

Abstract
(2,6-Dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl) methoxymethyl chloride [1, monomethoxydiphenylmethoxylmethyl chloroide (MDPM-Cl)] shows a significant relative stability and 1 reacts with uridine ureido nitrogen in the presence of DBU to form the corresponding protected uridine 8 in 95% yield. The MDPM-protected uridines are stable to a wide variety of conditions utilized for the synthesis of analogs of capuramycin and muraymycins. Significantly, the MDPM protecting group can conveniently be deprotected by using 30% TFA in CH2Cl2. In addition, polymer-bound MDPM-Cl 23 is useful for immobilization of uridine derivatives.
Keywords: Ureido nitrogen, Uridine, Monomethoxydiphenylmethoxylmethylgroup, Polymer-bound, linker
1. Introduction
Uridine is an essential biological compound for multiple biosynthetic processes and found in all cells.1 To date, a large number of uridine-containing natural products that show significant biological activities have been isolated.2 Uridine-containing antibiotics such as liposidomycin, caprazamycin, muraymycins, and capuramycin have been of increasing interest to the development of new antibacterial agents for MDR-bacterial infections.3
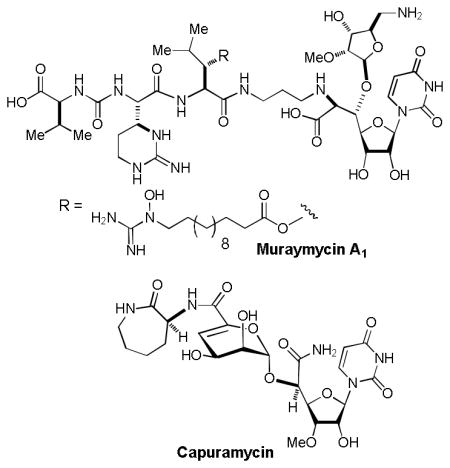
In our ongoing program of development of drug leads for MDR Mycobacterium tuberculosis, we have synthesized analogs of muraymycins A1 and capuramycin.4 Protection of the uridine ureido nitrogen was indispensable to achieve the synthesis of wide range of uridine-containing molecules. Benzyloxymethyl (BOM) group has been utilized as a convenient protecting group for the uridine ureido nitrogen (P1).5 However, we and other groups observed that BOM deprotection of uridine derivatives under hydrogenation conditions often resulted in poor yield with over-reduction product(s).6 In general, hydrogenolytic deprotection is the only method to remove the BOM group of the uridine ureido nitrogen.
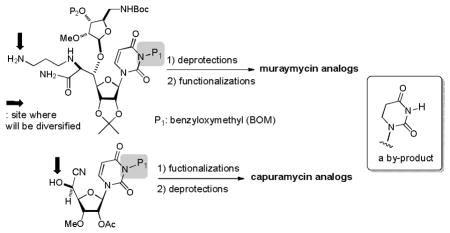
We have sought an alternative methoxymethyl-type protecting group for the uridine ureido nitrogen that 1) is stable to reaction conditions for the syntheses of a wide range of muraymycin and capuramycin analogs, but 2) can be cleaved with mild and volatile acids. In this paper we report our studies of (2,6-dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl) methoxymethyl chloride [1, monomethoxydiphenyl methoxylmethyl chloroide (MDPM-Cl) for the protection of the uridine ureido nitrogen and application of 1 to the polymer-bound MDPM-Cl for synthesis of analogs on polymer-support.
2. Results and discussion
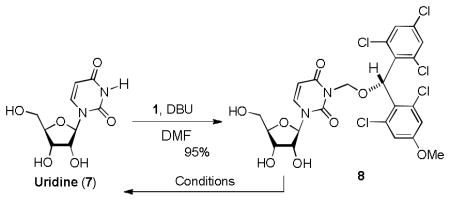
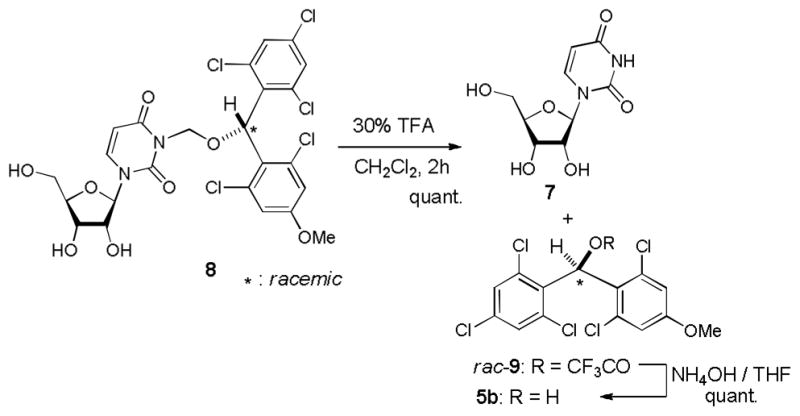
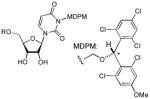
We have previously reported utilities of rac-5a and optically pure 5a for protections of alcohols, amines, and carboxylic acids, and as a chiral derivatizing agent.7 (2,6-Dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methanol (5a) could efficiently be synthesized via a Friedel-Crafts reaction followed by LiBH4 reduction. Similarly, a large quantity of (2,6-dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl) methanol (5b) could be synthesized efficiently. Stability tests of 5a and 5b against a wide variety of Lewis and Brønsted acids revealed that 5b exhibited longer half-life in the acidic conditions summarized in Table 1; 5a was converted to the corresponding TFA ester in 30 min. when exposed to 30% TFA in CH2Cl2 at room temperature, on the other hand, 5b required more than 1h to form its TFA ester under the same reaction conditions. In addition, we have recognized that rac-5a forms diastereomers in NMR spectra when reacted with chiral substrates. However, 5b has not formed noticeable diastereomers in NMR spectra. In addition, generated diastereomers have not been separated via HPLC.8 Because of the reasons stated above, we decided to utilize 5b for further investigation. As illustrated in Scheme 1 (2,6-dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl) methanol (5b) could be converted to the corresponding MDPM-Cl 1. The alcohol 5b was first transformed to the (alkoxymethyl)methyl sulfide 6 in 98% yield, which was then subjected to a Cl-displacement reaction with SO2Cl2 to yield 1 in greater than 95% yield.9 MDPM-Cl 1 was stable at room temperature and can be stored for several months without loss of purity. The uridine ureido nitrogen was efficiently protected with 1 in the presence of DBU in DMF to afford 8 in 95% yield (Table 1). The MDPM group of 8 exhibited excellent stability against a variety of Brønsted and Lewis acids such as 20% TFA, 10% TMSOTf, 10% HCl, 30% HF, 80% AcOH, 30% TsOH, La(OTf)3, BF3•OEt2, and TiCl4 at room temperature. The MDPM-protected uridine 8 also showed stability under basic conditions; 8 was intact under NH4OH (40% in aq MeOH), LiOH (10% in aq THF-MeOH), and DBU (10% in toluene) at room temperature for over 24 h. Selected examples are summarized in Table 1. Moreover, the MDPM protecting group of 8 was stable to the reduction conditions such as Al(Hg), nBu3SnH/AIBN, and Raney Ni. The MDPM-protected uridine 8 was stable to NBS, and was photolytically stable (200~350 nm for over 6 h). The MDPM protecting group of 8 was not cleaved by hydrogenation conditions using Pd-C or Pd black even at 100 psi. The MDPM group could not be cleaved by using the standard conditions for the deprotection of PMB (p-methoxybenzyl) ether groups, however, could be deprotected with 30% TFA in CH2Cl2 to afford uridine (7). The (2,4-dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methyl cation generated by the treatment of 8 with 30% TFA is a sterically hindered species and stabilized by the electron-withdrawing Cl atoms, being efficiently reacted with the trifluoroacetate ion to afford (2,4-trichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methyl 2,2,2-trifluoroacetate (9) in quantitative yield. The treatment of 9 with NH3/MeOH of 7 gave rise to the parent alcohol 5b in quantitative yield (Scheme 2). Thus, the MDPM-Cl 1 can be regenerated through the chemical steps illustrated in Scheme 1.
Table 1.
Protection of the uridine ureido nitrogen with 1 and its relativities against representative reagents.
 | |||
|---|---|---|---|
| Conditionsa | 8 (t1/2) | Conditionsa | 8 (t1/2) |
| 20% TFA, CH2Cl2 | >6h | DBU, Toluene | >24h |
| 10% TMSOTf, CH2Cl2 | >2h | Al(Hg), 1,4-Dioxane | >12h |
| 10% HCl, MeCN | >6h | Raney Ni 1,4-Dioxane | >12h |
| 10% HCl, 1,4-dioxane | >6h | nBu3SnH, AIBN Toluene, reflux | >6h |
| 30% HF, MeCN | >12h | NBS THF |
>6h |
| 80% AcOH | >12h | ||
| 30% TsOH, 1,4-dioxane | >12h | hv, MeCN | >6h |
| La(OTf)3, THF | >12h | H2/Pd-C, 1 atom MeOH | >12hb |
| 10% BF3-OEt2, CH2Cl2 | >12h | H2/Pd-C, 50 psi MeOH | >12hb |
| 10% TiCl4, CH2C12 | >4h | H2/Pd-C, 100 psi MeOH | >12hb |
| DDQ, CH2Cl2-water | >12h | H2/Pd black MeOH | >12hb |
Reaction was carried out at room temperature.,
the uracil double bond of 8 was reduced.
Scheme 1.

Syntheses of monomethoxydiphenylmethoxylmethyl chloroides 1.
Scheme 2.
Deprotection of MDPM group.
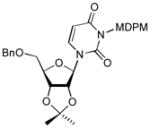
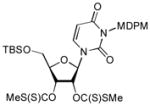
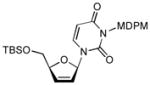
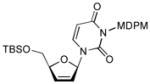
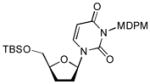
In order to demonstrate robustness of MDPM group as a protecting group for the uridine ureido nitrogen, the MDPM-protected uridine 8 was transformed to a wide range of uridine derivatives that can be utilized for the syntheses of analogs of muraymycins and capuramycin.4d Selected examples are summarized in Table 2. Ketal formations of 10 or 8 under acidic conditions provided the corresponding 2,3-protected derivatives in good to high yields (entries 1 and 5). The primary TBS and TIPS group of 11a or 13 could be deprotected selectively with 50% HF in CH3CN to furnish 12 and 8, respectively without cleavage of the MDPM group (entries 2 and 4). The trityl group of 11b was selectively removed by using BF3•OEt2 in the presence of TolSH without affecting the MDPM group (entry 3). Selective hydrogenolytic deprotection of the benzyl group of 15 was carried out via 10% Pd-C in iPrOH-water to furnish 13 without over-reduction of the uracil double bond (entry 6). Olefination of the carbonodithioate 16 was achieved by using nBu3SnH and AIBN in toluene at refluxing temperature to provide 2′,3′-didehydro-2′, 3′-dideoxy derivative 17 in a reasonable yield (entry 7). Hydrogenation of the double bond of 17 was also achieved under a standard hydrogenation condition with 10% Pd-C within 1 h to provide the MDPM-protected uridine-2′,3′-dideoxy derivative 18 in high yield (entry 8). Thus, it was experimentally proved that MDPM group is a robust protecting group for the uridine ureido nitrogen to synthesize a wide range of uridine derivatives.
Table 2.
Functionalizations of MDPM-protected uridines.
| Entry | Starting material | Conditions | Product | Yield (%)a |
|---|---|---|---|---|
| 1 |
 10a : R = TBS 10b : R = Tr |
 NBS, TMSOTf/ CH3CN, rt, 1h |
 11a : R = TBS 11b : R = Tr |
75 (95)b for 10a 90 (98)b for 10b |
| 2 |
 11a |
50% HF/ CH3CN, rt, 1h |
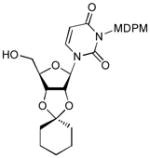 12 |
80 |
| 3 |
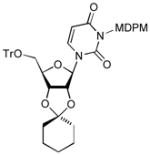 11b |
BF3-OEt2 (3 equiv), TolSH/ CH2Cl2, rt, 1h |
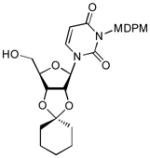 12 |
90 |
| 4 |
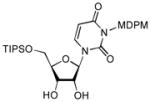 13 |
50% HF/ CH3CN, rt, 12h |
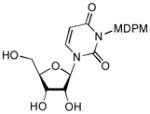 8 |
88 |
| 5 |
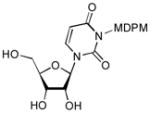 8 |
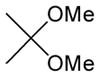 TsOH-H2O/ acetone, rt, 4h |
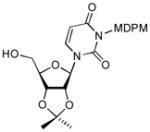 14 |
90 |
| 6 |
 15 |
H2 (1 atom) / 10% Pd-C iPrOH-watert, 2h |
 14 |
95c |
| 7 |
 16 |
nBuSnH, AIBN / toluene, reflux, 2h |
 17 |
75 |
| 8 |
 17 |
H2 (1 atom) / 10% Pd-C MeOH, 2h, |
 18 |
95c |
Product was isolated by SiO2 chromatography or PTLC.,
yield based on recovering starting material.,
no over-reduction was observed.
We have previously developed a novel ester linker 27, whose esters are stable against Brønsted and Lewis acids, Brønsted bases and a wide variety of nucleophiles.7c If the uridine ureido nitrogen can be immobilized onto polymer-resin, systematic syntheses of capuramycin and muraymycin analogs would be dramatically enhanced. As summarized in Scheme 3, the (2,6-dichloro-4-hydroxyphenyl) (2,4,6-trichlorophenyl) methanone (20)10 could efficiently be linked with hydroxymethylpolystyrene (PS) (~2 mmol/g) via a Mitsunobu reaction.11 The carbonyl group of 21 was reduced by LiBH4 in THF to afford the PS-alcohol 22. Available alcohol-linkers on the polymer surface were determined to be 1.8~2.0 mmol/g by coupling of the linkers with Fmoc-β-Ala-OH and subsequent release of Fmoc chromophore and elemental analyses of the Cl atoms for 22. According to the procedure summarized in Scheme 1, the PS-alcohol was transformed to the PS-MDPM-Cl 23, whose available chloromethoxy group was determined to be 1.5~1.8 mmol/g by its elemental analysis. Uridine (7) and 2,3-isopropylidene uridine 24 could be loaded onto the linker resin in 6 h via a 2 fold excess of 7 or 24 and DBU in DMF. The loaded uridine or 2,3-isopropyliden uridine were cleaved with 30% TFA in CH2Cl2 in 3 h to afford uridine (7) in greater than 85% yields.
Scheme 3.

The linker 23 for immobilizations of uridine derivatives.
3. Conclusion
In conclusion, we have developed a new protecting group, (2,6-dichloro-4-methoxyphenyl)(2,4,6-dichlorophenyl) methoxymethyl chloride (1) for the uridine ureido nitrogen. MDMP protecting group has significant advantages over BOM protecting group for the syntheses of muraymycin and capuramycin analogs systematically in that MDMP group 1) is stable to a wide variety of acids, 2) is also stable to hydrogenation conditions, and 3) can efficiently be deproteced by solvolytic cleavage with TFA (at 30% concentrations) at room temperature within 2 h without the addition of a cation scavenger.11 Similary, the MDMP resin 23 has been developed to immobilize uridine and a uridine derivative. In this article we have demonstrated robustness of the MDMP group in uridine derivatives and utility of the linker resin 23 with a limited number of molecules. Moreover, as BOM group has been widely utilized in organic syntheses, a new protecting group 1 and linker resin 23 described here will be valuable assets to protect not only for ureido nitrogens, but also for primary, secondary, and phenolic alcohols, and carboxylic acids.13 It is worth mentioning that immobilization of the uridine ureido nitrogen on polymer-support is not possible with previously reported linker resins. Utility of 1 and 23 in generation of optimized libraies of uridine-containing antibiotics in solution or on polymer-support will be reported elsewhere.
4. Experimental section
4.1. General
All glassware were oven dried, assembled hot and cooled under a stream of nitrogen before use. Reactions with air sensitive materials were carried out by standard syringe techniques. Commercially available reagents were used as received without further purification. Thin layer chromatography was performed using 0.25 mm silica gel 60 plates visualizing at 254 nm, or stained with anisaldehyde solution by heating with a hot-air gun. Specified products were purified by flash column chromatography using silica gel 60. IR absorptions were performed on NaCl plates. 1H NMR spectral data were recorded on 500 or 400 MHz NMR spectrometer. The residual solvent signal was utilized as an internal reference CDCl3 (7.26). 13C NMR spectral data were recorded at 125, 100 MHz instruments. The residual solvent signal was utilized as an internal reference CDCl3 (77.23). For all NMR spectra, δ values are given in ppm and J values in Hz.
4.2. (2,6-Dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl) methanone (4b)
Anhydrous AlCl3 (450 mg, 3.4 mmol) was added to PhNO2 (10 mL). The reaction mixture was cooled to 0 °C, and 2, 4, 6-trichlorobenzoyl chloride (0.53 mL, 3.4 mmol) and 3,5-dichloroanisole (500 mg, 2.82 mmol) were added. The reaction mixture was stirred at rt for 24 h, then it was diluted with Et2O (10 mL) at 0 °C and quenched by 1 N NaOH (~3 mL). The mixture was stirred vigorously until precipitate was formed. Filter and wash the precipitate with DCM. The organic layer was dried over Na2SO4 and concentrated to give the crude product. Purification by silica gel chromatography (hexanes/EtOAc = 20/1) provided 4b (867 mg, 80%). 1H NMR (CDCl3, 500 MHz) 7.36 (s, 2H), 6.88 (s, 2H), 3.85 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 194.25, 163.33, 141.89, 138.12, 136.31, 129.29, 127.78, 117.65, 57.03; IR (film): 1633, 1557, 1388, 1341 cm−1; HRMS (EI) m/z = 382.8967 calcd for C14H8Cl5O2 (MH+); found: 382.8975.
4.3. (2,6-Dichloro-4-methoxyphenyl)-(2,4,6-trichloro-phenyl) methanol (rac-5b)
A stirred solution of 4b (385 mg, 1 mmol) in THF (2 mL) was added LiBH4 (2 mL, 2.0 M in THF) dropwise at 0 °C. After 12 h at rt, the reaction mixture was quenched by sat. aq. NH4Cl (4 mL) at 0 °C. The water phase was extracted with Et2O. The combined extract was washed with brine, dried over Na2SO4 and evaporated in vacuo. Purification by silica gel chromatography (hexanes/EtOAc = 5/1) gave rac-5b (368 mg, 95%). 1H NMR (CDCl3, 400 MHz) 7.31 (s, 2H), 6.85 (s, 2H), 6.69 (d, J = 10 Hz, 1H), 3.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.23, 135.91, 135.04, 133.92, 129.57, 127.85, 115.55, 72.82, 55.88; IR (film): 3476, 1457, 1431, 1309 cm−1; HRMS (EI) m/z = 384.9123 calcd for C14H12Cl5O2 [M+H]; found: 384.9116.
4.4. (2,6-Dichloro-4-methoxyphenyl)-(2,4,6-trichlorophenyl) methoxymethyl methyl sulfide (6)
To a stirred suspension of NaH (31 mg, 60% in oil, 0.47 mmol) in THF (0.4 mL) rac-5b (100 mg, 0.26 mmol) in THF (0.3 mL) was added at 0 °C. After 30 min, chloromethyl methyl sulfide (0.044 mL, 0.52 mmol) was added. Reaction mixture was stirred at 0 °C for 3 h, and quenched by sat. aq. NH4Cl. The aqueous layer was extracted with Et2O, and the combined extract was washed with brine, dried over Na2SO4, and evaporated in vacuo. Purification by silica gel chromatography (hexanes/EtOAc = 10/1) gave 6 (114 mg, 98%). 1H NMR (CDCl3, 400 MHz) 7.31 (s, 2H), 6.86 (s, 2H), 6.68 (s, 1H), 4.72 (q, J = 11.6 Hz, 2H), 3.79 (s, 3H), 2.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.26, 136.77, 133.95, 132.79, 129.57, 125.47, 115.53, 75.67, 74.29, 55.70, 14.88; IR (film): 3469, 1544, 1368, 1351 cm−1; HRMS (EI) m/z = 446.8977 calcd for C16H13Cl5O2SNa ([M+Na]+); found: 446.8985.
4.5. (2,6-Dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)-methoxy methyl chloride (1)
To a stirred solution of 6 (447 mg, 1.0 mmol) in CH2Cl2 (2.5 mL) was added sulfuryl chloride (0.08 mL, 1.0 mmol) at rt. The reaction mixture was stirred for 1 h and all volatiles were evaporated to provide the cure product which was solidified by addition of hexanes (2 mL). The white solid was washed with hexanes twice to afford 1 (418 mg, 96%). 1H NMR (CDCl3, 400 MHz) 7.33 (s, 2H), 6.88 (s, 2H), 6.77 (s, 1H), 5.57 (q, J = 6.4 Hz, 2H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.56, 136.83, 136.63, 134.40, 131.73, 129.68, 124.31, 115.62, 80.11, 55.75; IR (film): 3473, 1445, 1309 cm−1; Anal. Calcd. C15H10Cl6O2, C 41.42; H 2.32; Cl 48.91; found C 41.81; H 2.41; Cl 48.97.
4.6. 3-[(2,6-Dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl) methoxymethyl]-1-(3,4-dihydroxy-5-hydroxymethyl tetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (8)
To a stirred solution of uridine (7, 1.83 g, 7.5 mmol) in DMF (15 mL) at 0 °C DBU (1.5 mL, 10.0 mmol) and 1 (2.18 g, 5.0 mmol) were added. After 1 h, MeOH (2 ml) was added, and the reaction mixture was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc, and the combined extract was washed with brine, dried over Na2SO4, and evaporated in vacuo. Purification by silica gel chromatography (DCM/MeOH = 15/1) afforded 8 (2.70 g, 84%). 1H NMR (CDCl3, 400 MHz) 7.67 (d, J = 6.8 Hz, 1H), 7.30 (d, J = 3.6 Hz, 2H), 6.83 (d, J = 4.8 Hz, 2H), 6.57 (s, 1H), 5.77 (d, J = 8.4 Hz, 1H), 5.59 (m, 3H), 4.32 (m, 2H), 4.24 (s, 1H), 3.97 (d, J = 12.0 Hz, 1H), 3.90 (s, 1H), 3.83 (m, 1H), 3.78 (s, 3H), 3.05 (s, 1H), 2.20 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 162.94, 159.36, 151.86, 140.07, 136.64, 134.06, 132.46, 129.49, 125.15, 115.49, 101.61, 93.18, 85.71,77.85,74.79, 70.39, 69.14, 61.69, 55.73, 36.61, 31.51; IR (film): 3435, 1719, 1665, 1440, 1081 cm−1; HRMS (EI) m/z = 640.9819 calcd for C24H22Cl5N2O8 [M+H]; found: 640.9825.
4.7. 1-[5-(tert-Butyldimethylsilanyloxymethyl)-3,4-dihydroxy-tetrahydrofuran-2-yl]-3-[(2,6-dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methoxymethyl]-1H-pyrimidine-2,4-dione (10a)
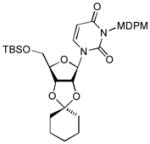
To a stirred solution of 8 (64 mg, 0.1 mmol) in CH2Cl2 (0.5 mL) was added imidazole (14 mg, 0.2 mmol) and TBSCl (18 mg, 0.12 mmol). After 4 h, the reaction mixture was quenched with water and extracted with EtOAc. The combined extract was dried over Na2SO4, evaporated in vacuo. Purified by silica gel chromatography (hexanes/EtOAc = 5/1) provide 10a (62 mg, 82 %). 1H NMR (CDCl3, 400 MHz) δ 7.89 (d, J = 8.4Hz, 1H), 7.28 (s, 2H), 6.81 (s, 2H), 6.56 (s, 1H), 5.76 (t, J = 4.0 Hz, 1H), 5.72 (d, J = 8.4 Hz, 1H), 5.55 (s, 1H), 4.18 (bs, 3H), 4.12 (m, 1H), 3.96 (d, J = 11.6 Hz, 1H), 3.79 (d, J = 11.6 Hz, 1H), 3.75 (s, 3H), 3.13 (bs, 1H), 0.90 (s, 9H), 0.13 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 162.80, 159.33, 152.24, 138.47, 136.65, 134.03, 132.48, 129.47, 125.22, 115.47, 101.46, 91.72, 85.99, 77.93, 76.44, 70.51, 69.17, 62.29, 55.69, 36.57, 31.6, −5.48, −5.56; [α]20D = +28 (c 2.5 in CHCl3); IR (film): 3420, 1701, 1659, 1439, 1255, 1088 cm−1; HRMS (EI) m/z = 755.0684 calcd for C30H36Cl5N2O8Si [M+H]; found: 755.0681.
4.8. 3-[(2,6-Dichloro-4-methoxy-phenyl)(2,4,6-trichlorophenyl) methoxymethyl]-1-(3,4-dihydroxy-5-trityloxymethyl tetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (10b)
To a stirred solution of 8 (128 mg, 0.2 mmol) in pyridine (0.7 mL) was added trityl chloride (67 mg, 0.24 mmol) and DMAP (2 mg). The reaction mixture was stirred at 60 °C for 4 h and cooled to rt. All volatiles were removed. The partition between EtOAc and water was conducted. EtOAc phase was washed with 1 NHCl and brine. The organic phase was dried over Na2SO4 and concentrated in vauo. Purification by silica gel chromatography (hexanes/EtOAc = 6/1) gave the 10b (155 mg, 88%). 1H NMR (500 MHz, CDCl3) δ 7.71 (t, J = 8.5 Hz, 1H), 7.31 (d, J = 7.5 Hz, 6H), 7.25 (m, 6H), 7.22 (m, 5H), 6.74 (s, 2H), 6.47 (s, 1H), 5.64 (s, 1H), 5.51 (m, 2H), 5.39 (d, J = 7.0 Hz, 1H), 4.27 (bs, 1H), 4.14 (bs, 2H), 4.10 (bs, 1H), 3.65 (d, J = 4.5 Hz, 3H), 3.42 (d, J = 11.0 Hz, 1H), 3.33 (m, 1H), 2.94 (bs, 1H),; 13C NMR (125 MHz, CDCl3) δ 162.70, 159.35, 152.09, 143.18, 138.42, 136.68, 134.05, 132.56, 129.51, 128.64, 128.09, 127.48, 125.25, 115.50, 101.65, 91.90, 87.56, 84.51, 77.86, 76.19, 70.61, 69.17, 62.40, 55.71, 25.64; [α]20D = +23 (c 1.5 in CHCl3); IR (film): 3425, 1718, 1669, 1446, 1096 cm−1; HRMS (EI) m/z = 883.0914 calcd for C43H36Cl5N2O8 [M+H]; found: 883.0916.
4.9. 3-[(2,6-Dichloro-4-methoxy-phenyl)(2,4,6-trichloro-phenyl)methoxymethyl]-1-(3,4-dihydroxy-5-triisopropylsilanyloxymethyltetrahydrofuran-2-yl)-1H-pyrimidine-2,4-dione (13)
The same procedure for the synthesis of 10a was applied, but TIPSCl was used. Yield: 90%. 1H NMR (500 MHz, CDCl3) δ 7.90 (q, J = 8.0 Hz, 1H) 7.30 (s, 2H), 6.83 (d, J = 1.5 Hz, 2H), 6.57 (s, 1H), 5.74 (m, 2H), 5.57 (m, 2H), 4.32 (bs, 1H), 4.19 (m, 2H), 4.06 (d, J = 12.5 Hz, 1H), 3.77 (s, 3H), 2.92 (bs, 1H), 1.16 (m, 3H), 1.08 (d, J = 6.5 Hz, 9H); 13C NMR (125 MHz, CDCl3) δ 162.68, 159.35, 152.37, 138.26, 136.69, 134.05, 132.58, 129.50, 125.25, 115.50, 101.48, 92.01, 86.31, 77.95, 76.64, 70.59, 69.17, 62.55, 55.72, 25.65, 18.02, 11.85, −3.58; [α]20D = +27 (c 5.5 in CHCl3); IR (film): 3422, 1700, 1657, 1441,1243, 1093 cm−1; HRMS (EI) m/z = 797.1153 calcd for C33H42Cl5N2O8Si [M+H]; found: 797.1159.
4.10. General procedure of desilylations
To a stirred solution of 13 (26 mg, 0.03 mmol) in MeCN (0.5 mL) at rt was added 50% HF (100 μL). After 5 h, the reaction mixture was quenched by aq. NaHCO3. The organic layer was washed with brine, and then dried over Na2SO4. Purification by silica gel chromatography (hexanes/EtOAc = 4/1) gave the product 8 (19 mg, 88%).
4.11. Cyclohexylidenation of 10a
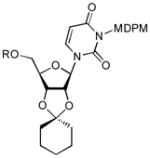
To a stirred solution of 10a (75.6 mg, 0.1 mmol) in MeCN (0.5 mL) were added dipent-4-enyl acetal (30 mg, 0.12 mmol), NBS (47 mg, 0.26 mmol), and BF3-OEt2 (0.01 mmol). The reaction mixture was stirred at rt and protected from light with an aluminum foil for 15 min. The reaction mixture was quenched with Et3N (50 μL), and extracted with CH2Cl2. The organic extract was washed with 10% Na2S2O3, and sat. aq. NaHCO3, and dried over Na2SO4. Purified by silica gel chromatography (hexanes/EtOAc = 10/1) afforded 11a (63 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 3.5 Hz, 2H), 6.82 (d, J = 3.5 Hz, 2H), 6.59 (d, J = 8.5 Hz, 1H), 5.94 (d, J = 4.0 Hz, 1H), 5.72 (d, J = 8.0 Hz, 1H), 5.59 (m, 2H), 4.73 (m, 1H), 4.65 (m, 1H), 4.29 (d, J = 2.5 Hz, 1H), 3.93 (d, J = 11.5 Hz, 1H), 3.79 (d, J = 11.5 Hz, 1H), 3.77 (s, 3H), 1.77 (m, 2H), 1.67 (m, 2H), 1.57 (m, 4H), 1.40 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 162.53, 159.24, 151.44, 141.79, 136.70, 133.94, 132.58, 129.47, 125.18, 115.50, 114.94, 102.13, 97.64, 87.33, 83.16, 79.85, 78.03, 69.25, 63.28, 55.68, 37.17, 34.89, 25.89, 24.91, 23.95, 23.63, 18.33, −5.40, −5.45; Yield:75%. [α]20D = +19 (c 2.0 in CHCl3); IR (film): 1726, 1703, 1656, 1442, 1261, 1088 cm−1;. HRMS (EI) m/z = 835.1310 calcd for C36H44Cl5N2O8Si [M+H]; found: 835.1316.
4.12. 3-[(2,6-Dichloro-4-methoxyphenyl)-(2,4,6-trichloro phenyl)methoxymethyl]-1-(2-cyclohexyl-6-trityloxymethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-1H-pyrimidine-2,4-dione (11b)
Procedure, see 4. 11. Yield: 90%. 1H NMR (400 MHz, CDCl3)δ 7.50 (dd, J = 4.0 Hz, 1H), 7.39 (d, J = 7.6 Hz, 7H), 7.29 (m, 9H), 6.82 (d, J = 3.2 Hz, 2H), 6.54 (d, J = 8.4 Hz, 1H), 5.85 (d, J = 3.2 Hz, 1H), 5.55 (t, J = 11.6 Hz, 1H), 5.46 (t, J = 10.0 Hz, 1H), 5.36 (bs, 1H), 4.78 (d, J = 6.4, 2H), 4.35 (s, 1H), 3.75 (s, 3H), 3.40 (bs, 2H), 1.74 (m, 2H), 1.59 (m, 2H), 1.25 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.60, 159.33, 152.07, 143.19, 138.02, 136.48, 134.15, 132.76, 129.50, 128.69, 128.09, 127.48, 125.28, 115.51, 101.66, 97.63, 91.91, 87.56, 84.51, 77.80, 76.19, 70.51, 69.19, 62.42, 55.71, 37.15, 34.60, 25.04, 23.55; . [α]20D = +17 (c 2.3 in CHCl3); IR (film): 1736, 1719, 1674, 1444, 1231 1075 cm−1; HRMS (EI) m/z = 963.1540 calcd for C49H44Cl5N2O8 [M+H]; found: 963.1544.
4.13. 3-[(2,6-Dichloro-4-methoxy-phenyl)-(2,4,6-trichloro-phenyl)-methoxymethyl]-1-(2-cyclohexyl-6-hydroxymethyl-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-1H-pyrimidine-2,4-dione (12)
To a stirred solution of 11b (35 mg, 0.04 mmol) in CH2Cl2 (0.4 mL) at 0 °C was added TolSH (14 mg, 0.12 mmol) and BF3•Et2O (100 μL). After 1 h at rt., the reaction was quenched with sat. aq. NaHCO3. The aqueous layer was extracted with CH2Cl2 three times. The combined organic extract was dried over Na2SO4, and concentrated in vacuo. The crude product was purified by silica gel chromatography (hexanes/EtOAc = 4/1) to provide 12 (26 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.30 (d, J = 5.2 Hz, 2H), 6.83 (d, J = 6.4 Hz, 2H), 6.58 (s, 1H), 5.77 (dd, J = 8.0 Hz, 1H), 5.57 (s, 2H), 5.47 (dd, J = 5.6 Hz, 1H), 5.00 (d, J = 2.8 Hz, 1H), 4.93(dd, J = 3.6 Hz, 1H), 4.28 (d, J = 2.8 Hz, 1H), 3.88 (d, J = 8.0 Hz, 1H), 3.79 (d, J = 8.0 Hz, 1H), 3.77 (s, 3H), 2.50 (bs, 1H), 1.67 (m, 2H), 1.64 (m, 2H), 1.59 (m, 4H), 1.41 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.56, 159.27, 151.34, 141.88, 136.70, 133.96, 132.55, 129.46, 125.17, 115.52, 115.04, 102.16, 97.66, 87.43, 83.15, 79.88, 78.00, 69.26, 62.86, 55.70, 37.12, 34.66, 24.92, 23.98, 23.54; [α]20D = +17 (c1.0 in CHCl3); IR (film): 3464, 1743, 1710, 1665, 1456, 1222 cm−1; HRMS (EI) m/z = 721.0445 calcd for C30H30Cl5N2O8 [M+H]; found: 721.0450.
4.14. Acetonization of 8
To a stirred solution of 8 (64 mg, 0.1 mmol) in acetone (0.6 mL) at 0 °C were added PTSA (2 mg) and 2,2-dimethoxypropane (15 μL, 0.12 mmol). The mixture was stirred for 4 h at the same temperature and concentrated in vacuo. Purification by silica gel chromatography (hexanes/EtOAc = 4/1) afford 14 (59 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 7.33 (s, 1H), 7.30 (d, J = 5.6 Hz, 2H), 6.83 (d, J = 6.0 Hz, 2H), 6.58 (s, 1H), 5.76 (d, J = 8.0 Hz, 1H), 5.55 (bs, 2H), 5.47 (dd, J = 5.6 Hz, 1H), 5.02 (d, J = 2.0 Hz, 1H), 4.94 (d, J = 5.6 Hz, 1H), 4.28 (d, J = 2.8 Hz, 1H), 3.90 (dd, J = 4.8 Hz, 1H), 3.89 (s, 1H), 3.88 (s, 3H), 2.52 (bs, 1H), 1.57 (s, 3H), 1.35 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.55, 159.28, 151.35, 141.74, 136.69, 133.97, 132.53, 129.46, 125.18, 115.46, 114.24, 102.13, 97.58, 87.30, 83.67, 80.34, 78.03, 69.24, 62.84, 55.70, 27.25, 25.25; [α]20D = +17 (c 1.5 in CHCl3); IR (film): 3462, 1740, 1713, 1666, 1448, 1221 cm−1; HRMS (EI) m/z = 681.0132 calcd for C27H26Cl5N2O8 [M+H]; found: 681.0138.
4.15. 1-(6-Benzyloxymethyl-2,2-dimethyl-tetrahydro-furo[3,4-d][1,3]dioxol-4-yl)-3-[(2,6-dichloro-4-methoxy-phenyl)-(2,4,6-trichloro-phenyl)methoxymethyl]-1H-pyrimidine-2,4-dione (15)
To a stirred suspension of NaH (5 mg, 60% in oil, 0.125 mmol) in DMF (0.5 mL) at 0 °C was added 14 (78 mg, 0.1 mmol) in DMF (0.5 mL). After 5 min. BnBr (36 μL, 0.3 mmol) was added. After 3 h, the reaction mixture was quenched with H2O, and extracted with EtOAc. The organic phase was washed with brine, dried over Na2SO4, and evaporated in vacuo to give the crude product. This was used for the study of debenzylation.
4.16. General procedure of hydrogenation
To a solution of 15 (76 mg, 0.1 mmol)in iPrOH/H2O (2:1, 3 mL) was added 10% Pd/C (10 mg) under N2. H2 was intruced by using a balloon. After 2 h, Pd/C was filtered with a celite pad. The filtrate was concentrated in vacuo. Purification by silica gel chromatography (hexanes/EtOAc = 4/1) afforded 14 (65 mg, 95%). Physical data for 14, see 4.14.
4.17 O,O′-((2R,3S,4S,5R)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-(3-(((2,6-dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methoxy)methyl)-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3,4-diyl) S,S′-dimethyl dicarbonodithioate (16)
To a stirred solution of 10a (160 mg, 0.21 mmol) in DMSO (0.3 mL) was added 5 N NaOH (0.2 mL). After 20 min., CS2 (0.2 mL) was added. After an additional 30 min., MeI (0.3 mL) was added. Then the reaction mixture was stirred for 2 h, and quenched by sat. aq. NH4Cl, and extracted with EtOAc. The combined organic extract was washed with brine, dried over Na2SO4, concentrated in vauo. Purification by silica gel chromatography (hexanes/EtOAc = 10/1) gave 15 (135 mg, 69%). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 11.6 Hz, 2H), 6.82 (d, J = 9.2 Hz, 2H), 6.60 (t, J = 7.6 Hz, 1H), 6.55 (d, J = 9.2 Hz, 1H), 6.25 (t, J = 5.2 Hz, 1H), 6.05 (m, 1H), 5.77 (t, J = 6.8 Hz, 1H), 5.60 (m, 2H), 4.45 (s, 1H), 4.03 (dd, J = 27.2 Hz, 2H), 3.76 (s, 3H), 2.59 (s, 3H), 2.52 (d, J = 15.2 Hz, 3H); 0.95 (s, 9H), 0.19 (d, J = 12.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 214.70, 162.61, 159.23, 151.29, 138.36, 136.69, 134.58, 133.93, 132.88, 129.38, 125.23, 115.53, 102.83, 85.97, 84.37, 80.01, 78.77, 77.81, 69.47, 63.35, 55.69, 26.01, 19.11, 18.40, −5.25, − 5.59; [α]20D = +23 (c 2.5 in CHCl3); IR (film): 1711, 1648, 1446, 1259, 1079 cm−1; HRMS (EI) m/z = 956.9699 calcd for C34H39Cl5N2O8S4SiNa ([M+Na]+); found: 956.9702.
4.18. 1-[5-(tert-Butyl-dimethylsilanyloxymethyl)-2,5-dihydrofuran-2-yl]-3-[(2,6-dichloro-4-methoxyphenyl)(2,4,6-trichloro-phenyl)methoxymethyl]-1H-pyrimidine-2,4-dione (17)
To a stirred solution of 15 (47 mg, 0.05 mmol) in toluene (0.5 mL) were added AIBN (4 mg) and nBu3SnH (0.07 mL, 0.25 mmol) at 100 °C. After 30 min, the reaction mixture was cooled to rt and all volatiles were evaporated in vacuo. Purification by silica gel chromatography (hexanes/EtOAc = 10/1) gave 16 (26 mg, 71%). 1H NMR (400 MHz, CDCl3) δ 7.78 (m, 1H), 7.31 (d, J = 2.0 Hz, 2H), 6.88 (bs, 1H), 6.75 (d, J = 4.8 Hz, 2H), 6.23 (d, J = 9.2 Hz, 1H), 5.81 (m, 1H), 5.69 (m, 1H), 5.61 (m, 2H), 4.88 (bs, 1H), 3.87 (m, 1H), 3.78 (m, 1H), 3.69 (s, 1H), 3.68 (s, 3H), 0.90 (s, 9H), 0.17 (d, J = 12.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 163.10, 159.26, 139.28, 136.78, 133.94, 132.83, 129.42, 126.83, 125.24, 115.43, 101.80, 90.59, 87.41, 77.80, 69.21, 64.14, 55.68, 25.92, 18.52, −5.36, −5.50; [α]20D = +20 (c 1.5 in CHCl3); IR (film): 1705, 1658, 1443, 1255, 1079 cm−1; HRMS (EI) m/z = 721.0629 calcd for C30H34Cl5N2O6Si [M+H]; found: 721.0623.
4.19. 1-[5-(tert-Butyl-dimethylsilanyloxymethyl)-tetrahydro furan-2-yl]-3-[(2,6-dichloro-4-methoxyphenyl)(2,4,6-trichlorophenyl)methoxymethyl]-1H-pyrimidine-2,4-dione (18)
The procedure described for 15 was applied. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 1H), 7.29 (d, J = 2.0 Hz, 2H), 6.77 (d, J = 4.8 Hz, 2H), 6.21 (d, J = 9.2 Hz, 1H), 5.85 (m, 1H), 5.78 (m, 1H), 5.63 (m, 1H), 4.80 (bs, 1H), 3.82 (m, 1H), 3.71 (m, 1H), 3.68 (s, 1H), 3.66 (s, 3H), 2.15 (m, 2H), 1.85 (m, 2H), 0.90 (s, 9H), 0.17 (d, J = 12.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 163.11, 159.22, 139.25, 136.79, 133.96, 132.85, 125.21, 115.40, 101.87, 90.64, 87.39, 77.82, 69.19, 64.11, 55.66, 28.01, 25.95, 21.33, 18.55, −5.35, −5.51; [α]20D = +21 (c 1.3 in CHCl3); IR (film): 1707, 1650, 1440, 1245, 1083 cm−1; HRMS (EI) m/z = 723.0785 calcd for C30H36Cl5N2O6Si [M+H]; found: 723.0789.
4.20. Polymer-supported (2,6-dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl)methanone 21
Hydromethylstyrene resin 19 (purchased from Aldrich, 1.0 g, ~2 mmol) was washed with THF. 19 was suspended in THF (20 mL) and TPP (5.24 g, 20 mmol), DIAD (3.93 ml, 20 mmol) and 20 (1.11 g, 3 mmol) were added. The reaction mixture was gently stirred for 12 h. The polymer-resins were filtered, and thoroughly washed with THF/H2O (4:1), THF, EtOAc, and hexanes, and dried dried under high vacuum to give 21 (1.75 g).
4.21. Polymer-supported (2,6-dichloro-4-methoxyphenyl) (2,4,6-trichlorophenyl)methanol 22
To a gently stirred polymer-resin 21 (1.75 g) in THF (8 mL) was added LiBH4 (10 ml, 2.0 M in THF) at 0 °C. After 12 h at rt, the reaction mixture was quenched with H2O (5 mL). The polymer-resins were thoroughly washed with THF/1% HCl (4:1), THF, EtOAc and hexanes, and dried dried under high vacuum to afford the polymer-resin 22 (1.66 g).
4.22. Polymer-supported MDPM-Cl 23
To a suspension of 22 (0.44 g) in THF (10 mL) was added NaHMDS (0.6 mL, 1.0 M in THF, 0.6 mmol). After 30 min, chloromethyl methyl sulfide (0.09 ml, 1.3 mmol) was added. After 8 h, the polymer was thoroughly washed with THF/H2O (4:1), THF, EtOAc, and hexanes, and dried in vacuo to give the polymer-supported methyl sulfide (464 mg). This was suspended in CH2Cl2 (2 mL) and sulfuryl chloride (0.17 mL, 4.1 mmol) was added. After 5 h, the polymer resins were washed thoroughly with CH2Cl2, and dried under high vacuum to afford 23. Anal. found. Cl 232mg/g.
4.23. Loading uridine on PS-MDPM-Cl resin 23
To a suspension of PS-MDPM-Cl 23 (800 mg) in DMF (1 mL) were added DBU (30μL, 0.2 mmol) and uridine (49 mg, 0.2 mmol). After 3 h, the polymer resins were thoroughly washed with THF/H2O (4:1), THF, EtOAc and hexanes, and then dried under high vacuum to afford 25 (55 mg).
4.24. Loading 24 on PS-MDPM-Cl resin 23
The procedure was same as described in 4.23.
4.25. Cleavage of linker
The resin 25 was washed with CH2Cl2, and added 30 % TFA in CH2Cl2 (2 mL). After 4 h at rt., the polymer resins were washed thoroughly with CH2Cl2. The filtrate was concentrated in vacuo. Purification by silica gel chromatography (CHCl3:MeOH = 10:1) furnished uridine (7) (19 mg, 90%). The resin 26 was cleaved via the same procedure to afford 7 (21 mg, 85%)
Acknowledgments
The authors thank the National Institutes of Health (NIAID grant AI084411-02) and The University of Tennessee for generous financial supports. NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
References and notes
- 1.(a) Yamamoto T, Koyama H, Kurajoh M, Shoji T, Tsutsumi Z, Moriwaki Y. Clinica Chimica Acta. 2011;412:1712–1724. doi: 10.1016/j.cca.2011.06.006. [DOI] [PubMed] [Google Scholar]; (b) Guillemette C, Levesque E, Harvey M, Bellemare J, Menard V. Drug Metabo Rev. 2010;42:24–44. doi: 10.3109/03602530903210682. [DOI] [PubMed] [Google Scholar]
- 2.(a) Bryskier A, Dini AC. Agents: Antibacter Antifung. 2005:377–398. [Google Scholar]; (b) Timothy DH, Lloyd AJ, Roper DI. Infect Disorders: Drug Targets. 2006;6:85–106. doi: 10.2174/187152606784112128. [DOI] [PubMed] [Google Scholar]
- 3.(a) Reddy VM, Einck L, Nacy CA. Antimicro Agents Chemother. 2008;52:719–721. doi: 10.1128/AAC.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ichikawa S. Chem Pharm Bull. 2008;56:1059–1072. doi: 10.1248/cpb.56.1059. [DOI] [PubMed] [Google Scholar]; (c) Dini Christophe. Curr Top Med Chem. 2005;5:1221–1236. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]; (d) Dini C, Drochon N, Feteanu S, Guillot JC, Peixoto C, Aszodi J. Bioorg Med Chem Lett. 2001;11:529–531. doi: 10.1016/s0960-894x(00)00715-0. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kurosu M, Li K, Crick DC. Org Lett. 2009;11:2393–2397. doi: 10.1021/ol900458w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurosu M, Li K. J Org Chem. 2008;73:9767–9771. doi: 10.1021/jo801408x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kurosu M, Biswas K, Crick DC. Org Lett. 2007;9:1141–1145. doi: 10.1021/ol070150f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kurosu M, Narayanasamy P, Crick DC. Heterocycles. 2007;72:339–352. [Google Scholar]; (e) Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Tetrahedron Lett. 2007;48:799–803. [Google Scholar]
- 5.(a) Hanessian S, Kloss J, Sugawara T. J Am Chem Soc. 1986;108:2758–2759. [Google Scholar]; (b) Goff DA, Harris RN, Bottaro JC, Bedford CD. J Org Chem. 1986;51:4711–4714. [Google Scholar]; (c) Connor DS, Klein GW, Taylor GN, Boeckman RK, Medwid JB. Org Syn. 1972;52:C10–19. [Google Scholar]
- 6.Spork AP, Wiegmann D, Granitzka M, Stalke D, Ducho C. J Org Chem. 2011;76:10083–10098. doi: 10.1021/jo201935w. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kurosu M, Li K. Org Lett. 2009;11:911–914. doi: 10.1021/ol8028719. [DOI] [PubMed] [Google Scholar]; (b) Kurosu M, Li K. Synthesis. 2009;21:3633–3641. [Google Scholar]; (c) Kurosu M, Biswas K, Crick DC. Org Lett. 2007;9:1141–1144. doi: 10.1021/ol070150f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kurosu M, Biswas K, Narayanasamy P, Crick DC. Synthesis. 2007;16:2513–2516. [Google Scholar]
- 8.The esters and carbamates synthesized with 5b showed single isomers in their 1H-NMRs and single peaks in their HPLC chromatography analyses.
- 9.Goómez C, Macià B, Lillo VJ, Yus M. Tetrahedron. 2006;62:9832–9839. [Google Scholar]
- 10.(2,6-Dichloro-4-hydroxyphenyl) (2,4,6-trichlorophenyl) methanone (20) was synthesized by demethylation of 4b with HBr in AcOH.
- 11.Mitsunobu O. Synthesis. 1981;29:1–28. [Google Scholar]
- 12.Deprotections of alkoxybenzyl ethers have resulted in good yields when a cation scavenger was added into reaction mixture.
- 13.The BOM group has been utilized to protect alcohols and carboxylic acids. We realized that 1 and 23 could protect or immobilize primary alcohols, phenolic alcohols, and carboxylic acids.