

Abstract
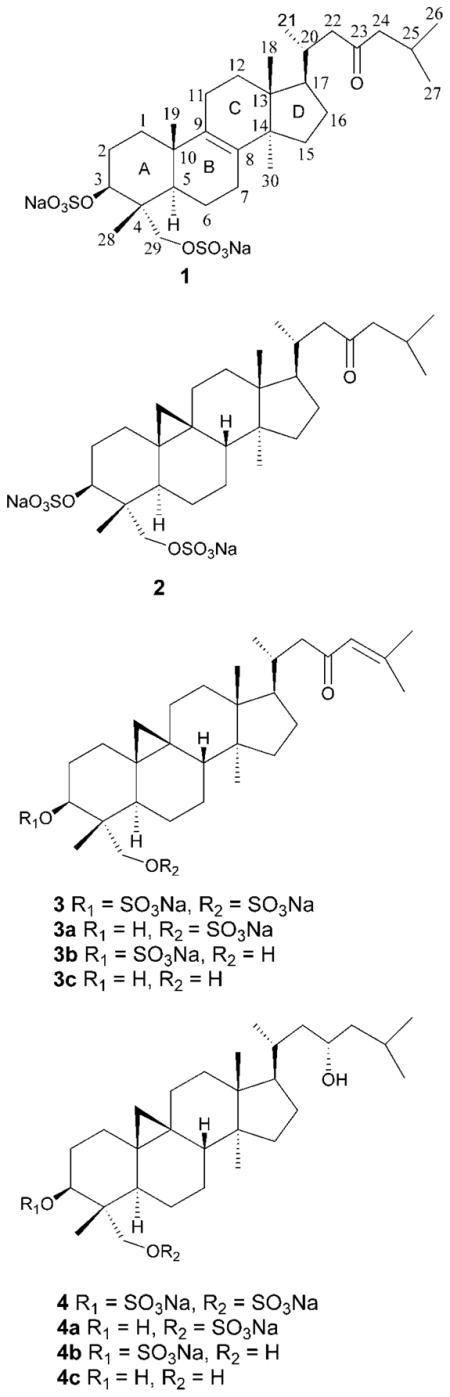
Cytotoxicity-guided fractionation of the green macroalga Tydemania expeditionis led to isolation of four sulfate-conjugated triterpenoids including one new lanostane-type triterpenoid disulfate, lanosta-8-en-3,29-diol-23-oxo-3,29-disodium sulfate (1), and three known cycloartane-type triterpenoid disulfates, cycloartan-3,29-diol-23-one 3,29-disodium sulfate (2), cycloart-24-en-3,29-diol-23-one 3,29-disodium sulfate (3), and cycloartan-3,23,29-triol 3,29-disodium sulfate (4). Extensive 1D and 2D NMR analyses in combination with X-ray crystallography established the structure and absolute configuration of 1 and allowed determination of the absolute configurations of 2–4 with a revision of previously assigned configuration at C-5. Each natural product was moderately cytotoxic in tumor cell and invertebrate toxicity assays. Of the natural products, only 4 exhibited significant antifungal activity at whole-tissue natural concentrations against the marine pathogen Lindra thalassiae. Comparison of the biological activities of natural products with their desulfated derivatives indicated that sulfation does not appear to confer cytotoxicity or antifungal activity.
Tydemania expeditionis Weber-van Bosse (Udoteaceae) is a weakly calcified green alga distributed in the tropical Pacific and Indian Oceans. Previous studies on this species from Guam led to the isolation of three norcyloartene triterpenoids1 and a linear diterpenoid,2 while investigation of another population from Micronesia revealed three cycloartanol sulfates with inhibitory activity against the pp60v-rsc protein tyrosine kinase.3 However, none of these studies were related to chemical ecology. In our ongoing effort to isolate and identify ecological leads for drug discovery from marine algae, we investigated the chemical components of T. expeditionis collected in Fiji. Herein, we report the structure elucidation and absolute configuration of triterpenoid sulfates (1–4) from T. expeditionis and their ecological effects on Lindra thalassiae, a marine fungal pathogen known to infect some macroalgae.4
The high-resolution ESI mass spectrum of lanosta-8-en-3,29-diol-23-oxo-3,29-disodium sulfate (1) showed a quasimolecular ion [M−Na]− at m/z 639.2651, corresponding to C30H48O9S2Na with six degrees of unsaturation. The presence of sulfur was suggested by the low density mass offset of +2 amu (34S) for both parent and fragment ions and the presence of sulfate confirmed by the characteristic loss of 102 amu at m/z 537.3262 [M − SO3Na + H − Na]−.
The 13C NMR spectrum displayed 30 signals consistent with a triterpenoid skeleton and, in combination with DEPT analysis, suggested one carbonyl, seven methyls, 11 methylenes, five methines, and six quaternary carbon atoms (Supporting Information). The low-field region of the 13C NMR spectrum contained resonances for an oxo group at δ 213.0 (s, C-23), a tetrasubstituted olefin at δ 135.1 (s, C-8) and 134.4 (s, C-9), one oxymethine at δ 79.6 (d, C-3), and one oxymethylene at δ 69.0 (t, C-29). Inspection of the 1H NMR spectrum revealed three secondary methyl groups at δ 0.89 (d, J = 6.5 Hz, H3-21), 0.90 (d, J = 7.0 Hz, H3-27), and 0.91 (d, J = 7.0 Hz, H3-26) and four quaternary methyl groups at δ 0.77 (s, H3-18), 0.81 (s, H3-28), 0.92 (s, H3-30), and 1.06 (s, H3-19). The resonances for one oxygenated methine and one isolated oxygenated methylene were observed at δ 4.37 (dd, J = 4.5, 11.5 Hz, H-3), 3.96 (d, J = 9.5 Hz, H-29β), and 3.83 (d, J = 9.5 Hz, H-29α). The downfield-shifted resonances of H-3/C-3 and H2-29/C-29 relative to typical hydroxylated methines and methylenes,5 respectively, suggested sulfate groups at these positions.
The assignment and connectivity of 1 were established by 1H–1H COSY, HSQC, and HMBC spectra. The 1H–1H COSY data indicated two spin systems within the side chain, H-21↔H-20↔H-22 and H-24↔H-25↔H-26 (↔H-27). The HMBC cross-peaks H-22→C-23 and H-24→C-23 indicated that these two spin systems were connected by a carbonyl carbon (C-23), supporting a 2-methylheptan-4-one side chain. The HMBC correlation H-17→C-20 and the 1H–1H COSY cross-peak H-17↔H-20 revealed C-17↔C-20 connectivity. The location of the Δ8,9 double bond was confirmed by HMBC correlations H-19→C-9 and H-30→C-8. The HSQC correlation H-3 (δ 4.37)→C-3 (δ 79.6) in combination with the HMBC correlations H-3→C-4, H-3→C-28, H-1→C-3, and H-2→C-3 indicated the location of a sulfate moiety at C-3. Similarly, the HSQC correlation H2-29 (δ 3.96 and δ 3.83)→C-29 (δ 69.0) in combination with the HMBC correlations H-29→C-3, C-4, C-5, and C-28 confirmed another sulfate moiety at C-29.
The absolute configuration of 1 was established by single-crystal X-ray analysis,6 which revealed a stable complex constructed from two fundamental units: a triterpenoid sulfate anion and a sodium cation coordinated by one water and two methanol molecules (Figure 1 and Supporting Information). In the crystalline state, the sodium sulfate moiety formed an inorganic layer of a face-sharing NaO6 coordination network along the (110) plane and the triterpenoid units of 1 were extended, at the junctions of O-1 and O-5, on both sides of inorganic layers to form a scaffold-like structure. Because 1 contained two sodium and two sulfur atoms exhibiting sufficient anomalous dispersion, the final refinement resulted in a Flack parameter of 0.18(5), allowing an unambiguous assignment of the absolute configuration (3S, 4R, 5R, 10S, 13R, 14R, 17R, 20R).
Figure 1.

ORTEP view of 1 · 2MeOH · H2O from X-ray diffraction data. Atoms are shown as 30% thermal ellipsoids. Selected interatomic distances and angles: Na1–O2 2.328 Å, Na2–O8 2.525 Å, Na1–O2–S1 131.3°, Na2–O8–S2 121.4°.
Triterpenoid sulfates, previously reported primarily from marine macroalgae, vary in both their triterpene cores and patterns of sulfation. Examination of previous natural product structures reveal four common types: (i) cycloartane or norcycloartane skeletons bearing one sulfate group at C-3, e.g., methyl 3β,23-dihydroxycy-cloart-24-en-28-oate-3-sulfate from the red alga Tricleocarpa fragilis;7 (ii) cycloartane skeletons bearing one sulfate group at C-29, e.g., capisterone A from the green alga Penicillus capitatus;8 (iii) cycloartane skeletons bearing two sulfate groups at C-3 and C-29, e.g., sulfated triterpenoids from green algae of the Tuemoya genus9 and from Tydemania expeditionis3 (also see below), and (iv) norlanostane skeletons bearing one sulfate group at C-3, e.g., methyl 3β,23-dihydroxy-29-norlanosta-8,24-dien-28-oate 3-sulfate from Tricleocarpa fragilis.7 The discovery of 1 represents a fifth type, the first lanostane-type triterpenoid bearing 3,29-disodium sulfate moieties.
The structures of three additional compounds were identified by comparing their spectroscopic data (UV, ESIMS, 1H and 13C NMR) with a published report on the chemistry of Tydemania expeditionis,3 leading to identification of cycloart-24-en-3,29-diol-23-one 3,29-disodium sulfate (2), cycloartan-3,29-diol-23-one 3,29-disodium sulfate (3), and cycloartan-3,23,29-triol 3,29-disodium sulfate (4). H-5 in 2–4 was previously reported to be β-oriented,3 despite its characteristic axial coupling (dd, J = 2, 7 Hz) at δ 1.83 resulting from interactions with two vicinal protons, one axial (H-6α) and the other equatorial (H-6β), with dihedral angles close to 180° and 60°, respectively. Accordingly, H-5 is α-oriented in compounds 2–4.
X-ray diffraction analysis of 2 confirmed an α-oriented H-5 and revealed the presence of a three-membered ring at C-9 and C-10 with an average bond distance of 1.514 Å and bond angle of 60°, characteristic of cycloartanes (Figure 2 and Supporting Information). Similar to 1, 2 contained two sodium and two sulfur atoms, and the final refinement resulted in a suitably low Flack parameter,10 permitting assignment of its absolute configuration as 3S, 4R, 5R, 8S, 9S, 10R, 13R, 14S, 17R, 20R. We predict that 3 and 4, which share a carbon skeleton with 2, possess the same absolute configuration. The α-oriented hydroxy group at C-23 in cycloartanol sulfates was previously established by Mosher’s method.7 The 23-hydroxy group in 4 was inferred to share the same α-orientation by considering the biogenetic relationship in this class of compounds.
Figure 2.

ORTEP view of 2 · MeOH · 2H2O from X-ray diffraction data.
With LC-MS, using prominent MS ion signals at discrete retention times, linear calibration curves were obtained for pure samples of 1–4 (r2 = 0.99 for all, n = 6 concentrations each). Comparison of standard curve data with ion intensities for each compound within the crude extract resulted in the following estimate of natural whole-tissue concentrations: 1.38 mM (1), 1.20 mM (2), 1.07 mM (3), and 1.59 mM (4). Since this measurement was taken from only one crude extract, an assessment of variation among algal specimens was not possible.
In order to investigate the biological role of sulfation, PTSA-catalyzed sulfate ester hydrolysis was undertaken on 3 and 4 (Supporting Information). The 1H NMR spectra of 3a–3c were similar to that of 3 except for the high-field shifts of H-3 in 3a (δ 3.76, dd, J = 5.3, 10.8 Hz), H-29 in 3b (H-29β: δ 3.58, d, J = 12.0 Hz; H-29α: δ 3.34, d, J = 12.0 Hz), and both H-3 and H-29 in 3c (H-3: δ 3.71, dd, J = 5.0, 11.5 Hz; H-29β: δ 3.58, d, J = 11.0 Hz; H-29α: δ 3.34, d, J = 11.0 Hz), which indicated that 3-O-sulfate, 29-O-sulfate, and 3,29-O-disulfate were converted to hydroxy groups in 3a, 3b, and 3c, respectively. Accordingly, these three compounds were identified as cycloart-24-en-3,29-diol-23-one 29-sodium sulfate (3a), cycloart-24-en-3,29-diol-23-one 3-sodium sulfate (3b), and cycloart-24-en-3,29-diol-23-one (3c). Pseudo-molecular ions with m/z 535 [M − Na]−, 535 [M − Na]−, and 457 [M + H]+ for 3a, 3b, and 3c, respectively, confirmed their structures. Similarly, monosulfates from the hydrolysis of 4 were identified as cycloartan-3,23,29-triol 29-sodium sulfate (4a) and cycloartan-3,23,29-triol 3-sodium sulfate (4b). A small amount of desulfate 4c was suggested by LC-MS, but was not purified nor fully characterized.
When 1, 2, 3, 3a, 3b, 3c, and 4 were tested for inhibitory activity against a panel of 12 breast, colon, lung, prostate, and ovarian tumor cell lines, monosulfated derivatives 3a and 3b and desulfated 3c showed moderate activities with mean IC50 values of 6.0 to 11 μM. Disulfated natural products 1–4 exhibited weaker antitumor effects with mean IC50 values ranging from 31 to 38 μM. Inhibition of invertebrate (rotifer) feeding, a proxy for cytotoxicity,12 ranged from 9–87% when each compound was tested at 10 ppm.
In contrast to fairly uniform cytotoxic effects of 1–4 described above, only 4 exhibited significant antifungal activity against the marine pathogen Lindra thalassiae when tested at 1 mM, a concentration close to the whole-tissue natural concentrations of 1–4 of 1.07–1.59 mM (Figure 3A). This suggested the C-23 hydroxy group (as in 4) was essential to antifungal activity since 2, with a carbonyl group in this position, was not significantly active. In contrast, capisterones A and B, bearing C-23 carbonyls and differing from 2–4 in A-ring substitution and sulfation patterns, were active against L. thalassiae with low micromolar IC50 values.8
Figure 3.

Ecological antifungal activity: (A) growth inhibitory effects of 1–4 at 0.67 mg/mL (1 mM) on Lindra thalassiae, showing that 4 is the only significantly active antifungal agent (bars represent one standard deviation; ** denotes p < 0.01, 1-way ANOVA with Dunnett’s post test); (b) dose–response curves for 4, 4a, and 4b.
An antifungal IC50 value of 26 μM (17 μg/mL) was determined for 4, indicating that Tydemania expeditionis contains approximately 60 times more 4 than required to suppress growth of this pathogen (Figure 3B). Monosulfates 4a and 4b exhibited antifungal IC50 values of 55 and 30 μM, respectively, similar to the disulfated parent compound 4. As with cytotoxic activities described above, sulfation did not appear to confer antifungal activity.
Experimental Section
General Experimental Procedures
HPLC separations were completed and UV spectra recorded with a Waters 2695 HPLC system and Waters 2996 diode-array detector. Optical rotations were measured with a Jasco P-1010 spectropolarimeter. NMR spectra (1H, 13C, DEPT, COSY, HSQC, HMBC, and NOESY) were acquired on a Bruker DRX-500 instrument, using a 5 mm broadband or inverse detection probe. HRMS were generated by electrospray ionization with an Applied Biosystems QSTAR-XL hybrid quadrupole time-of-flight tandem mass spectrometer and Analyst QS software.
Collection
T. expeditionis (tuft form) was collected from depths of 7–20 m at Herald Pass, Kadavu Province, Fiji (18°46.370′ S, 178°27.746′ E) and frozen at −20 °C until extraction (collection G-2004-06-45). A voucher specimen was identified by Dr. Posa Skelton at the University of the South Pacific by comparison with previously described morphological traits,11 stored in aqueous formaldehyde, and deposited at the University of the South Pacific.
Isolation Guided by Rotifer Ingestion Toxicity Assay
The whole plant (500 g, wet mass) was exhaustively extracted with MeOH and MeOH–CH2Cl2 to afford a crude extract, which was partitioned between MeOH–H2O (9:1) and hexanes. The aqueous fraction was successively partitioned against EtOAc and n-BuOH to afford hexanes-, EtOAc-, n-BuOH- and H2O-soluble fractions. The ethyl acetate-soluble fraction showed the most potent toxicity. Further fractionation was accomplished by semipreparative reversed-phase HPLC, yielding four compounds: 1 (15 mg), 2 (9 mg), 3 (12 mg), and 4 (15 mg). Crystals of 1 and 2 were obtained by recrystallization from MeOH (for details see Supporting Information).
Determination of Natural Concentrations of 1–4
Pure 1–4 were solubilized in MeOH, diluted to six concentration levels, and analyzed by LC-MS (Supporting Information). Calibration curves were constructed from peak areas of appropriate molecular ions. Concentrations of 1–4 in the crude extract were calculated from corresponding standard curves.
Rotifer Ingestion Toxicity Assay
Experiments were performed in 24-well microtiter plates using previously described procedures.12
Antitumor Assay
Compounds were tested against a panel of tumor cell lines: breast cancer (BT-549, DU4475, MDA-MB-468, MDA-MB-231), colon cancer (HCT116), lung cancer (NCI-H446 and SHP-77), prostate cancer (PC-3, LNCaP-FGC, Du145), ovarian cancer (A2780/DDP-S), and leukemia (CCRF-CEM). Cells were seeded in 96-well plates and compounds added 24 h later. After 72 h of exposure, cells were stained with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt and quantified spectrophotometrically. Conversion of the MTS dye in the mitochondria was directly proportional to live cells. Dose–response curves were generated and IC50 values calculated for each compound and cell line, with a mean IC50 value determined for the 12 cell line panel.
Antifungal Assay
The antifungal activity of isolated compounds was evaluated against a marine pathogen, Lindra thalassiae (ATCC 56663), using a procedure similar to that described previously.13
Lanosta-8-en-3,29-diol-23-oxo-3,29-disodium sulfate (1)
Colorless powder; [α]20D +50.0 (c 0.02 in CH3OH); UV δmaxMeOH 210 nm; 1H NMR (CD3OD) 4.37 (1H, dd, J = 4.5, 11.5 Hz, H-3), 3.96 (1H, d, J = 9.5 Hz, H-29β), 3.83 (1H, d, J = 9.5 Hz, H-29α), 2.48 (1H, dd, J = 2.5, 16 Hz, H-22β), 2.30 (2H, d, J = 7 Hz, H-24), 2.21 (1H, m, H-2α), 2.17 (1H, dd, J = 1.0, 16 Hz, H-22α), 2.09 (1H, m, H-25), 2.07 (1H, m, H-11α), 2.01 (2H, m, H-7), 1.98 (1H, m, H-20), 1.93 (1H, m, H-16α), 1.82 (1H, m, H-6β), 1.79 (1H, m, H-11β), 1.78 (1H, m, H-2β), 1.77 (2H, m, H-12), 1.74 (1H, m, H-1α), 1.70 (1H, dd, J = 2, 10.5 Hz, H-5), 1.65 (1H, m, H-15β), 1.56 (1H, m, H-17), 1.52 (1H, m, H-6α), 1.30 (1H, m, H-16β), 1.22 (1H, m, H-1β), 1.19 (1H, m, H-15α), 1.06 (3H, s, H-19), 0.92 (3H, s, H-30), 0.91 (3H, d, J = 7.0 Hz, H-26), 0.90 (3H, d, J = 7.5 Hz, H-27), 0.89 (3H, d, J = 7.0 Hz, H-21), 0.81 (3H, s, H-28), 0.77 (3H, s, H-18); 13C NMR (CH3OH-d4) 213.0 (s, C-23), 135.1 (s, C-8), 134.4 (s, C-9), 79.6 (d, C-3), 69.0 (t, C-29), 52.3 (t, C-24), 50.7 (d, C-17), 50.5 (t, C-22), 50.2 (s, C-14), 44.8 (s, C-13), 43.2 (d, C-5), 41.8 (s, C-4), 36.9 (s, C-10), 35.2 (t, C-1), 33.4 (d, C-20), 31.2 (t, C-12), 30.9 (t, C-15), 28.4 (t, C-16), 26.0 (t, C-7), 24.6 (d, C-25), 24.2 (t, C-2), 23.5 (q, C-30), 21.9 (q, C-26), 21.8 (q, C-27), 21.0 (t, C-6), 19.2 (q, C-21), 18.9 (q, C-19), 17.8 (t, C-11), 15.4 (q, C-18), 12.4 (q, C-28); ESI-MS m/z 639 [M − Na]−, 617 [M − 2Na + H]−, 537 [M − SO3Na + H − Na]−, 519 [M − SO3Na + H − Na − H2O]−, 308 [M − 2Na]2−; HRESIMS m/z [M − Na]− 639.2651, calcd for C30H48O9S2Na 639.2643.
p-Toluenesulfonic Acid (PTSA)-Catalyzed Sulfate Ester Hydrolysis
Compounds 3 and 4 were hydrolyzed according to the reported procedures,14 as reported in the Supporting Information.
Crystal data of 1 and 2 in standard CIF format have been deposited with the Cambridge Crystallographic Data Centre with CCDC numbers as shown in refs 6 and 10. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033; deposit@ccdc.cam.ac.uk).
Supplementary Material
Acknowledgments
This research was supported by the U.S. National Institutes of Health’s International Cooperative Biodiversity Groups program (grant U01-TW007401-O1) and by an NSF-IGERT predoctoral fellowship to A.L.L. We thank the Roko Tui Kadavu and Tui Dravuni for their hospitality and for permission to collect in the reefs of Kadavu Province. We also thank the government of the Fiji Islands for permission to export samples. The authors are grateful for technical assistance provided by M. Sharma, K. Feussner, P. Skelton, E. P. Stout, T. Myers, A. Prusak, and A. Chequer.
Footnotes
Supporting Information Available: Overview of natural sulfate bioconjugates; isolation details; X-ray analysis; derivatization of natural products; LC-MS analysis; 1H and 13C NMR spectra for 1; X-ray crystal structure diagrams; mass spectrum of 3; and proposed fragmentation pathway. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Paul VJ, Fenical W, Raffii S, Clardy J. Tetrahedron Lett. 1982;23:3459–3462. [Google Scholar]
- 2.Paul VJ, Fenical W. Phytochemistry. 1985;24:2239–2243. [Google Scholar]
- 3.Govindan M, Abbas SA, Schmitz FJ, Lee RH, Papkoff JS, Slate D. J Nat Prod. 1994;57:74–78. doi: 10.1021/np50103a010. [DOI] [PubMed] [Google Scholar]
- 4.Kohlmeyer J. Mar Biol. 1971;8:344–350. [Google Scholar]
- 5.Crews P, Rodríguez J, Jaspars M. Organic Structure Analysis. Oxford University Press; Oxford: 1998. p. 75. [Google Scholar]
- 6.Crystal data for compound 1. C30H48O9S2Na2 · 2CH3OH · H2O, colorless plates (0.28 × 0.21 × 0.11 mm) were recrystallized from MeOH solution, orthorhombic, P212121, a = 7.1495(5) Å, b = 10.4548(7) Å, c = 49.990(3) Å, V = 3736.6(4) Å3, Z = 4, dx = 1.317 g cm−3, F(000) = 1584, λ(CuKα) 1.54178 Å, Flack parameter 0.18(5). Data collection was performed on a SMART 1000 CCD; 5594 [R(int) 0.0409] unique reflections were collected to θmax = 65.9°, in which 5110 reflections were observed [F2 > 4σ(F2)]. The crystal structure was resolved by direct methods using SHELXS-97. In the final stage, R = 0.105 and S = 1.094. CCDC no. 686956.
- 7.Horgen D, Sakamoto B, Scheuer PJ. J Nat Prod. 2000;63:210–216. doi: 10.1021/np990448h. [DOI] [PubMed] [Google Scholar]
- 8.Puglisi MP, Tan LT, Jensen PR, Fenical W. Tetrahedron. 2004;60:7035–7039. [Google Scholar]
- 9.Clement JA, Zhou BN, Johnson RK, Kingston DGI. Magn Reson Chem. 2003;41:644–646. [Google Scholar]
- 10.Crystal data for compound 2. C30H46O9S2Na2 · CH3OH · 2H2O, colorless plates (0.25 × 0.21 × 0.08 mm) were recrystallized from MeOH solution, monoclinic, P21, a = 6.899(1) Å, b = 9.952(1) Å, c = 26.382(3) Å, β= 91.73(1)°, V = 1810.4(4) Å3, Z = 2, dx = 1.331 g cm−3, F(000) = 774, λ (Cu Kα) 1.54178 Å, Flack parameter 0.05(3). Data collection was performed on a SMART 1000 CCD; 4376 [R(int)) 0.0348] unique reflections were collected to θmax = 66.11°, in which 4069 reflections were observed [F2 > 4σ(F2)]. The crystal structure was resolved by direct methods using SHELXS-97. In the final stage, R = 0.061 and S = 1.080. CCDC no. 686957.
- 11.Littler DS, Littler MM. South Pacific Reef Plants. Offshore Graphics, Inc; Washington, D.C.: 2003. pp. 258–259. [Google Scholar]
- 12.Snell TW. Rotifer Ingestion Test for Rapid Assessment of Toxicity. In: Blaise C, Ferard JF, editors. Small-Scale Freshwater Environment Toxicity Test Methods. Kluwer; Dordrecht: 2005. pp. 323–335. [Google Scholar]
- 13.Kubanek J, Jensen PR, Keifer PA, Sullards MC, Collins DO, Fenical W. Proc Natl Acad Sci USA. 2003;100:6916–6921. doi: 10.1073/pnas.1131855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh SB. Tetrahedron Lett. 2000;41:6973–6976. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.