

Abstract
Four new bromophycolides, R–U (1–4), were isolated from the Fijian red alga Callophycus serratus and were identified by 1D and 2D NMR and mass spectroscopic analyses. These compounds expand the known structural variety of diterpene-benzoate macrolides and exhibited modest cytotoxicity toward selected human cancer cell lines. Bromophycolide S (2) also showed submicromolar activity against the human malaria parasite Plasmodium falciparum.
Callophycus serratus is a marine red alga that lives on rocky floors of caves and on undercut walls at depths of 3–20 m throughout the tropical and subtropical Pacific Ocean.1Recent reviews have demonstrated that secondary metabolites from red algae are dominated by terpenes and halogenated polyphenols, which exhibit multiple types of biological activity.2,3Our previous investigation of C. serratus from Fijian coral reefs resulted in the discovery of 18 novel diterpene-benzoate macrolides (bromophycolides A–Q and debromophycolide A), eight diterpene-benzoic acids (callophycoic acids A–H), and two diterpene phenols (callophycols A, B), several of which possessed antimalarial, antibacterial, antitubercular, anticancer, and antifungal activities.4-8
As part of our continuing investigation of new bioactive natural products from Fijian marine organisms, extracts of C. serratus were separated by liquid–liquid partition to give four fractions (see Extraction and Isolation). The CHCl3-soluble fraction was further separated by reversed-phased HPLC to yield four new bromophycolides (R–U, 1–4), which were identified by NMR and mass spectrometric analyses.
Bromophycolide R (1), isolated as a white powder, showed a HR-ESIMS molecular ion peak at m/z [M + H]+of 503.1824, corresponding to a molecular formula of C27H35BrO4 and supported by a monobrominated isotopic splitting pattern. The UV spectrum had an absorption maximum at 262 nm common to reported bromophycolides.4,5,7In the 1H NMR spectrum of 1, an ABX coupling system at δ 8.30 (br s), 7.66 (dd, J ) 8.5, 1.5 Hz), and 6.72 (d, J ) 8.0 Hz) was in good agreement with the p-hydroxybenzoate ester portion of previously isolated bromophycolides.4,5,713C NMR signals at δ 111.6 (C-26) and 144.3 (C-15) suggested a vinyl group. Both H2-26 vinyl protons (δ 4.90, 5.02) correlated to C-27 (δ 18.4) and C-14 (δ 78.7) in the HMBC spectrum, revealing an isopropenyl diterpene head. Four isoprene units were suggested by HMBC correlations from Me-27 (δ 1.78) to C-14, C-15, and C-26; from Me-25 (δ 1.34) to C-10 (δ 67.2), C-11 (δ 64.1), and C-12 (δ 32.9); from Me-24 (δ 0.96) to C-7 (δ 43.8), C-8 (δ 36.0), and C-22 (δ 63.3); and from both H2-23 protons (δ 4.76, 4.81) to C-6 (δ 46.0) and C-20 (δ 37.9). The head-to-tail linkages of the isoprene units were established through COSY correlations between H-13b (δ 2.05) and H-12a (δ 1.16); H-9a (δ
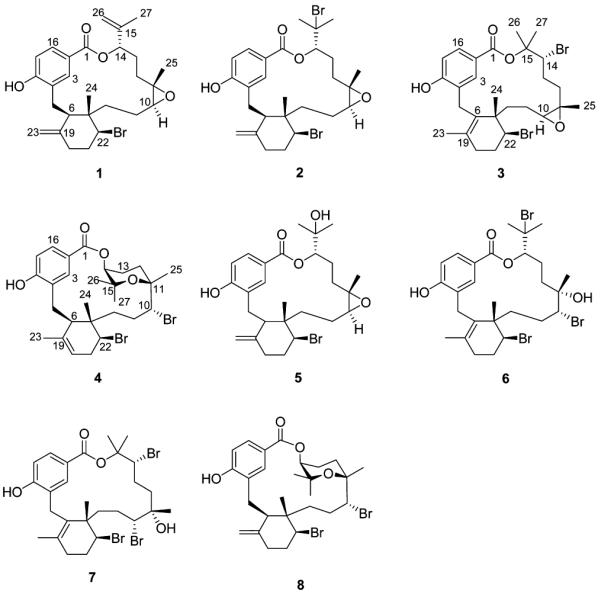 1.63) and H-8a (δ 1.91); and H-21a (δ 2.08) and H-20a (δ 2.23). An HMBC correlation from Me-24 to C-6 closed the C-6–C-7 linkage of the six-membered ring. Given the fact that only one oxygen atom was not already assigned, the two upfield shifted carbinol signals at C-10 and C-11 suggested an epoxide and accounted for the final degree of unsaturation. The remaining bromine was assigned at C-22, consistent with other previously identified bromophycolides.5Bromophycolide R (1) is the dehydrated form of previously reported bromophycolide F (5),5resulting in the isopropenyl terpene head of 1.
1.63) and H-8a (δ 1.91); and H-21a (δ 2.08) and H-20a (δ 2.23). An HMBC correlation from Me-24 to C-6 closed the C-6–C-7 linkage of the six-membered ring. Given the fact that only one oxygen atom was not already assigned, the two upfield shifted carbinol signals at C-10 and C-11 suggested an epoxide and accounted for the final degree of unsaturation. The remaining bromine was assigned at C-22, consistent with other previously identified bromophycolides.5Bromophycolide R (1) is the dehydrated form of previously reported bromophycolide F (5),5resulting in the isopropenyl terpene head of 1.
Configurational assignments for 1 were facilitated by ROESY spectroscopic data and previously reported X-ray crystallographic data of bromophycolide A (6).4NOE correlations between H-6 (δ 3.45) and H-22 (δ 4.61), together with correlations between Me-24 and both H2-5 protons (δ 2.70, 3.16), suggested 6R, 7S, 22S stereocenters. Likewise, NOE correlations between Me-25 and H-9a, along with correlations between H-10 (δ 2.83) and H-12a, supported a trans configuration at C-10 and C-11. Additional NOEs observed between H-9a and Me-24 together with NOE correlations between H-10 and H-3, H-3, and H-6 and a weak NOE between H-6 and H-10 linked the epoxide to confirmed chiral centers C-6 and C-7 and suggested a 10S, 11S configuration. H-14 (δ 4.74) showed an NOE correlation with overlapping signals at δ 1.96–1.97, assigned to H-13a and H-12b. However, NOE correlations observed between Me-25 and H-13b and between H-14 and H-13b indicated that Me-25, H-13b, and H-14 were on the same face of the molecule. This placed H-13a and H-12b in an anti relationship with respect to each other. Considering the large (J ) 11 Hz) coupling of the broad doublet observed for H-14 indicating an anti relationship between H-14 and H-13a, the NOE observed between H-14 and an overlapping proton at δ 1.96–1.97 must be to H-12b (and not H-13a). Thus, a 14S configuration was concluded, which is consistent with previously reported bromophycolides for which X-ray crystallography established the absolute configuration.4,5
Bromophycolide S (2) possessed a molecular formula of C27H36Br2O4 from the ion with m/z 605.0929 [M + Na]+, supported by a dibrominated isotopic splitting pattern, suggesting the addition of HBr relative to 1. All 13C NMR chemical shifts for 2 differed by less than 3 ppm from those of 1 except for the signals at C-14 (δ 80.4), C-15 (δ 66.9), Me-26 (δ 32.7), and Me-27 (δ 29.7), revealing a 15-membered lactone framework with a p-hydroxybenzoate structure as 1.4One fewer olefin and an additional methyl signal were found in 2, suggesting that the differences between 1 and 2 were at the diterpene head. HMBC correlations from Me-26 (δ 1.83) and Me-27 (δ 1.78) to C-15, C-14, and each other revealed two methyls at brominated carbon C-15, identical to 6.4Due to the structural resemblances and similar observed NOEs between 1, 2, and 6, the configuration of 2 was assigned as 6R,7S,10S,11S,−14S,22S.
The high-resolution mass spectrum of bromophycolide T (3) displayed a molecular ion peak at m/z 605.0881 [M + Na]+, appropriate for a molecular formula of C27H36Br2O4, isomeric with 2. Comparing the 1H and 13C NMR spectra for 3 to those of 1 and 2, the p-hydroxybenzoate and the epoxy functions remained intact. HMBC correlations from H-5a (δ 3.17) to olefinic carbon C-6 (δ 131.9), as well as from Me-23 (δ 1.90) to C-19 (δ 132.8) and C-6, revealed regioisomerization of the double bond in 3 relative to 1 and 2. The connectivity between the benzoate and diterpene head was also altered, with the downfield shifted oxygenated quaternary carbon C-15 (δ 82.4) as the site of connection, as previously shown for bromophycolide B (7).4Both diterpene head methyl groups Me-26 (δ 1.86) and Me-27 (δ 1.76) correlated with C-14 (δ 66.8), C-15, and each other in the HMBC spectrum. C-14 was further linked to the epoxide, as shown through COSY correlations between H-14 (δ 4.00) and H-13a (δ 2.10) and between H-13b (δ 2.22) and H-12a (δ 1.68) and by HMBC correlations from Me-25 (δ 1.27) to C-12 (δ 37.4).
From previously reported X-ray crystallographic data for 7 and examination of ROESY spectroscopic data, a 7S,22S configuration for 3 was suggested on the basis of NOEs observed between H-22 and H-9a and between H-9a and H-10, along with NOE correlations between H-25 and H-9b; H-9b and H-24; H-25 and H-8a; and H-8a and H-24. Similarly, NOEs observed between H-10 (δ 2.97) and H-12a and between Me-25 (δ 1.27) and H-9b (δ 1.71) suggested a 10S,11S configuration similar to that of 1. Given the structural similarities with bromophycolide H and 7,4,5and NOE correlations between H-14 and both Me-26 and Me-27, a 14R configuration was inferred.
The molecular formula for bromophycolide U (4) was confirmed as C27H36Br2O4 from the parent ion at m/z 605.0871 [M + Na]+, isobaric with 2 and 3. The 1H NMR spectroscopic data for 4 were similar to bromophycolides P (8) and Q in our previous report.7The cyclohexenyl double bond of 4 appeared to display a C-19–C-20 unsaturation identical to bromophycolide O7and could be confirmed by COSY correlations from H-21b (δ 2.57) to H-22 (δ 4.29) and H-20 (δ 5.17), along with HMBC correlations from Me-23 (δ 1.50) to C-6 (δ 48.1), C-19 (δ 137.0), and C-20 (δ 119.9). Considering that this is a common double-bond rearrangement with otherwise similar NMR spectroscopic data to 8 and bromophycolide Q,7the configuration for 4 was proposed to be 6R,7S,10R,11R,−14S,22S.
Bromophycolides R–U (1–4) would be expected to follow a similar biosynthetic pathway to that proposed for previously reported bromophycolides.4,5,7The epoxide in 1–3 would be expected to arise from an SN2 nucleophilic attack of the C-11 hydroxy at C-10, displacing bromide and resulting in an inversion of configuration at C-10 and a trans epoxide. While we cannot completely rule out the possibility that epoxide formation occurred during the isolation process, experimental evidence suggests otherwise. We have found that the epoxide will not form from the bromohydrin in 6 at temperatures less than 50 °C nor without the presence of base. Thus, given that our isolation occurred at or below 25 °C and at relatively neutral pH, the likelihood that the epoxides were formed during the isolation process is low.
Bioactive compounds 1–4 exhibited moderate activity against the human malarial parasite Plasmodium falciparum with IC50 values ranging from 0.9 to 8.4 μM (Table 2). The antimalarial activity of 2 (IC50 0.9 μM) was comparable to the most active bromophycolides reported previously.7Within the 15-membered lactone framework, less polar groups at the diterpene head appear to be associated with potency, because substitution of a bromine (as in 2 and bromophycolide D) for a hydroxy at C-15 was associated with 2–6-fold increase in activity, and bromophycolides possessing an isopropenyl group at the diterpene head (as in 1 and bromophycolide E) displayed intermediate antimalarial activity. The tetrahydropyran ring of 4, 8, and bromophycolide Q (IC50 1.4–2.9 μM) contributed to reduced activity compared to open forms.7In both the 15- and 16-membered lactone frameworks, the epoxide at C-10 (1–3) contributed to reduced antimalarial activity compared to the bromohydrin function (7, bromophycolides D and E) at C-10–C-11.7The antibacterial, antifungal, and anticancer activities of 1–4 were also analyzed (Table 2). The results were similar to those of previously reported bromophycolides.4,5,7
Table 2.
Pharmacological Activities of Bromophycolides R–U (1–4)
| antimalarial activity |
antibacterial activity (μM) |
antifungal activitya |
anticancer activity (μM) |
||||
|---|---|---|---|---|---|---|---|
| cmpd | IC50 (μM) | MRSA IC50 | VREF IC50 |
M. tuberculosis MIC |
IC50 (μM) | meanb | cell line selectivity (IC50 max/IC50 min) |
| 1 | 1.7 | >15 | >15 | >50 | >15 | 19 | 2.9 |
| 2 | 0.9 | >15 | 3.8 | 23 | >15 | 16 | 2.2 |
| 3 | 8.4 | >15 | >15 | >50 | >15 | 24 | 1.6 |
| 4 | 2.1 | 0.9 | 0.9 | 22 | >15 | 16 3 | 1 |
Using amphotericin-resistant Candida albicans.
Mean of 12 cancer cell lines (see the Experimental Section for details). MRSA = methicillin-resistant Staphylococcus aureus; VREF = vancomycin-resistant Enterococcus faecium.
Here, we describe four new bromophycolides and their biological activities. This study not only expands the number of discovered diterpene-benzoate macrolides from C. serratus but also explores the structure–activity relationship for antimalarial activity and suggests possibilities for the design and synthesis of antimalarial drugs.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco P-1010 spectropolarimeter. UV spectra were recorded in methanol with a Spectronic 21D spectrophotometer. IR spectra were recorded on a Shimadzu FTIR 8400S spectrophotometer. NMR spectra were acquired on a Bruker DRX-500 instrument, using a 5 mm broadband or inverse detection probe for 1H, 13C, 1H–1H COSY, HSQC, HMBC, NOESY, and ROESY experiments. For some compounds, quaternary carbon chemical shifts were inferred from HMBC data. High-resolution mass spectra were generated using electrospray ionization with an Applied Biosystems QSTAR-XL hybrid quadrupole time-of-flight tandem mass spectrometer and Analyst QS software. LC-MS analyses were conducted using a Waters 2695 HPLC with Waters spectrometer with 2996 diode-array UV detection and Micromass ZQ 200 mass spectrometer with electrospray ionization. LC-MS chromatography was achieved with an Xterra NS-C18 3.5 μm column measuring 2.1 × 15 mm and gradient mobile phases of aqueous methanol with 0.1% acetic acid. Semipreparative HPLC was performed using a Waters 2690 pump, with a Waters 996 diode-array UV detector, controlled by Waters Millenium software. Compound purification by HPLC was achieved using Agilent Zorbax SB-C18 (5 μm, 9.4 250 mm) and Phenomenex Develosil C30 RPAQUEOUS (5 μm, × 4.6 250 mm) columns. All commercial chemicals were reagent grade×except for solvents used for HPLC and LC-MS, which were HPLC or Optima grade (Fisher Scientific Co.). NMR solvents were purchased from Cambridge Isotope Laboratories.
Algal Material
Callophycus serratus (Harvey ex Kutzing 1957) (family Solieriaceae, order Gigartinales, class Rhodophyceae, phylum Rhodophyta) was collected from coral reefs offshore from Yanuca Island in Fiji (18°23′57″ S, 177°57′59″ E). Fresh samples were immediately frozen at −20 °C until further processed for extraction in the laboratory. Voucher specimens were identified by comparison with previously described morphological traits7and preserved in 10% aqueous formalin. Vouchers with identification ICBG-G-0004 and ICBG-G-0593 were deposited at the University of the South Pacific in Suva, Fiji, and at Georgia Institute of Technology, Atlanta, GA.
Extraction and Isolation
Freeze-dried C. serratus was extracted with MeOH five times. The extracts were combined, reduced under vacuum, and sequentially partitioned between MeOH/H2O (9:1) and petroleum ether. The aqueous fraction was adjusted to MeOH/H2O (3:2) and partitioned against CHCl3. The CHCl3-soluble fraction was separated by column chromatography using HP20ss resin, starting with MeOH/H2O (3:2) and eluting with MeOH (100%). Further purification by C30 reversed-phase HPLC with 84% aqueous MeOH afforded bromophycolides R–U (1–4). Pure compounds were analyzed by LC-MS to determine λmax and molecular mass and quantified by 1H NMR spectroscopy using 2,5-dimethylfuran as internal standard.
Bromophycolide R (1)
white, amorphous solid (0.57 mg, 0.0029% dry mass); [α]23 D +118 (c 0.038, MeOH); UV (MeOH) λmax (log ε) 260 (3.52) nm; IR (NaCl) νmax 3356, 2926, 1717, 1603, 1456, 1273, 1119 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, Table 1; 2D NMR data, Supporting Information; HRESIMS [M + H]+m/z 503.1824 (calcd for C27H36BrO4, 503.1797).
Table 1.
NMR Spectroscopic Data (500 MHz, CDCl3) for Bromophycolides R–U (1–4)a
| bromophycolide R (1) |
bromophycolide S (2) |
bromophycolide T (3) |
bromophycolide U (4) |
|||||
|---|---|---|---|---|---|---|---|---|
| no. | δ C | δH (J in Hz) | δ C | δH (J in Hz) | δ C | δH (J in Hz) | δ C | δH (J in Hz) |
| 1 | 166.8 | 168.0 | 164.2 | 164.9 | ||||
| 2 | 124.0 | 124.1 | 124.0 | 122.8 | ||||
| 3 | 131.4 | 8.30, br s | 129.7 | 8.03, br s | 133.9 | 7.96, d 2.0 | 131.9 | 7.94, s |
| 4 | 127.4 | 127.2 | 125.8 | 129.8 | ||||
| 5a | 24.2 | 2.70, d (15.5) | 24.6 | 2.66, d (16.0) | 33.3 | 3.17, d (16.5) | 26.7 | 2.65, d (16.0) |
| 5b | 3.16, dd (15.5, 12.0) | 3.25, dd (16.0, 11.5) | 4.01, d (16.5) | 2.84, dd (16.0, 12.5) | ||||
| 6 | 46.0 | 3.45, d (11.5) | 43.7 | 3.54, d (11.0) | 131.9 | 48.1 | 2.61, m | |
| 7 | 43.8 | 43.8 | 43.2 | 41.3 | ||||
| 8a | 36.0 | 1.91, m | 36.0 | 1.90, m | 31.8 | 1.54, m | 38.5 | 1.54, m |
| 8b | 1.96, m | 1.90, m | 1.91, m | 2.08, m | ||||
| 9a | 25.0 | 1.63, m | 25.0 | 1.59, m | 23.4 | 1.38, m | 24.1 | 2.19, m |
| 9b | 1.98, m | 1.96, m | 1.71, m | 2.41, m | ||||
| 10 | 67.2 | 2.83, d (5.0) | 67.4 | 2.71, d (9.5) | 61.9 | 2.97, dd (9.5, 5.5) | 63.8 | 4.59, dd (7.0, 2.5) |
| 11 | 64.1 | 64.6 | 73.0 | 76.1 | ||||
| 12a | 32.9 | 1.16, m | 32.2 | 0.88, m | 37.4 | 1.68, m | 26.4 | 1.81, m |
| 12b | 1.97, m | 1.92, m | 2.01, m | 2.47, m | ||||
| 13a | 28.9 | 1.96, m | 26.1 | 2.22, m | 30.8 | 2.10, m | 21.7 | 1.84, m |
| 13b | 2.05, m | 2.42, m | 2.22, m | 2.32, m | ||||
| 14 | 78.7 | 4.74, br d (11.0) | 80.4 | 4.80, dd (12.0, 2.0) | 66.8 | 4.00, dd (11.0, 3.0) | 70.5 | 5.15, d (6.5) |
| 15 | 144.3 | 66.9 | 82.4 | 74.5 | ||||
| 16 | 128.8 | 7.66, dd (8.5, 1.5) | 129.1 | 7.61, d (8.0) | 130.2 | 7.83, dd (8.0, 2.0) | 128.8 | 7.75, dd (8.5, 1.5) |
| 17 | 115.4 | 6.72, d (8.0) | 115.5 | 6.77, d (8.0) | 116.2 | 6.82, d (8.0) | 115.7 | 6.77, d (8.5) |
| 18 | 156.7 | 157.0 | 159.8 | 156.5 | ||||
| 19 | 146.2 | 146.2 | 132.8 | 137.0 | ||||
| 20a | 37.9 | 2.23, m | 37.8 | 2.30, m | 33.6 | 2.21, m | 119.9 | 5.17, m |
| 20b | 2.26, m | 2.30, m | 2.28, m | |||||
| 21a | 35.3 | 2.08, m | 35.1 | 2.09, m | 29.4 | 2.20, m | 34.4 | 2.53, m |
| 21b | 2.28, m | 2.29, m | 2.26, m | 2.57, m | ||||
| 22 | 63.3 | 4.61, dd (12.5, 4.0) | 62.9 | 4.96, dd (12.5, 4.0) | 60.8 | 4.26, dd (11.0, 4.0) | 60.0 | 4.29, dd (10.5, 5.5) |
| 23a | 109.5 | 4.76, s | 109.3 | 4.72, s | 20.5 | 1.90, s | 20.9 | 1.50, s |
| 23b | 4.81, s | 4.77, s | ||||||
| 24 | 17.3 | 0.96, s | 17.3 | 0.95, s | 23.5 | 0.73, s | 17.6 | 1.08, s |
| 25 | 17.1 | 1.34, s | 16.6 | 1.35, s | 17.3 | 1.27, s | 26.4 | 1.43, s |
| 26a | 111.6 | 4.90, s | 32.7 | 1.83, s | 27.6 | 1.86, s | 28.0 | 1.07, s |
| 26b | 5.02, s | |||||||
| 27 | 18.4 | 1.78, s | 29.7 | 1.78, s | 23.9 | 1.76, s | 26.5 | 1.08, s |
| OH | 5.29, br s | 5.36, br s | 5.57, br s | 5.33, br s | ||||
br = broad; s = singlet; d = doublet; dd = doublet of doublets; m = multiplet.
Bromophycolide S (2)
white, amorphous solid (1.2 mg, 0.0062% dry mass); [α]23 D +66 (c 0.076, MeOH); UV (MeOH) λmax (log ε) 260 (3.53) nm; IR (NaCl) νmax 3366, 2930, 1715, 1600, 1454, 1273, 1111 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, Table 1; 2D NMR data, Supporting Information; HRESIMS [M + Na]+m/z 605.0929 (calcd for C27H36Br2O4Na, 605.0878).
Bromophycolide T (3)
white, amorphous solid (0.58 mg, 0.0030% dry mass); [α]23 D +141 (c 0.039, MeOH); UV (MeOH) λmax (log ε) 260 (3.79) nm; IR (NaCl) νmax 3375, 2918, 1717, 1601, 1458, 1279, 1115 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, Table 1; 2D NMR data, Supporting Information; HRESIMS [M + Na]+m/z 605.0881 (calcd for C27H36Br2O4Na, 605.0878).
Bromophycolide U (4)
white, amorphous solid (0.95 mg, 0.0049% dry mass); [α]23 D +84 (c 0.063, MeOH); UV (MeOH) λmax (log ε) 260 (3.72) nm; IR (NaCl) νmax 3352, 2926, 1715, 1605, 1456, 1273, 1111 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz) data, Table 1; 2D NMR data, Supporting Information; HRESIMS [M + Na]+m/z 605.0871 (calcd for C27H36Br2O4Na, 605.0878).
Pharmacological Assays
The pharmacological assays were run as in our previous reports.4-8Antimalarial activity was determined with a SYBR Green-based parasite proliferation assay.7-9Antibacterial assays were performed using methicillin-resistant Staphylococcus aureus (MRSA, ATCC 33591) and vancomycin-resistant Enterococcus faecium (VREF, ATCC 700221) as test pathogens.4−7Antifungal assays were performed using amphotericin B-resistant Candida albicans (ATCC 90873).6−8Antitubercular activity was assessed against Mycobacterium tuberculosis strain H37Rv (ATCC 27294) using the microplate alamar blue assay (MABA).10Anticancer assays were conducted using 12 human cancer cell lines including breast (BT-549, DU4475, MDAMD-468, and MDA-MB-231), colon (HCT-116), lung (SHP-77 and A549), prostate (PC-3, LNCaP-FGC, and DU145), ovarian (A2780/ DDP-S), and leukemia (CCRF-CEM) cancer cell lines. In vitro cytotoxicity was tested by using MTS methods described previously.11
Supplementary Material
Acknowledgment
This research was supported by the U.S. National Institutes of Health’s International Cooperative Biodiversity Groups program (Grant No. U01 TW007401). We thank the Government of Fiji for the permission to perform research in their territorial waters and for permission to export samples. We especially thank the Roto Tui Serua and the people of Yanuca Island for facilitating this work. We thank M. Sharma and K. Feussner for extractions; T. Davenport and S. Engel for most antimicrobial assays; C. Redshaw for LC-MS assistance; M. C. Sullards and D. Bostwick for mass spectroscopic analyses; L. Gelbaum for NMR assistance; A. Bommarius and T. Rogers for use of their spectropolarimeter; and S. M. Brombosz and U. H. F. Bunz for help in use their IR spectrophotometer.
Supporting Information Available: COSY, HMBC, and NOE data tables and 1H and 13C NMR spectra for 1–4 are available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Littler DS, Littler MM. South Pacific Reef Plants. Offshore Graphics, Inc.; Washington, D.C.: 2003. p. 90. [Google Scholar]
- (2).Blunt JW, Copp BR, Hu WP, Munro MHG, Northcote PT, Prinsep MR. Nat. Prod. Rep. 2009;26:170–244. doi: 10.1039/b805113p. [DOI] [PubMed] [Google Scholar]
- (3).Stout EP, Kubanek J. Marine Macroalgal Natural Products. 2nd ed. Vol. 2. Elsevier; New York: 2010. ComprehensiVe Natural Products Chemistry. in press. [Google Scholar]
- (4).Kubanek J, Prusak AC, Snell TW, Giese RA, Hardcastle KI, Fairchild CR, Aalbersberg W, Raventos-Suarez C, Hay ME. Org. Lett. 2005;23:5261–5264. doi: 10.1021/ol052121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kubanek J, Prusak AC, Snell TW, Giese RA, Fairchild CR, Aalbersberg W, Hay ME. J. Nat. Prod. 2006;69:731–735. doi: 10.1021/np050463o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lane AL, Stout EP, Hay ME, Prusak AC, Hardcastle K, Fairchild CR, Franzblau SG, Roch KL, Prudhomme J, Aalbersberg W, Kubanek J. J. Org. Chem. 2007;72:7343–7351. doi: 10.1021/jo071210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lane AL, Stout EP, Lin A-S, Prudhomme J, Roch KL, Fairchild CR, Franzblau SG, Hay ME, Aalbersberg W, Kubanek J. J. Org. Chem. 2009;74:2736–2742. doi: 10.1021/jo900008w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lane AL, Nyadong L, Galhena AS, Shearer TL, Stout EP, Parry RM, Kwasnik M, Wang MD, Hay ME, Fernandez FM, Kubanek J. Proc. Natl. Acad. Sci. U.S.A. 2009;106:7314–7319. doi: 10.1073/pnas.0812020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob. Agents Chemother. 2005;49:1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lee FYF, Borzilleri R, Fairchild CR, Kim SH, Long BH, Reventos-Suarez C, Vite GD, Rose WC, Kramer RA. Clin. Cancer Res. 2001;7:1429–1437. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.