

Abstract
Proliferating germ cells in Caenorhabditis elegans provide a useful model system for deciphering fundamental mechanisms underlying the balance between proliferation and differentiation. Using gene expression profiling, we identified approximately 200 genes upregulated in the proliferating germ cells of C. elegans. Functional characterization using RNA-mediated interference demonstrated that over forty of these factors are required for normal germline proliferation and development. Detailed analysis of two of these factors defined an important regulatory relationship controlling germ cell proliferation. We established that the kinase VRK-1 is required for normal germ cell proliferation, and that it acts in part to regulate CEP-1(p53) activity. Loss of cep-1 significantly rescued the proliferation defects of vrk-1 mutants. We suggest that VRK-1 prevents CEP-1 from triggering an inappropriate cell cycle arrest, thereby promoting germ cell proliferation. This finding reveals a previously unsuspected mechanism for negative regulation of p53 activity in germ cells to control proliferation.
Keywords: C. elegans, Germ cells, Proliferation, VRK-1, Kinase, p53
Introduction
Germline stem cells balance the ability to self-renew with the ability to produce daughter cells that properly undergo meiosis and form gametes. The rate of germ cell proliferation must be coordinated with differentiation so that ample precursor cells are present to ensure appropriate numbers of mature gametes. Insufficient proliferation can result in sterility, while excessive or unregulated proliferation can result in tumorigenesis, as exemplified by testicular germ cell tumors, the most common cancer found in young men (Bosl and Motzer, 1997). An understanding of the fundamental mechanisms regulating germ cell proliferation is therefore relevant to both stem cell biology and cancer biology. Because these regulatory mechanisms have an instrumental role in homeostasis, they are highly likely to be conserved.
The nematode Caenorhabditis elegans provides an excellent model system to investigate how germ cell division is regulated. Although C. elegans germ nuclei are incompletely surrounded by the cell membrane and share a common cytoplasm, they exhibit mostly autonomous function, and are typically referred to as germ cells. These germ cells are arrayed in orderly distal-to-proximal fashion, with proliferating germ cells harbored in the distal region of the gonad, under the control of a somatic niche called the distal tip cell (DTC) (reviewed in (Byrd and Kimble, 2009)). As germ cells move away from the influence of the distal tip cell and toward the proximal gonad, they cease proliferation, enter into meiosis, and differentiate into gametes. The first wave of maturing germ cells differentiate into sperm at the L4 larval stage, while later germ cells mature into oocytes in the adult, permitting C. elegans to be a self-fertile hermaphrodite. Thus, how changes in proliferation rate affect germ cell differentiation into either sperm or oocytes can be assessed within the same animal.
In the adult hermaphrodite, the population of proliferating germ cells is maintained in large part by Notch signaling. The DTC expresses a ligand, LAG-2 (Delta), that interacts with the GLP-1(Notch) receptor present on the surface of the germ cells. This interaction results in the activation of downstream transcription factors to promote proliferation and/or prevent entry into meiosis (Petcherski and Kimble, 2000). A gain-of-function mutation, glp-1(oz112gf), causes germ cells to activate Notch signaling independently of LAG-2 and proliferate outside the niche. Thus a glp-1(oz112) mutant has a gonad filled with proliferating cells at the expense of meiotic and differentiating germ cells (Berry et al., 1997).
While the master regulators of the proliferation vs. meiotic entry decision have been well studied, surprisingly little is known about the basic mechanisms controlling germ cell division in C. elegans. Previous experiments have determined that germ cell divisions do not show any obvious asymmetry, although the length of the cell cycle does vary depending on the location of the germ cell in the gonad (Crittenden et al., 2006; Maciejowski et al., 2006; Jaramillo-Lambert et al., 2007). However, the mechanisms that control proliferation rates, and how those mechanisms might interface with the core cell cycle machinery, the decision to enter meiosis, or responses to environmental influences remain unknown.
Understanding how germ cells retain their proliferative capacity and flexibility in differentiation requires comprehensive knowledge of the genes expressed in these cells and how they function. To that end, we used microarray-based profiling of glp-1(oz112) mutants to identify genes preferentially expressed in proliferating germ cells, and performed an initial assessment of the function of these genes in germline development using RNAi. We then focused on developing a deeper understanding of the function of one of these genes, vrk-1, which encodes a conserved kinase with a fundamental but poorly understood role in cell cycle regulation. We discovered that VRK-1 activity is required for successful proliferation, and its loss results in diverse defects in the cell cycle. We also demonstrated that much of these defects are caused by the unregulated activity of cep-1, the C. elegans ortholog of p53. Our studies have therefore uncovered a novel mechanism for Vrk1 in regulating p53 activity in proliferating germ cells. Importantly, this regulation appears to be independent of DNA damage.
Methods
Strains and maintenance
C. elegans strains were maintained using standard techniques (Brenner, 1974). The C. elegans N2 strain was used as the wild type in addition to the following genetic backgrounds: LG I: cep-1(gk138), cep-1(lg12501), hus-1(op241), glp-4(bn2ts); LG II: vrk-1(ok1181), mIn1[mIs14 dpy-10(e128)]; LG III: unc-119(ed3), unc-32(e189), glp-1 (oz112), clk-2(qm37), ced-4(n1162), dpy-17(e164), mrt-2(ok1260); LG IV: fem-1(hc17), him-8(e1489); LG V: him-5, dpy-5(e61), him-5 (e1490). Transgenes: IsIs17[pGZ295: P-pie-1::gfp:pcna, pDP#MM051: unc-119(+)]; ltIs37 [pAA64: P-pie-1::mCherry::his-58, unc-119(+)]. For temperature-sensitive mutants, 15 °C or 20 °C was the permissive temperature and 25 °C was the restrictive temperature.
Microarray analysis
Animals homozygous for either unc-32(e189) alone or both unc-32 (e189) glp-1(oz112) were collected as young adults after being grown at 25 °C from the L1 stage. More than 50 animals of each genotype were collected from each of four independent sample preparations. RNA isolation and amplification was carried out as described elsewhere (Chi and Reinke, 2006). Microarrays carrying PCR products corresponding to approximately 90% of protein coding genes were hybridized, and Genepix software was used to capture hybridization intensities as described previously (Reinke et al., 2004). The data were analyzed as described previously (Reinke et al., 2004), with a cut-off of >1.8×, p<0.001 (Student’s t test) used to define the 202 genes with enriched expression in the glp-1(oz112) mutant relative to controls. Raw data are available in GEO at GSE21531.
RNAi screen
RNAi was performed based on the protocols of Maeda et al. (2001). PCR products representing each gene of interest were created using primers containing both gene-specific and T7 sequences. These products were then used as templates in a second round of PCR with T7 primers, in order to fully include T7 sequences in the PCR products. Standard PCR conditions and 35 cycles were used for both rounds. Second-round PCR products (4 μl of a 25 μl reaction) were used as templates in 20 μl in vitro transcription reactions using T7 RNA polymerase (New England Biolabs) in 96-well plates. The RNA was then precipitated using NaOAc and ethanol, dried thoroughly, and resuspended in 5 μl of RNAse-free water. PCR and RNA synthesis failures occurred for 36 genes, resulting in 166 of the 202 genes actually being analyzed. Worms at the L3 stage were starved for 2 h to remove bacteria from the gut, collected and washed in 0.1 M NaCl. Worms were counted, spun down, and resuspended in 2× soaking buffer [1×: 10.9 mM Na2HPO4, 5.5 mM KH2PO4, 2.1 mM NaCl, 4.7 mM NH4Cl, 3 mM spermidine, and 0.05% gelatin; (Maeda et al., 2001)]. Between 50 and 100 worms/well were then distributed to the wells of the plate containing the dsRNA, and incubated overnight at 20 °C. 50 μl of S basal containing DH5α bacteria (OD600=2.2), were added to each well and incubated at 20 °C for 36 h. Worms were then washed several times in S basal, then fixed in ice cold methanol in the 96-well plates. Fixed worms were then placed on a Fisher poly-lysine-coated slide, stained with DAPI, and mounted under a cover slip. Samples were viewed using a Zeiss Axioplan epifluorescence microscope with attached CCD camera.
BrdU detection
BrdU incorporation was assayed essentially as described by (Crittenden et al., 2006). Briefly, thymidine-deficient MG1693 Escherichia coli were cultured in 0.4% dextrose, 1 mM MgSO4, 1.25 μg/mL vitamin B1, 0.5 μM thymidine and 10 μM Bromodeoxyuridine (BrdU) (BD Pharmagen). Staged C. elegans hermaphrodites were fed BrdU-positive MG1693 on NGM plates supplemented with 100 μg/mL ampicillin. After at least 20 h, adults were dissected and gonads were fixed to poly-lysine-coated slides by the freeze-crack method. Slides were stored in methanol at −20 °C for up to 2 weeks before being soaked in PBS with 1 mg/mL BSA (1× PBSB) for 20 min, 1% paraformaldehyde in PBSB for 30 min, 2 N HCl for 10 min, and 0.1 M Sodium Borate (B4Na2O7), pH 8.5 for 10 min before being washed twice with PBSB for 15 min each. Slides were incubated with mouse FastImmune anti-BrdU with DNase (Becton Dickinson) overnight. Slides were washed once in PBS for 15 min before being stained with 0.5 μg/mL DAPI. After two 15-min washes in PBS, slides were incubated with a fluorescent secondary antibody (1:500, Molecular Probes). Slides were washed another two times in PBS for 15 min each, mounted in DABCO (Sigma), and sealed under a coverslip.
Immunohistochemistry
Gonads were dissected from animals and fixed as previously described (Golden et al., 2000; Seydoux and Dunn, 1997; Strome and Wood, 1982). The following antibodies and dilutions were used: rabbit polyclonal anti-histone H3 phospho-S10 (1:200) (Upstate) and rabbit polyclonal anti-VRK-1(1:200) (gift from P. Askjaer). Samples for immunohistochemistry with anti-VRK-1 were fixed using standard methods (as used for anti-pH3), except no paraformaldehyde was used. Samples were incubated at room temperature for 2–3 h with a fluorescent secondary antibody (1:500, Molecular Probes). Slides were mounted with DABCO (Sigma). Slides were viewed using a Zeiss Axioplan 2 epifluorescence microscope.
Transgene production
The genomic region containing both the coding and 3′ UTR sequences of vrk-1 was PCR amplified and cloned in frame downstream of GFP into pID3.01, which contains the pie-1 promoter, gfp, the pie-1 3′UTR and a rescuing unc-119 allele. The construct was bombarded into unc-119(ed3) animals by microparticle bombardment as described (Praitis et al., 2001). unc-119(ed3) worms were grown for bombardment at 23 °C and transformants were selected at 25 °C. Lines bearing 100% non-Unc animals, presumed to be integrated, were examined for GFP expression.
Results
Identification of transcripts enriched in proliferating germ cells
We took a functional genomics approach to investigate the properties of C. elegans proliferating germ cells in vivo. We used whole-genome DNA microarrays to directly compare the gene expression profiles of adult wild type animals to glp-1(oz112) animals, which have ectopically proliferating germ cells. This analysis identified 202 genes with increased expression in glp-1(oz112) animals (>1.8-fold, p<0.01; based on four independently prepared sets of samples) (Supplemental File 1).
To exclude the possibility that upregulation of these 202 genes was a non-specific consequence of ectopic glp-1 activity, we investigated whether they are expressed in normally proliferating germ cells. First, we examined the expression profiles of these genes in independent microarray analyses of germline development, which have defined sets of genes with enriched expression in undifferentiated germ cells in both hermaphrodites and males, as well as during gametogenesis (Reinke et al., 2004). This study also includes a temporal expression analysis in wild type animals from the L3 larval stage to adult, when the germ line is undergoing dynamic development. Genes expressed in proliferating germ cells should be expressed in both males and hermaphrodites, prior to spermatogenesis.
We found that 80% of the 202 candidate factors were previously shown to have enriched expression in the hermaphrodite germ line relative to the soma (5.7× over-representation) (Fig. 1A). Moreover, 46% of the candidates have strongly enriched expression in the male germ line (15× over-representation), and many other candidates show at least moderate expression in the male germ line (Fig. 1A). Almost none have enriched expression in cells undergoing differentiation into sperm, suggesting that their enrichment in both the male and hermaphrodite germ line is not due to a role in spermatogenesis. (Their apparent increased expression in oocytes could be due to the fact that, in this comparison, the strain bearing sperm has a decreased amount of immature germ cells compared to the strain bearing oocytes; see Reinke et al (2004) for detailed explanation.) Additionally, the temporal expression profile of these genes indicates that many are initially expressed during the larval stages, when the germ line is primarily composed of proliferating, non-differentiating germ cells (Fig. 1B).
Fig. 1.
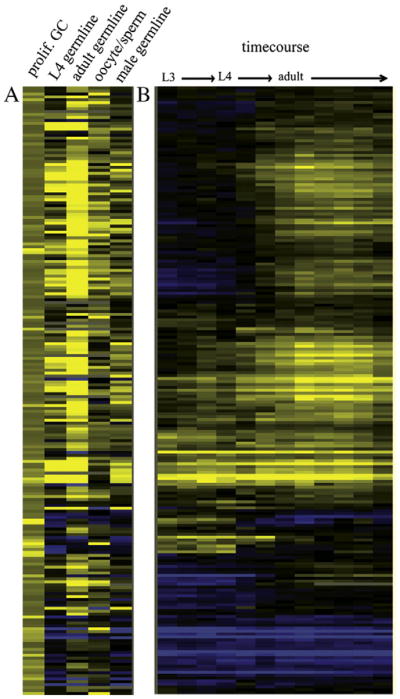
Identification of genes expressed in proliferating germ cells. (A) A clustergram of the 202 genes with expression >1.8×, p<.01 in glp-1(gf) animals relative to wild type (left column, prolif. GC), using microarray data from Reinke et al. (2004) to further characterize the expression profiles of these genes in germline development (all columns to the right). Each column header describes the stage and/or tissue in which expression of that gene is expected. Yellow indicates higher expression, blue indicates lower expression. For the oocyte/sperm comparison, yellow indicates high expression in oocytes, blue is high expression in sperm. (B) The developmental expression in whole animals of the same 202 genes. Data also from Reinke et al. (2004).
Second, we examined spatial expression patterns for the 202 candidate factors. Of the 87 genes for which in situ hybridization patterns are available (http://nematode.lab.nig.ac.jp/), 81 have detectable expression in the distal germ line in the adult, and 65 of these are detectably expressed in the larval germ line as well (Supplemental File 1). Forty of these genes show strong staining in the distal germ line that diminishes in the proximal germ line, suggesting that these mRNAs are not retained or expressed during oogenesis. As a control, we determined the frequency of such a distal-restricted expression pattern more generally among germline-expressed genes, by examining in situ hybridization images for 100 genes expressed in the germ line but not enriched in our glp-1(gf) microarray analysis. We saw a distal-restricted germline expression pattern in adults in only six. Thus, our experiments identifying factors expressed in proliferating germ cells enrich for genes preferentially expressed in the distal germ line relative to the germ line as a whole.
In sum, of the 202 candidate factors, 180 are analyzed in independent microarray and/or in situ analysis datasets. Of these, 165 (91%) are normally expressed in the distal wild type hermaphrodite germ line, where proliferating germ cells reside. Thus, the expression profiles and patterns in wild type animals indicate that most of these genes are normally expressed in the right time and place to function in germ cell proliferation, and their increased expression in the glp-1(oz112) mutant is not simply an artifact of ectopic GLP-1 activity. However, we do not exclude the likely possibility that many of these candidates are also expressed in other cell types, based on their predicted functions.
Candidate factors encode diverse regulatory proteins
Using BLAST, we examined conservation of the 202 candidates with human genes and found that 52% have significant conservation (based on a value of≤e−10). Many of the highly conserved genes are unstudied in both mammals and worms, indicating that investigation of these novel genes could yield further insights into conserved mechanisms governing germ cell proliferation.
We next examined the predicted protein functions of the candidate factors (Fig. 2; Supplemental File 1). Approximately 16% of the transcripts enriched in proliferating germ cells encode proteins predicted to function in RNA metabolism, as they contain RNA binding, RNA processing, and/or RNA helicase domains. A network of proteins involved in RNA control mechanisms that includes FBF-1/2, GLD-1, GLD-2, and GLD-3 mediate the balance between stem cell self-renewal and entry into meiosis (reviewed in Crittenden et al., 2003). Some of the candidate RNA metabolism proteins we identified could also be playing a similar role. An additional 10% of the candidates encode proteins predicted to localize to the nucleolus and affect ribosome biogenesis or other nucleolar functions. Most of the predicted nucleolar factors that we have identified (e.g. nst-1, npg-1, lpd-7) are likely to play regulatory, rather than structural, roles in ribosome biogenesis and nucleolar organization, based on their functional domains and known roles of orthologs in yeast.
Fig. 2.
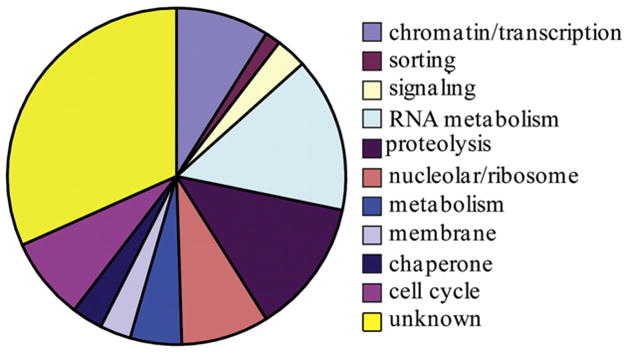
Functional categories of genes expressed in proliferating germ cells. Pie chart showing the predicted functions of the 202 genes. See sheet two of Supplemental File 1 for assignment of functional categories to individual genes.
Several candidates (13%) are predicted to encode factors that function in proteasome-mediated protein degradation, including several containing BTB (Pintard et al., 2003; Xu et al., 2003) or F-box domains that act as specificity factors for degradation by various cullin-containing complexes, as well as ubiquitin and proteasome components.
Nine percent of the factors are predicted to affect gene expression at the level of transcription, including proteins with C2H2 zinc finger domains or domains found in components of the basal transcription machinery. Five encode SET or chromo domains; these factors are predicted to affect chromatin status and could regulate local or global gene expression, or direct the location of origins of replication (Aggarwal and Calvi, 2004). Of particular interest, we found that the transcription factor cep-1, the ortholog of the tumor suppressor protein p53, has enriched expression in proliferating germ cells.
Finally, the candidate factors also include several with previously identified roles in germ cell proliferation, such as puf-8, fbf-1/fbf-2, smc-4, and gpr-1/2 (Crittenden et al., 2003; Hagstrom et al., 2002; Srinivasan et al., 2003; Subramaniam and Seydoux, 1999), as well as a few genes predicted by homology to function in cell division, such as cdc-6 and rfc-4. Included in this group is the gene encoding the vaccinia-related kinase, VRK-1. VRK-1 orthologs have important roles in mitosis and meiosis in several organisms, presumably through their ability to phosphorylate multiple proteins implicated in cell cycle regulation, including histone H2A, the nuclear envelope protein BAF, and several transcription factors including p53. The function of this kinase has been implicated in germline development in C. elegans (Klerkx et al., 2009), but the mechanism of its involvement is not well understood.
An RNAi screen of candidate genes highlights the importance of ribosome biogenesis in germ cell proliferation
To identify which factors might be required for germ cell function, we performed a systematic RNAi screen of 166 of the 202 genes, in which we delivered dsRNA to early third-stage (L3) larvae by soaking. After recovery and growth to adulthood, we scored plates of animals for highly-penetrant sterility (>80%). This experimental design allowed us to avoid potential embryonic lethality or larval arrest and focus on adult germline phenotypes.
A total of 39 genes reproducibly gave sterile phenotypes upon functional depletion (Supplemental File 2). We fixed affected animals and stained with DAPI to visualize germ cell nuclei using fluorescence microscopy. For each gene, we collected multiple digital images and examined them for germline defects, including under-proliferation, aberrant gamete differentiation and abnormal nuclear morphology (examples in Fig. 3). Functional depletion of the vast majority of these genes consistently produced a gonad with reduced germ cell number relative to wild type, suggestive of defects in cell proliferation. In most cases, the underproliferation phenotype was accompanied by defects in the nuclear morphology of both mitotic and early meiotic (pachytene) germ cells, as well as defects in gametogenesis, described below. The meiotic and gametogenesis defects could be a downstream consequence of the effects of reduced gene product in proliferating cells.
Fig. 3.

Example germline RNAi phenotypes for proliferating germ cell factors. Whole animal DAPI staining showing one gonad arm from a wild type control and six animals undergoing RNAi for genes from different functional categories (nucleolar: C15H11.9, E02H1.1, K12H4.3; unknown: C50B8.3, Y47G6A.9; and chaperone: daf-21). Visible region of gonad outlined in white; asterisk indicates distal tip. Scale bars represent 20 μm.
Defects in both types of gametes were apparent for many genes. In DAPI-stained wild type animals, sperm nuclei are distinctive by their extremely tightly condensed nuclei while oocytes are easily distinguished by the presence of sets of bivalent chromosomes in the proximal gonad. Examination of nuclear morphology showed that RNAi of virtually every gene resulted in an absence of oocytes; however, sperm were present in most cases, although clearly abnormal or incompletely differentiated in some. Oogenesis could be more severely affected because it occurs subsequent to spermatogenesis, after RNAi had more time to take effect, or because RNAi of these factors specifically causes defects in regulating the switch from spermatogenesis to oogenesis. Indeed, RNAi of several genes produced a phenotype of excess sperm, similar to that seen in animals lacking FBF, a known stem cell factor that also regulates the sperm/oocyte switch (Crittenden et al., 2003; Zhang et al., 1997). In summary, we identified 39 factors that appear to influence the balance between proliferation and differentiation in germ cells. In many cases, loss of the corresponding gene product affects both proliferation and differentiation.
Several genes with known mutant phenotypes in the germ line were not captured in our RNAi screen, most likely because the application of RNAi at the L3 stage was too late to cause effective functional depletion by young adulthood. The majority of the genes that did produce phenotypes are implicated by homology or other functional analysis in ribosome biogenesis. The preferential identification of this class of genes in the RNAi screen is presumably because the germ line is extremely sensitive to alterations in translation capacity. While an interesting observation, it is difficult to distinguish the essential role of ribosome biogenesis in maintaining living cells from specific functions these factors might have in germ cell proliferation, and we elected not to pursue further functional analysis of these genes.
We therefore decided to instead focus on increasing our understanding of how the kinase VRK-1 controls germ cell proliferation. We were particularly interested in this question because the glp-1(gf) expression data indicated that both vrk-1 and cep-1(p53) are expressed in proliferating germ cells. Previous work in mammals had demonstrated that p53 was a phosphorylation substrate of Vrk1 (Lopez-Borges and Lazo, 2000). In mammalian cells, p53 levels are low unless cells undergo DNA damage, which promotes the stabilization and activation p53. Activated p53 induces a cell cycle arrest in proliferating cells (Boehme and Blattner, 2009). Similarly, C. elegans p53, CEP-1, also can initiate a DNA-damage-induced cell cycle arrest in proliferating germ cells (Derry et al., 2007). Unlike mammalian systems however, CEP-1 is present at relatively high levels in proliferating germ cells in the absence of DNA damage (Schumacher et al., 2005). The continued proliferation of these cells despite high levels of CEP-1 indicates that CEP-1 is unable to induce a cell cycle arrest. We therefore speculated that VRK-1 might negatively regulate CEP-1 activity post-translationally to permit germline proliferation in C. elegans.
Loss of vrk-1 causes defects in germ cell number and morphology
To begin to test this hypothesis, we first investigated the function of vrk-1 in germ cell development, taking advantage of a mutant allele, vrk-1(ok1181). This mutant lacks a large portion of the open reading frame, and the truncated protein is not detectable by immunoblot analysis (Klerkx et al., 2009). The two most obvious defects of vrk-1 (ok1181) adults are vulva malformations and sterility. While the vulval defects have been analyzed in detail (Klerkx et al., 2009), the mechanism of sterility has not. We first dissected gonads from wild type and vrk-1 mutant adult hermaphrodites and directly examined the number and morphology of germ cells (Fig. 4A). At this stage, germ cells in vrk-1 mutants were vastly decreased in number compared to wild type. Moreover, the DNA morphology was abnormal and appears both condensed and also somewhat fragmented. We did not see nuclei with recognizable chromosome morphology corresponding to the pachytene, diplotene, or diakinesis stages of meiosis I. Moreover, while sperm were often present in the proximal gonad, oocytes were not. In males, similar reductions in germ cell number were also apparent. These defects can be attributed to loss of vrk-1 function because similar defects are also seen when RNAi is used to deplete vrk-1 levels post-embryonically (data not shown). Additionally, performing vrk-1(RNAi) in an rrf-1 mutant background, which restricts RNAi to the germ line, resulted in germ cell defects that are as extensive as those seen in vrk-1(ok1181) mutants (Fig. 4B). This result indicates that vrk-1 primarily functions within the germ line, rather than the soma, to ensure normal germ cell number and morphology.
Fig. 4.

Loss of vrk-1 disrupts germline development. (A) Gonads dissected from wild type and vrk-1 mutant adult animals were stained with DAPI. Chromatin morphology is magnified in bottom panel to show condensed chromatin in vrk-1 mutant germ cells. Scale bars represent 20 μm. (B) Gonads dissected from rrf-1 mutants undergoing either control (L4440) or vrk-1 RNAi, and stained with DAPI. Scale bars same as in A. (C) The average number of germ cells per gonad arm was quantified in developing hermaphrodites from wild type and vrk-1 mutants. Error bars represent standard deviation. Student’s t test p values for L3, L4, and adult data are <2.0×10−5 (asterisks).
We next wished to determine the developmental stage at which the decreased cell number was first apparent. We counted the number of germ cells per gonad arm at multiple stages of larval development in wild type and vrk-1(ok1181) mutants (Fig. 4C). Slight differences in germ cell number were seen as early as the L3 stage, with significant differences apparent by the L4 stage. The slight increase in germ cell number in vrk-1 mutants between L4 and adult is likely due to increasing DNA fragmentation resulting in extra cell counts, as evidence presented below indicates that proliferation ceases prior to the adult stage.
Reduced proliferation, not apoptosis, contributes to vrk-1(ok1181) germ cell defects
Two possible explanations could account for the germ cell defects seen in the vrk-1(ok1181) mutant, either increased apoptosis or decreased proliferation. These two possibilities are not necessarily mutually exclusive. We first tested whether apoptosis contributed to the decreased germ cell number of vrk-1 mutants. In wild type animals, apoptosis occurs in a subset of germ cells at the pachytene stage of meiosis I due to physiological cell death (Gumienny et al., 1999), resulting in a background level of approximately five SYTO12-positive apoptotic cells per germ line. However, in vrk-1 mutants, we rarely detected even single apoptotic cells (Fig. 5A). Moreover, when germ cells were prevented from undergoing apoptosis by crossing a mutation in the pro-apoptotic gene ced-4 into the vrk-1 background, we did not observe any rescue of the vrk-1 mutant phenotype (Fig. 5B). These data indicate that apoptosis plays no significant role in the reduction of germ cell number seen in vrk-1 mutants.
Fig. 5.

Apoptosis does not account for the vrk-1 mutant germ cell defects. (A) SYTO-12 staining was quantified in wild type and vrk-1 mutant germ cells. Error bars represent standard error values. Student’s t test p<2.0×10−5 (asterisks). (B) Dissected gonads from ced-4, vrk-1, and ced-4;vrk-1 mutants were stained with DAPI. Gonads outlined in white. Scale bars represent 20 μm.
To examine proliferation, we used an antibody to Ser10-phosphorylated histone H3 (pH3) to identify cells in early M phase in wild type and vrk-1(ok1181) mutant gonads. Quantification of pH3-positive cells at both L4 and young adult stages showed a significant decrease in the number of cells in mitosis in the vrk-1 mutant (Fig. 6A). Moreover, the failure of pH3 to accumulate in vrk-1 mutant germ cells indicates that the condensed germ nuclei are not uniformly arrested in the early stages of M phase, despite their compact morphology.
Fig. 6.
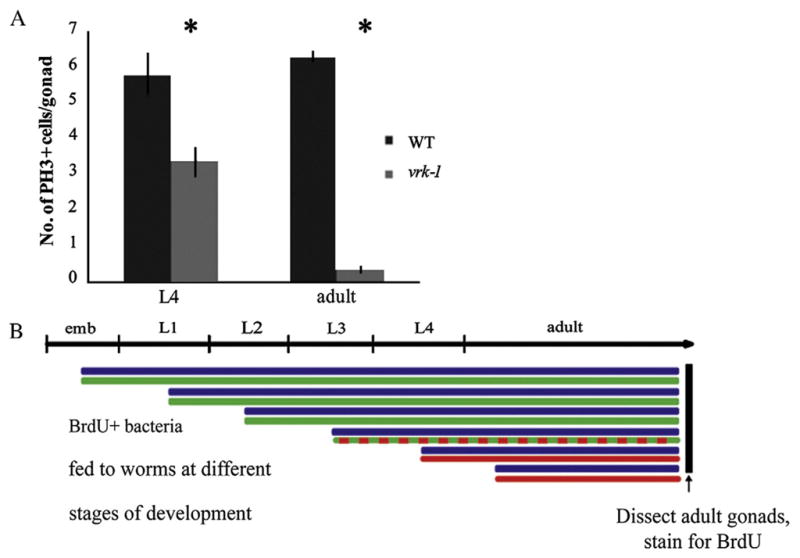

Proliferation is reduced in the germ line of vrk-1 mutants. (A) The number of nuclei in mitosis was determined by counting pH3-positive nuclei in dissected gonads from wild type and vrk-1 mutants (n>20). Error bars represent standard error values. Student’s t test p values are <0.005 (asterisks). (B) BrdU-positive E. coli were fed to hermaphrodites at progressively later stages of development (blue lines). Green lines indicate stage at which BrdU was first fed and successfully incorporated in the vrk-1 mutant, while red indicates the stages at which feeding commenced but BrdU was no longer incorporated. (C) Gonads from adult wild type (left column) and vrk-1 mutant (right column) hermaphrodites exposed to BrdU-positive bacteria at each developmental stage were dissected and stained with antibodies against BrdU (red) and DAPI (DNA). Total number of gonads analyzed, and percentage with BrdU incorporation, is listed in the bottom right corner of each panel. Scale bars represent 20 μm.
We next wanted to determine when during development the proliferation defects first begin. A timed BrdU incorporation assay allows the tracking of S phase at different times in development, depending on when BrdU is first provided (Crittenden et al., 2006). We fed bacteria containing BrdU to wild type and vrk-1 mutant animals at successive stages from L1 through adult, as outlined in Fig. 6B. The extent of BrdU incorporation was determined by staining dissected gonads in adult animals with antibodies to BrdU. Wild type germ cells in the distal region displayed extensive BrdU incorporation, regardless of when BrdU was first delivered (Fig. 6C, left column). By contrast, vrk-1 mutant germ cells efficiently incorporated BrdU only if exposure occurred at or prior to the L3 stage (Fig. 6C, right column). Exposure to BrdU at the L4 or adult stages, resulted in few, if any, cells staining, and even at the L3 stage, the extent and intensity of staining was decreased relative to wild type. Thus, our analysis of S phase points to a cell division rate that begins to slow as early as the L3 stage, and ceases almost entirely by the L4 stage in vrk-1 mutants.
To further investigate the cell cycle defects in vrk-1 mutants, we followed GFP:PCNA localization during the onset of the vrk-1 mutant phenotype, from L3 through adulthood (Fig. 7). We chose to monitor PCNA because it displays differential localization at different phases of the cell cycle: it is found in the nucleus during interphase, and dissipates into the cytoplasm upon nuclear envelope breakdown (Brauchle et al., 2003). The first obvious defects in vrk-1 mutants at L3 were enlarged nuclei and a higher incidence of PCNA-positive nuclei relative to wild type. These defects suggest that the cells have difficulty progressing through interphase and might be delayed in S phase, when DNA replication could account for the enlarged nuclei. In L4 and adult vrk-1 mutants, although some enlarged PCNA-positive nuclei were still present, there was an increasing accumulation of highly condensed, fragmented nuclei in L4 and adult animals that do not stain with PCNA. There are two possible explanations for these observations. The enlarged nuclei could eventually resolve into condensed nuclei over time. Alternatively, the different phenotypes could reflect different levels of vrk-1 activity. At early stages, vrk-1 mutant gonads might still have some residual maternal vrk-1 activity (although it is sub-threshold), causing the cells to arrest as enlarged PCNA-positive nuclei, whereas at later stages, vrk-1 activity is lower and the nuclei adopt a hyper-condensed, PCNA-negative conformation.
Fig. 7.

vrk-1 mutant germ nuclei have defects in both mitosis and interphase. A transgenic strain expressing both GFP:PCNA (green) and H2B:mCherry (pink) to mark chromatin was crossed to vrk-1 mutants. Live animals were mounted for examination of GFP:PCNA localization at L3, L4, and adult. Gonads are outlined in white, and the distal tip is indicated by an asterisk. The far right panels are insets from the adult stage, indicated by the dotted square. In the vrk-1 mutant germ line, examples of enlarged interphase cells are indicated by arrows, whereas a nucleus with more condensed DNA is marked by an arrowhead, in the inset. Scale bars represent 20 μm.
VRK-1 is broadly expressed in germ cells and displays cell cycle-dependent subcellular localization
We next examined VRK-1 localization in the wild type germ line using an antibody raised against VRK-1 (Klerkx et al., 2009). We found that VRK-1 was detectable in the nuclei of germ cells at all stages of progression from the mitotic zone to mature oocytes, except in maturing spermatids (Fig. 8A). This broad expression is surprising given its role in dividing cells. However, VRK-1 could have a function in meiosis that we are unable to assay because the mitotic defects preclude analysis of later stages. Alternatively, VRK-1 expression may persist into oocytes to promote cell division in the early embryo (Gorjanacz et al., 2007).
Fig. 8.

VRK-1 is expressed in the nuclei of almost all germ cells. (A) Gonads dissected from adult wild type animals were stained with DAPI (blue) to visualize DNA and antibodies against VRK-1 (red). The gonad is outlined in white. Spermatids lacking VRK-1 staining are indicated with an arrow, and the distal tip is marked by an asterisk. The scale bar represents 20 μm. (B) High power images of the distal region of adult gonads (white outline) expressing functional GFP:VRK-1 (green) and H2B:mCherry (red). Arrow shows discrete puncta of H2B:mCherry and GFP:VRK-1 colocalization. The arrowhead points to a ring of GFP:VRK-1 surrounding the DNA, presumably the nuclear envelope. Scale bars represent 5 μm.
To better follow subcellular localization of VRK-1 in living animals, we generated a GFP:VRK-1 fusion protein expressed specifically in the germ line under the pie-1 promoter. This strain rescued vrk-1 mutants to fertility, indicating that the fusion protein is functional. The brood size was about 16% of wild type, likely because vulval defects, which limit progeny number, are not rescued by the expression of VRK-1 exclusively in the germ line. We found that VRK-1 is largely nuclear, based on co-localization with H2B:mCherry, throughout most of the cell cycle (Fig. 8B). On occasion it appears concentrated at chromatin-associated foci at the nuclear periphery during interphase; we do not know the function or genomic location of these foci. Additionally, we saw VRK-1 localized to structures resembling the nuclear envelope in cells that by DNA morphology appear to be in early prophase. Overall, VRK-1 localization in germ cells displays similar patterns of cell cycle-associated localization as described for early embryos (Gorjanacz et al., 2007).
The VRK-1 phenotype is modified by CEP-1/p53
Vrk1-mediated phosphorylation of the tumor suppressor and transcriptional activator p53 in mammalian cell culture stabilizes p53 by blocking association of the degradation factor MDM2 (Lopez-Borges and Lazo, 2000). However, whether p53 function is activated or inhibited by this phosphorylation event has not been assessed in vivo. Because VRK-1 and CEP-1 are both expressed in C. elegans proliferating germ cells (Fig. 8A; Schumacher et al., 2005), we hypothesized that VRK-1 might inhibit the cell cycle arrest activity of CEP-1, thereby permitting germ cell proliferation.
To test this hypothesis in vivo, we constructed a strain carrying both vrk-1(ok1181) and the loss-of-function allele cep-1(gk138). cep-1 (gk138) mutants do not display obvious defects under normal conditions, but fail to undergo a cell cycle arrest in germ cells exposed to DNA damage (Derry et al., 2007; Derry et al., 2001). When we examined adult cep-1; vrk-1 gonads, we saw a substantial increase in germ cell number relative to vrk-1 mutants (Fig. 9A). Many cep-1;vrk-1 germ cells had apparently entered into the pachytene stage of meiosis I (Fig. 9B), and in some cases, nuclei exhibited a diakinesis chromosome morphology indicative of early oogenesis. Moreover, the proliferation rate was increased in both larvae and adults, based on pH3 staining and BrdU incorporation assays (Fig. 9C, D). Ultimately, however, these animals were still sterile, indicating that cep-1; vrk-1 mutant germ cells have additional defects that prevent normal fertilization. Thus, reduced cep-1 activity partially rescued the vrk-1 phenotype. These data are consistent with a role for VRK-1 in inhibiting CEP-1 activity.
Fig. 9.

Loss of cep-1 rescues germline proliferation defects of vrk-1 mutant hermaphrodites. (A) DAPI-stained gonads from adult wild type, cep-1, vrk-1, and cep-1;vrk-1 hermaphrodites. Scale bars represent 50 μm. (B) High power view showing chromatin morphology. Scale bars represent 5 μm. (C) The number of pH3-positive nuclei in adults was quantified for each genotype (n>20/genotype). Student’s t test p values between cep-1 and cep-1;vrk-1 backgrounds are not significant (NS); p value between vrk-1 and cep-1;vrk-1 backgrounds is <1.8×10−9. (D) Wild type, cep-1, vrk-1 and cep-1;vrk-1 mutants were exposed to BrdU at the L4 stage and stained for BrdU incorporation (red) as adults. DNA was stained with DAPI (blue). Scale bars represent 10 μm.
To confirm the interaction between vrk-1 and cep-1, we tested whether a second allele of cep-1, lg12501 (Schumacher et al., 2005), could also rescue the vrk-1 phenotype. The lg12501 allele is predicted to include more of the DNA binding domain of CEP-1 than gk138, and might retain some function that is lost by the gk138 allele. In contrast to gk138, the lg12501 allele of cep-1 did not display significant rescue; germ cell morphology of the cep-1(lg12501); vrk-1(ok1181) mutant looked virtually identical to vrk-1(ok1181) alone (Fig. 10A). Although it is possible that the background of one of the two alleles might contain an independent mutation that affects the vrk-1 mutant phenotype, extensive backcrossing of both cep-1 alleles did not alter their respective ability to rescue. To distinguish whether background differences or allele differences account for the different rescue potential of gk138 and lg12501, we constructed a trans-heterozygous strain, cep-1(gk138)/cep-1(lg12501);vrk-1(ok1181). With this strain, we saw intermediate levels of rescue (Fig. 10B). As a heterozygote, cep-1(gk138)/+ did not display any rescue, suggesting that the background of the gk138 allele did not contribute significant rescuing ability (Fig. 10C). Thus, these data indicate that functional differences exist between the two alleles of cep-1, which could account for the discrepancy in their ability to rescue the vrk-1 mutant phenotype. Possibly, the cep-1(lg12501) allele includes enough of the DNA binding domain to retain some residual function. Overall, our results are consistent with a severe loss of cep-1 activity partially rescuing germ cell proliferation in the vrk-1 mutant.
Fig. 10.

A trans-heterozygous cep-1 mutant partially rescues the vrk-1 mutant phenotype. DAPI staining of dissected gonads from hermaphrodite adults of the genotype indicated in each panel. Gonads are outlined in white and regions within dashed boxes are magnified in the right column. (A) cep-1(lg12501) did not improve the germline size or DNA morphology of vrk-1 mutants. (B) A trans-heterozygous cep-1(lg12501)/cep1(gk138) background displayed partial rescue of vrk-1 germline defects. (C) A single copy of cep-1 (gk138) in a heterozygous background failed to rescue vrk-1 mutant defects. Scale bars represent 20 μm.
Finally, we tested whether the relationship between vrk-1 and cep-1 was an indirect consequence of the vrk-1 mutant phenotype. Possibly, the chromosomal defects induced in vrk-1 mutants are interpreted by the cell as DNA damage, which would then signal through the canonical DNA damage signaling pathway to trigger activation of CEP-1 and induce a cell cycle arrest. If true, then vrk-1 mutant germ cells that could not detect DNA damage would fail to initiate a cep-1-dependent cell cycle arrest. We therefore examined whether disrupting the DNA damage-sensing pathway upstream of cep-1 could also rescue the vrk-1 phenotype. We created strains containing mutations in both vrk-1 and one of three genes each required for transmitting the DNA damage signal to cep-1. We found that hus-1;vrk-1, vrk-1;clk-2, and vrk-1;mrt-2 mutant germ cells all displayed defects equally severe as those seen in vrk-1 mutants (Fig. 11). This result indicates that the rescue of the vrk-1 phenotype by cep-1 is not an indirect consequence of triggering the DNA damage pathway, and instead argues for a more direct functional relationship between vrk-1 and cep-1.
Fig. 11.

The DNA damage checkpoint is not activated in vrk-1 mutants. Whole animals were fixed and stained with DAPI to visualize DNA. The vrk-1 mutant germ line phenotype is not improved by the loss of each of the required members of the DNA damage checkpoint pathway: mrt-2, clk-2, or hus-2. The cep-1; vrk-1 mutant is shown as a control for rescue of the vrk-1 mutant phenotype. Gonads are outlined in white; asterisks indicate the distal tip in each image.
Discussion
Understanding how germ cell proliferation is controlled in order to balance cell number with differentiation rates requires a comprehensive knowledge of the genes involved in this process. Here we have performed a global expression analysis to identify genes with enriched expression in proliferating germ cells, followed by an initial functional characterization of their role in germline development. From these data, we then hypothesized and demonstrated a negative regulatory relationship between the vaccinia-related kinase vrk-1 and the tumor suppressor p53 in controlling germ cell proliferation. Thus our work provides both a broad overview and mechanistic knowledge about key pathways controlling germ cell proliferation.
Our genome-wide analysis of gene expression in glp-1(gf) animals identified 202 genes with higher expression in gonads with excess proliferating germ cells. Because these profiling experiments were carried out in a mutant with ectopic GLP-1(Notch) activity, we carefully examined whether these genes were normally expressed in proliferating germ cells by a combination of temporal and spatial expression data from other studies. Most tellingly, the in situ hybridization database provided independent evidence that many of these genes are indeed preferentially expressed in the proliferating population of germ cells in the distal germ line, compared to the proximal germ line, in wild type animals. Intriguingly, most of these genes do not seem to encode core components of the cell cycle machinery, but rather proteins involved in various aspects of RNA regulation, including translation, as well as in the regulation of proteolysis. Many genes involved in the cell cycle were not identified in our analysis, most likely because they are required for embryonic as well as germ cell divisions, and are expressed in the proximal as well as the distal germ line, whereas our glp-1(gf) microarray experiments preferentially enriched for genes expressed in the distal germ line.
We followed up our expression analysis with an RNAi screen of many of these candidates, and identified 39 genes required for normal germ cell development. A potential caveat to this screen is that, due to the relatively late timing of dsRNA delivery (L3), we did not fully deplete target gene products. The extent of reduction in transcript levels, the half-life of pre-existing protein product, and sensitivity of cells to reduced levels of gene products are highly variable between target genes. Perhaps because of their high expression and fundamental importance to cell viability, the vast majority of factors that displayed phenotypes in the RNAi screen encode proteins implicated in ribosome biogenesis. Previous studies have linked the mutation of specific regulators of ribosome biogenesis to defective germ cell proliferation (Killian and Hubbard, 2004; Kudron and Reinke, 2008; Voutev et al., 2006), suggesting that both the somatic gonad and the germ line are especially sensitive to changes in ribosome production. In support of this possibility, microarray experiments directly comparing mitotic and meiotic germ cell populations in dissected distal gonad tips identified genes encoding factors regulating translation, including essentially all ribosome components, as strongly enriched in the mitotic germ cells (W. Chi and V.R., unpublished). Thus, proliferating germ cells highly express the core translation machinery. One possible reason for this high level of expression is to replace ribosomes inherited from the mother, either to improve translation efficiency in the germ line, or possibly to alter ribosome composition to accommodate germline-specific translational regulatory mechanisms. Alternatively, these cells express high levels of the key components of ribosome biogenesis in order to load large amounts of ribosomes into progeny via the oocyte.
Since our primary focus in conducting these studies was to better understand the specific regulatory mechanisms controlling germ cell proliferation, we decided to investigate vrk-1, which encodes an intriguing kinase implicated in proliferation in other systems. In particular, Drosophila mutants lacking NHK-1, the ortholog of VRK-1, have mitotic and meiotic defects in the germ line, including chromosome hyper-condensation and lagging chromosomes (Cullen et al., 2005; Ivanovska et al., 2005). In C. elegans, VRK-1 has been studied in embryos and during vulval development, but not in germ cell proliferation. Our analysis of the C. elegans vrk-1 mutant phenotype in germ cells reveals that proliferation is indeed abnormal beginning at the third larval stage, in the absence of any increase in apoptosis. The fact that the mitotic marker pH3 does not accumulate in mutant germ cells indicates that the failure to proliferate in vrk-1 mutants is not due to a uniform arrest in early mitotic prophase. Indeed, the germ cells in vrk-1 mutant animals do not appear to undergo a uniform arrest at any specific point in the cell cycle. Rather, a variety of cellular phenotypes are observed, ranging from enlarged nuclei to hyper-condensed and fragmented chromosomes, many of which are similar to those seen in nhk-1 mutants in Drosophila. Therefore, the loss of vrk-1 activity causes a variety of defects that culminate to prevent successful progression through the cell cycle.
The complexity of the vrk-1 mutant phenotype makes it all the more remarkable that it can be significantly rescued by loss of cep-1 (p53). Our results imply that VRK-1 normally acts to limit CEP-1 activity, so that in the absence of VRK-1, unregulated CEP-1 disrupts germ cell proliferation. In mammals, p53 has been implicated in both the G1 and G2/M arrest, as well as in S phase progression (Giono and Manfredi, 2006). Thus, loss of the negative regulation of p53 could result in a mixture of phenotypes at multiple stages of the cell cycle. Even though p53 is normally activated by DNA damage, our data rule out a DNA damage-dependent mechanism of CEP-1 regulation by VRK-1. This observation indicates that the inhibition of CEP-1 by VRK-1 might occur through a more direct interaction. A current key question that remains is whether CEP-1 is actually a phosphorylation target of VRK-1. Unfortunately, the sequence conservation between mammalian p53 and C. elegans CEP-1 is very low, and the Vrk1 phosphorylation site that has been mapped in mammalian p53 is not readily identifiable in CEP-1. Indeed, CEP-1 has many potential VRK-1 phosphorylation sites. We attempted to perform in vitro phosphor-ylation assays to directly test whether CEP-1 could serve as a phosphorylation substrate of VRK-1, but could not purify sufficient amounts of CEP-1 for the assay.
In mammals, high levels of p53 are tolerated in the absence of DNA damage in proliferating cells such as embryonic stem cells, and the normal mode of degradation through the inhibitor MDM2 does not occur (Aladjem et al., 1998). We suggest that in this and similar situations requiring cell proliferation, Vrk1 might act as a post-translational negative regulator of p53. This mechanism of post-translational regulation would allow stem cells to retain high levels of inactive p53, permitting a rapid response to DNA damage. High levels of p53 may also enable cells to modulate proliferation rates in order to balance cell numbers with differentiation rates. Because we hypothesized that VRK-1 primarily affects CEP-1 function rather than altering protein levels, we attempted to determine whether CEP-1 levels were altered in vrk-1 mutants. However, multiple distinct CEP-1:GFP transgenes went silent during strain construction, so we were unable to directly test our hypothesis.
The variety of cellular phenotypes seen in vrk-1 mutants may also reflect a role of VRK-1 as a kinase with multiple phosphorylation substrates. In support of this possibility, cep-1;vrk-1 double mutants, while showing extensive rescue of proliferation, were not fertile and displayed other defects in germ cell differentiation. This observation indicates that while CEP-1 might be a critical target in controlling proliferation, it is likely not to be the only target of VRK-1. Additional targets likely exist that have roles in regulating the differentiation and maturation of germ cells, as well. Future studies investigating the relationship of Vrk1 and p53, as well as identifying how additional phosphorylation substrates of Vrk1 are involved in germ cell proliferation and differentiation, will help to clarify how this kinase functions as a major coordinator of these essential processes.
Supplementary Material
Acknowledgments
The authors thank Lynn Cooley, Michelle Kudron, and Fabio Piano for providing critical experimental suggestions. We are grateful to Peter Askjaer for the VRK-1 antibody, Geraldine Seydoux for the pie-1 transgene vector, and Sarah Crittenden and Judith Kimble for advice on the BrdU protocol. We also thank the Reinke lab for comments on the manuscript, and the CGC for strains. This work was funded by the Pew Foundation and the Connecticut Stem Cell Research Grants Program (08-SCA-YALE010).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ydbio.2010.06.022.
References
- Aggarwal BD, Calvi BR. Chromatin regulates origin activity in Drosophila follicle cells. Nature. 2004;430:372–376. doi: 10.1038/nature02694. [DOI] [PubMed] [Google Scholar]
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol. 1998;8:145–155. doi: 10.1016/s0960-9822(98)70061-2. [DOI] [PubMed] [Google Scholar]
- Berry LW, Westlund B, Schedl T. Germ-line tumor formation caused by activation of glp-1, a Caenorhabditis elegans member of the Notch family of receptors. Development. 1997;124:925–936. doi: 10.1242/dev.124.4.925. [DOI] [PubMed] [Google Scholar]
- Boehme KA, Blattner C. Regulation of p53–insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44:367–392. doi: 10.3109/10409230903401507. [DOI] [PubMed] [Google Scholar]
- Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337:242–253. doi: 10.1056/NEJM199707243370406. [DOI] [PubMed] [Google Scholar]
- Brauchle M, Baumer K, Gonczy P. Differential activation of the DNA replication checkpoint contributes to asynchrony of cell division in C. elegans embryos. Curr Biol. 2003;13:819–827. doi: 10.1016/s0960-9822(03)00295-1. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DT, Kimble J. Scratching the niche that controls Caenorhabditis elegans germline stem cells. Semin Cell Dev Biol. 2009;20:1107–1113. doi: 10.1016/j.semcdb.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W, Reinke V. Promotion of oogenesis and embryogenesis in the C. elegans gonad by EFL-1/DPL-1 (E2F) does not require LIN-35 (pRB) Development. 2006;133:3147–3157. doi: 10.1242/dev.02490. [DOI] [PubMed] [Google Scholar]
- Crittenden SL, Eckmann CR, Wang L, Bernstein DS, Wickens M, Kimble J. Regulation of the mitosis/meiosis decision in the Caenorhabditis elegans germline. Philos Trans R Soc Lond B Biol Sci. 2003;358:1359–1362. doi: 10.1098/rstb.2003.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden SL, Leonhard KA, Byrd DT, Kimble J. Cellular analyses of the mitotic region in the Caenorhabditis elegans adult germ line. Mol. Biol. Cell. 2006;17:3051–3061. doi: 10.1091/mbc.E06-03-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen CF, Brittle AL, Ito T, Ohkura H. The conserved kinase NHK-1 is essential for mitotic progression and unifying acentrosomal meiotic spindles in Drosophila melanogaster. J Cell Biol. 2005;171:593–602. doi: 10.1083/jcb.200508127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;294:591–595. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- Derry WB, Bierings R, van Iersel M, Satkunendran T, Reinke V, Rothman JH. Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ. 2007;14:662–670. doi: 10.1038/sj.cdd.4402075. [DOI] [PubMed] [Google Scholar]
- Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- Golden A, Sadler PL, Wallenfang MR, Schumacher JM, Hamill DR, Bates G, Bowerman B, Seydoux G, Shakes DC. Metaphase to anaphase (mat) transition-defective mutants in Caenorhabditis elegans. J Cell Biol. 2000;151:1469–1482. doi: 10.1083/jcb.151.7.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorjanacz M, Klerkx EP, Galy V, Santarella R, Lopez-Iglesias C, Askjaer P, Mattaj IW. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. EMBO J. 2007;26:132–143. doi: 10.1038/sj.emboj.7601470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development. 1999;126:1011–1022. doi: 10.1242/dev.126.5.1011. [DOI] [PubMed] [Google Scholar]
- Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev. 2002;16:729–742. doi: 10.1101/gad.968302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovska I, Khandan T, Ito T, Orr-Weaver TL. A histone code in meiosis: the histone kinase, NHK-1, is required for proper chromosomal architecture in Drosophila oocytes. Genes Dev. 2005;19:2571–2582. doi: 10.1101/gad.1348905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Lambert A, Ellefson M, Villeneuve AM, Engebrecht J. Differential timing of S phases, X chromosomal replication, and meiotic prophase in the C.elegans germ line. Dev Biol. 2007;308:206–221. doi: 10.1016/j.ydbio.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Killian DJ, Hubbard EJ. C. elegans pro-1 activity is required for soma/germline interactions that influence proliferation and differentiation in the germ line. Development. 2004;131:1267–1278. doi: 10.1242/dev.01002. [DOI] [PubMed] [Google Scholar]
- Klerkx EP, Alarcon P, Waters K, Reinke V, Sternberg PW, Askjaer P. Protein kinase VRK-1 regulates cell invasion and EGL-17/FGF signaling in Caenorhabditis elegans. Dev Biol. 2009;335:12–21. doi: 10.1016/j.ydbio.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudron MM, Reinke V. C. elegans nucleostemin is required for larval growth and germline stem cell division. PLoS Genet. 2008;4:e1000181. doi: 10.1371/journal.pgen.1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Borges S, Lazo PA. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene. 2000;19:3656–3664. doi: 10.1038/sj.onc.1203709. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, Ugel N, Mishra B, Isopi M, Hubbard EJA. Quantitative analysis of germline mitosis in adult C. elegans. Dev Biol. 2006;292:142–151. doi: 10.1016/j.ydbio.2005.12.046. [DOI] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr Biol. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Petcherski AG, Kimble J. LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature. 2000;405:364–368. doi: 10.1038/35012645. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitinligase. Nature. 2003;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development. 2004;131:311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann ER, Hengartner M, Schedl T, Gartner A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell. 2005;120:357–368. doi: 10.1016/j.cell.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Dunn MA. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development. 1997;124:2191–2201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- Srinivasan DG, Fisk RM, Xu H, van den Heuvel S. A complex of LIN-5 and GPR proteins regulates G protein signaling and spindle function in C elegans. Genes Dev. 2003;17:1225–1239. doi: 10.1101/gad.1081203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Wood WB. Immunofluorescence visualization of germ-line-specific cytoplasmic granules in embryos, larvae, and adults of Caenorhabditis elegans. Proc Natl Acad Sci USA. 1982;79:1558–1562. doi: 10.1073/pnas.79.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam K, Seydoux G. nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development. 1999;126:4861–4871. doi: 10.1242/dev.126.21.4861. [DOI] [PubMed] [Google Scholar]
- Voutev R, Killian DJ, Ahn JH, Hubbard EJ. Alterations in ribosome biogenesis cause specific defects in C. elegans hermaphrodite gonadogenesis. Dev Biol. 2006;298:45–58. doi: 10.1016/j.ydbio.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gallegos M, Puoti A, Durkin E, Fields S, Kimble J, Wickens MP. A conserved RNA-binding protein that regulates sexual fates in the C. elegans hermaphrodite germ line. Nature. 1997;390:477–484. doi: 10.1038/37297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.