-value = 0.49). However, they are all top ranked ones reported by VERSE, demonstrating the enhanced statistical power of VERSE.
In addition, we also observed that the advantage brought by VERSE is different across different human tissues. In tissues like adipose, breast, testes and several cancer cell lines, more than half of the identified motifs by VERSE were also claimed by linear regressions and such high overlapping rates were observed in both upstream and downstream regions (high intersect/union ratio in Table 1), indicating the consistency of both algorithms. However, in other tissues, such as brain, liver, and colon, very limited number of SRES were found to be shared by the two methods. These indicate the different roles of the baseline preferences in different tissues.
Table 1.
Comparison of Predicted SREs from VERSE and the Linear Regression. Where P-Value Cutoffs Are Set at 0.0005.
| |
Upstream |
Downstream |
||||||
|---|---|---|---|---|---|---|---|---|
| Tissue | VERSE | LR | Union | Intersect | VERSE | LR | Union | Intersect |
| BT474 | 86 | 57 | 91 | 52 | 59 | 29 | 63 | 25 |
| lymph | 285 | 191 | 301 | 175 | 206 | 140 | 220 | 126 |
| testes | 350 | 224 | 363 | 211 | 274 | 207 | 287 | 194 |
| adipose | 182 | 132 | 206 | 108 | 195 | 134 | 212 | 117 |
| colon | 28 | 2 | 28 | 2 | 26 | 6 | 27 | 5 |
| muscle | 146 | 48 | 149 | 45 | 199 | 102 | 209 | 92 |
| heart | 65 | 31 | 66 | 30 | 69 | 26 | 71 | 24 |
| liver | 47 | 15 | 50 | 12 | 32 | 5 | 32 | 5 |
| maquhr | 96 | 52 | 98 | 50 | 63 | 44 | 73 | 34 |
| maqhc | 40 | 3 | 40 | 3 | 43 | 5 | 43 | 5 |
| T47D | 122 | 73 | 130 | 65 | 110 | 75 | 120 | 65 |
| MB435 | 165 | 102 | 176 | 91 | 125 | 87 | 136 | 76 |
| MCF7 | 124 | 87 | 137 | 74 | 128 | 71 | 139 | 60 |
| breast | 405 | 307 | 427 | 285 | 325 | 262 | 344 | 243 |
| HME | 189 | 142 | 197 | 134 | 192 | 121 | 208 | 105 |
| brain | 87 | 6 | 87 | 6 | 79 | 11 | 81 | 9 |
2.3. General and unique SREs display significant GC disparity
Two types of the predicted SREs were defined: the specific ones were defined as those declared by VERSE in only one tissue, while the general ones were identified in at least half of the 16 human tissues/cell lines. Because VERSE considers tissue-specific alternative splicing events, the majority of the predicted motifs were identified in a single tissue. For example, in the upstream region, 632 out of 1135 SREs (55.68%) were found to be significant only in one tissue, while only 37 SREs (3.26%) were declared as the general SREs (dark bars in Fig. 4). Similar results were also observed in downstream regions: 585 specific SREs (56.80%) and 33 general SREs (3.20%). p-values for the chi-square tests were both less than 2.2 × 10−16, indicating the significant tissue preference of our predicted SREs in alternative splicing regulation. Interestingly, we discovered that the general motifs are mostly AT enriched, while the specific ones demonstrate substantial GC enrichment. Specifically, the average GC content for the specific SREs was 0.484 and 0.471 for upstream and downstream regions respectively, but it dropped to 0.099 and 0.081 for general SREs (p-values for the unpaired Wilcoxon tests were 2.2 × 10−16 and 4.1 × 10−16, respectively).
FIG. 4.

Tissue specific and general SREs. The x-axis is the number of tissues within which SREs are identified as functional by VERSE, and the y-axis is the total number of SREs falling into this category. The p-value cut-off is set at 0.0005 for motif identification, and regression bandwidth is 1.0.
Besides, tissue specificity was defined as the ratio of the number of specific motifs to the general ones identified by VERSE. As shown in Table 2, the brain tissue showed the highest specificity, demonstrating the distinct splicing regulation rules. It is worthwhile to mention that the mixed tissue maquhr (Stratagene's universal human reference RNA, which is composed of total RNA from 10 different human cell lines) demonstrated the lowest specificity at 0.414 and 0.850 in both flanking intron regions, as we expected.
Table 2.
Tissue Specificity of 16 Human Tissues in Both Up- and Downstream Intron Flanks
| |
Upstream |
Downstream |
||||
|---|---|---|---|---|---|---|
| Tissue | Specific | General | Specificity | specific | General | Specificity |
| BT474 | 22 | 21 | 1.048 | 21 | 7 | 3.000 |
| lymph | 84 | 34 | 2.471 | 46 | 28 | 1.643 |
| testes | 92 | 34 | 2.706 | 67 | 33 | 2.030 |
| adipose | 34 | 29 | 1.172 | 47 | 26 | 1.808 |
| colon | 16 | 3 | 5.333 | 6 | 6 | 1.000 |
| muscle | 33 | 24 | 1.375 | 55 | 24 | 2.292 |
| heart | 12 | 15 | 0.800 | 22 | 14 | 1.571 |
| liver | 18 | 4 | 4.500 | 11 | 3 | 3.667 |
| maquhr | 12 | 29 | 0.414 | 17 | 20 | 0.850 |
| maqhc | 11 | 2 | 5.500 | 17 | 9 | 1.889 |
| T47D | 33 | 28 | 1.179 | 21 | 26 | 0.808 |
| MB435 | 37 | 34 | 1.088 | 30 | 26 | 1.154 |
| MCF7 | 31 | 18 | 1.722 | 38 | 15 | 2.533 |
| breast | 113 | 36 | 3.139 | 95 | 33 | 2.879 |
| HME | 44 | 28 | 1.571 | 52 | 24 | 2.167 |
| brain | 40 | 4 | 10.00 | 40 | 4 | 10.00 |
2.4. Position and conservation preferences of predicted SREs
Lines of references mentioned that the function of SREs depends on their relative positions to splicing junctions, and some may even perform opposite roles when located in different regions. For example, the poly pyrimidine like motif CTCTCT or TCTCTC would help the recruitment of PTB before splice sites most of time, while (GGG)n runs might regulate exon recognition both before and after exon junctions. Such position dependency reveals the complexity of the splicing regulation process, so we analyzed this preference of our predicted SREs in each tissue separately.
In general, only 603 out of 1562 SREs in 16 human tissues/cell lines (around 38.60%) are shared in both upstream and downstream regions, indicating the significant position preference. We also observed substantial tissue variations in the percentage of shared SREs between upstream and downstream intronic flanks. For instance, in breast and testes tissues, the shared percentages were up to 23.31% and 22.83%, respectively, while in brain and liver tissues, the percentage dropped to 3.75% and 1.28%. Such high discrepancy across multiple tissues does not only indicate the fact that the positional dependency would affect the regulation process to different extents, but also corresponds very well with our previous discovery that brains showed high complexity in the splicing regulation process.
3. Discussion
In this work, we developed VERSE through a varying coefficient regression model to associate the inclusion rate of exons with hexamers occurrences in intronic regions. The advantage of this model is that non-motif based biological features such as the conservation score can be incorporated into the model as the baseline preference and thus provides more precise splicing contribution in a quantitative way. Using VERSE, we successfully identified 1562 6-mer intronic SREs in 16 human tissues (as in Supplementary Table 1). The discovery of well-known motifs such as (T)GCATG for FOX-1 family, CT enriched poly pyrimidine sequences for PTB, (GGG)n runs for hnRNP A/B, and hnRNP F/H proved the reliability of our predictions. In addition to the overall association significance, our VERSE model also provided detailed contribution functions of these SREs according to their conservation scores, further expanding our knowledge to splicing regulation rules.
3.1. Integration of conservation scores improves accuracy of SRE prediction
Inspired by the report that the assembly of multiple features would greatly improve the prediction precision of splicing pattern (Barash et al., 2010), we adopted the conservation score as a prior information to predict SREs. By permitting changes of the motif occurrence coefficients, VERSE successfully accommodates non-linear relationship between SRE occurrence and splicing ratios. Phylogenetic conservation information has been reported in many literatures to be an important mRNA feature to infer biological functionality (Minovitsky et al., 2005; Sugnet et al., 2006). It is worth noting that the phyloP score is able to represent both conservation and evolution, and thus we could discover SREs that are under negative or positive selection. With the assistance of this prior information, VERSE not only identified almost all the motifs reported by the linear regression, but also discovered many novel ones with the conservation dependency. Some of these new ones were frequently mentioned as important splicing factor binding sites in numerous literatures. For example, the phylogenetically conserved SRE TGCATG was not reported as significant in neither brains nor muscles by the simple linear regression, but additional conservation information makes them top ranked by our VERSE model. This result indicates that if utilized in a rationale way, the baseline preference is very informative in SRE identification and deserves further attention.
3.2. Tissue, conservation, and position preferences of SREs demonstrate the complexity of splicing regulations
First, tissue preference of the SREs may affect splice site usage and promote regulation complexity in a much larger extent than ever expected. Among the 1562 putative motifs discovered in the intron regions, the majority were only declared to be functional in one tissue (as shown in the first bars in Fig. 4), while only around 3% were found in the majority of tissues (as shown in the last bars of Fig. 4). Additionally, our tissue SRE similarity study further supports the conclusion that regulatory elements might help splicing factor recruitment only under particular biological environment. These discoveries not only suggest that SREs contribute greatly to the tissue specification process through alternative splicing, but also imply that splice site recognition process is so complicated that splicing regulation rules and SRE identification deserve more consideration in a tissue specific manner.
Interestingly, a sharp GC difference was observed between the specific and general SREs. The general motifs show strong AT bias, while the specific ones are mostly GC enriched. It has been computationally predicted that many intronic SREs for constitutive splicing are AT enriched (Voelker and Berglund, 2007). Our discovery opens another possibility that the AT enriched ones would also be functional for general alternative splicing, instead of promoting tissue specific alternative splicing events. In addition, one of our pervious articles indicates that GC content is indeed involved in the splicing regulation process probably through maintaining stable mRNA secondary structures, and such involvement might exist since ancient times (Zhang et al., 2011). Our VERSE predicted results also indicate that GC content plays a role in the binding of splicing factors.
In addition, VERSE enables us to make the first genome wide prediction of conservation dependent SREs from RNA-seq data. The high percentage of hexamers that were uniquely discovered by VERSE through adding conservation scores as the prior information confirmed the existence of conservation dependent SREs. More importantly, we found that these conservation dependent SREs would preferentially function in a tissue specific manner rather than work as universal ones. Our estimated coefficient function analysis shows that up to 90% of the conserved SREs tend to promote alternative splicing, either by enhancing or repressing the expression of cassette exons, while the newly evolved counterparts usually perform differently from the conserved ones even within the same tissue. It strongly implies that these cis-acting SREs are under strict negative selection to maintain they contribution to enhance transcript diversity from ancient times. Even within the same tissue and under similar conservation constraint, motifs may act oppositely due to the location difference. For instance, only 38.60% percent of motifs are shared by both upstream and downstream regions. All together, these preferences of tissues, conservation, and positions, and the coupling of these factors indicate that splicing regulation is a complicated biological process, and the consideration of such information into its regulation mechanism exploration would significantly extend our current understanding.
3.3. Brains demonstrate the highest regulation complexity through SREs
It has been reported in lines of references that brains exhibited a large number of tissue specific alternative spliced exons. Consistent with such results, we also observed the highest level of regulation complexity in terms of SRE characteristics in brains. First of all, we observed an extraordinary large number of specific SREs but very limited general ones in brain tissues. This phenomenon was further supported by the tissue similarity analysis since brains shared very small percentage of motifs with other tissues. Moreover, even within the brain tissue, only 3.75% of the total identified SREs were found to be shared in the upstream and downstream intronic regions, compared with more than 10.00% in some other tissues, indicating a strong positional preference. Furthermore, a large number of SREs were found to regulate splicing in brain tissues with conservation constraints and most of these conservation-dependent SREs only work in a tissue specific manner. All of these results provide convincing evidence that brains might have the most complicated splicing regulation mechanism that needs tissue, conservation, and regional specificity to ensure a reliable and efficient regulation mechanism for the greatest level of tissue specific alternative splicing.
In conclusion, our VERSE model is powerful to detect SREs based on RNA-seq data. Besides the conservation scores, other types of non-motif features can be incorporated in the model in a similar manner. With the continuing improvement of high throughput sequencing techniques, more refined analysis of SREs would be feasible in the near future.
4. Methods
4.1. Data collection
RNA-seq data set (SRA010153 for the MAQC data, SRP000727 for the human tissue data) for 16 tissues or cell lines were downloaded from the NCBI sequence read archive (http://www.ncbi.nlm.nih.gov/sra/). We first aligned the reads to the human genome using Bowtie version 0.12.1 (Langmead et al. 2009) with the default settings, and then further aligned the unmapped ones against the human refseq RNA sequences (downloaded from the NCBI website, version 36) to discover the junction reads. Only the uniquely mappable reads were used and we only considered genes on autosomes or X chromosomes. The position-level read count was the number of body or junction reads starting from an exonic position of a gene (or an exon) without considering the strand information.
4.2. Exons considered in VERSE
A non-redundant exon list was assembled from the Refseq gene annotations. Overlapped regions were further split into disjointed regions. Then, in each tissue, the exon/gene expression levels were estimated by the average of total reads mapped in this region, and the exon inclusion rate was calculated as the ratio of exon expression level to its corresponding gene expression level. In order to discover the tissue specific alternative splicing events, exon inclusion rates were normalized across all the 16 tissues, and exons with a normalized inclusion rate greater than 5 or less than 0.3 were considered in VERSE. To make a control list, we also selected constitutive exons with the normalized exon inclusion rate between 0.95 and 1.05 in VERSE. Note that the selection of alternative or constitutive exons is only a rough selection and VERSE can further distinguish them according to the estimated coefficients.
4.3. VERSE
In order to differentiate the positional preference of the SREs, the upstream and downstream intronic regions that flanked the exons were discussed separately, and workflow was shown in Figure 1. We considered exons with flanking introns with at least 400bp in length. Specifically, the upstream and downstream 200bp intronic regions immediately flanking the exon junctions were considered as the target regions to search for splicing factor motif sites. Besides, to exclude the bias due to the consensus sequences near the two junction sites, the 10bp regions directly adjunct to the exon junctions were excluded in our analysis, resulting a 190bp target sequence in both directions. For each hexamer w, let 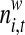 represent its occurrences in a target region around exon i in tissue t. yi,t denotes the absolute log value of the normalized exon inclusion rate in this tissue. For each exon i, the baseline preference of the jth occurrence is described by ui,j, which is averaged across the all the nucleotides within each hexamer. The phyloP conservation scores from 44 mammal species were downloaded from the UCSC genome browser to represent the baseline preferences (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/phastCons44way/placentalMammals/). All phyloP scores greater than 3 or less than −3 were transformed to 1 or −1, and the rest were normalized by 3. Hence the support of ui,j is [−1, 1]. The following semi-parametric varying coefficient regression is fitted with bandwidth h.
represent its occurrences in a target region around exon i in tissue t. yi,t denotes the absolute log value of the normalized exon inclusion rate in this tissue. For each exon i, the baseline preference of the jth occurrence is described by ui,j, which is averaged across the all the nucleotides within each hexamer. The phyloP conservation scores from 44 mammal species were downloaded from the UCSC genome browser to represent the baseline preferences (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/phastCons44way/placentalMammals/). All phyloP scores greater than 3 or less than −3 were transformed to 1 or −1, and the rest were normalized by 3. Hence the support of ui,j is [−1, 1]. The following semi-parametric varying coefficient regression is fitted with bandwidth h.
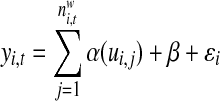 |
α(□)is the varying coefficient function to model the changing contribution of motif hits to the exon inclusion rate as a function of the baseline preference, and β is the parametric part of our model to indicate the baseline inclusion rate for each exon. ɛi is an i.i.d Gaussian distributed random variable with zero mean. A kernel function k(□) is used to smooth the errors and conduct local linear regressions. Define kh(□) = k(□/h)/h, 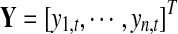 ,
, 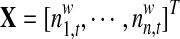 ,
, 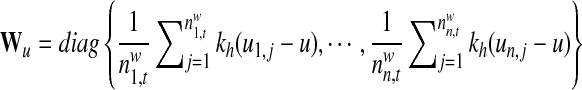 ,
, 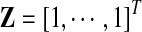 , and
, and
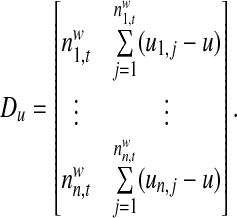 |
Many approaches have been proposed to estimate the coefficient β and the unknown coefficient function α(□), and here we adopted the profile least square estimation (Fan and Huang 2005) as the following:
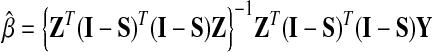 |
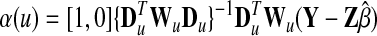 |
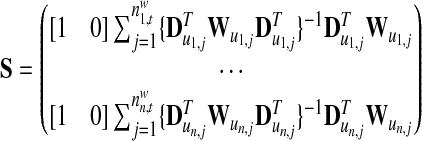 |
I is the identity function with dimension n. The bandwidth parameter h determines the radius of the neighborhood in the local linear regression and thus reflects the degree of freedom of the model. A larger bandwidth would benefit from the variance side, but lose on the precision side. We selected 1.0, which is half of the whole support of the baseline preference u, to perform the least square estimation for the varying coefficient model. To evaluate the significance of estimated varying coefficient function α(□), a chi-square test was carried out.
To make a comparison with VERSE, a linear regression model with the constant effect, was fitted without the consideration of the baseline preference and F test was used to evaluate the performance of each regression.
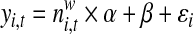 |
Supplementary Material
Acknowledgments
This research was supported in part by the National Institutes of Health (grants R01GM097230 and P50HG002790).
Disclosure Statement
No competing financial interests exist.
References
- Barash Y. Calarco J.A. Gao W., et al. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- Brudno M. Gelfand M.S. Spengler S., et al. Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Res. 2001;29:2338–2348. doi: 10.1093/nar/29.11.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemaker H.J. Li H. Siggia E.D. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- Chan R.C. Black D.L. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol. Cell. Biol. 1997;17:2970. doi: 10.1128/mcb.17.5.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasin L.A. Searching for splicing motifs. Adv. Exp. Med. Biol. 2007;623:85–106. doi: 10.1007/978-0-387-77374-2_6. [DOI] [PubMed] [Google Scholar]
- Das D. Clark T.A. Schweitzer A., et al. A correlation with exon expression approach to identify cis-regulatory elements for tissue-specific alternative splicing. Nucleic Acids Res. 2007;35:4845–4857. doi: 10.1093/nar/gkm485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother W.G. Yeh R.F. Sharp P.A., et al. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Fan J. Huang T. Profile likelihood inferences on semiparametric varying-coefficient partially linear models. Bernoulli. 2005;11:1031–1057. [Google Scholar]
- Gooding C. Roberts G.C. Smith C.W. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated alpha-tropomyosin exon. RNA. 1998;4:85–100. [PMC free article] [PubMed] [Google Scholar]
- Hafner M. Landthaler M. Burger L., et al. PAR-CliP—a method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010;41:2034. doi: 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K. Yeo G. An P., et al. A combinatorial code for splicing silencing: UAGG and GGGG motifs. PLoS Biol. 2005;3:e158. doi: 10.1371/journal.pbio.0030158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. Shi H. Lee D.K., et al. Specific SR protein-dependent splicing substrates identified through genomic SELEX. Nucleic Acids Res. 2003;31:1955–1961. doi: 10.1093/nar/gkg286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J. Zarnack K. Rot G., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. Trapnell C. Pop M., et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.X. Zhang M. Krainer A.R. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998;12:1998–2012. doi: 10.1101/gad.12.13.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.X. Chew S.L. Cartegni L., et al. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol. Cell. Biol. 2000;20:1063–1071. doi: 10.1128/mcb.20.3.1063-1071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R. Fisette J.F. Nasim F.U., et al. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4:e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minovitsky S. Gee S.L. Schokrpur S., et al. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 2005;33:714–724. doi: 10.1093/nar/gki210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen T.W. Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q. Shai O. Lee L.J., et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Pollard K.S. Hubisz M.J. Rosenbloom K.R., et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. Valcarcel J. Green M.R. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- Sugnet C.W. Srinivasan K. Clark T.A., et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput. Biol. 2006;2:e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke R. Manley J.L. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995;14:3540–3551. doi: 10.1002/j.1460-2075.1995.tb07360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J. Jensen K.B. Ruggiu M., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- Ule J. Jensen K. Mele A., et al. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Voelker R.B. Berglund J.A. A comprehensive computational characterization of conserved mammalian intronic sequences reveals conserved motifs associated with constitutive and alternative splicing. Genome Res. 2007;17:1023–1033. doi: 10.1101/gr.6017807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T. Sandberg R. Luo S., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008a;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Burge C.B. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008b;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Rolish M.E. Yeo G., et al. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Wen J. Chiba A. Cai X. Computational identification of tissue-specific alternative splicing elements in mouse genes from RNA-Seq. Nucleic Acids Res. 2010;38:7895–7907. doi: 10.1093/nar/gkq679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G.W. Coufal N.G. Liang T.Y., et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H. Chasin L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Kuo C.C. Chen L. GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics. 2011;12:90. doi: 10.1186/1471-2164-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W. Zeng P. Ma P., et al. RSIR: regularized sliced inverse regression for motif discovery. Bioinformatics. 2005;21:4169–4175. doi: 10.1093/bioinformatics/bti680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.