

Abstract
Polymorphoneuclear leukocytes or neutrophils, a major component of white blood cells, contribute to the innate immune response in humans. Upon sensing changes in the microenvironment, neutrophils adhere to the vascular wall, migrate through the endothelial cell (EC)-pericyte bilayer, and subsequently through the extracellular matrix to reach the site of inflammation. These cells are capable of destroying microbes, cell debris, and foreign proteins by oxidative and non-oxidative processes. While primarily mediators of tissue homeostasis, there are an increasing number of studies indicating that neutrophil recruitment and transmigration can also lead to host-tissue injury and subsequently inflammation-related diseases. Neutrophil-induced tissue injury is highly regulated by the microenvironment of the infiltrated tissue, which includes cytokines, chemokines, and the provisional extracellular matrix, remodeled through increased vascular permeability and other cellular infiltrates. Thus, investigation of the effects of matrix proteins on neutrophil-EC interaction and neutrophil transmigration may help identify the proteins that induce pro- or anti-inflammatory responses. This area of research presents an opportunity to identify therapeutic targets in inflammation-related diseases. This review will summarize recent literature on the role of neutrophils and the effects of matrix proteins on neutrophil-EC interactions, with focus on three different disease models: 1) atherosclerosis, 2) COPD, and 3) tumor growth and progression. For each disease model, inflammatory molecules released by neutrophils, important regulatory matrix proteins, current anti-inflammatory treatments, and the scope for further research will be summarized.
Keywords: inflammation, neutrophils, reactive oxygen species, extracellular matrix, endothelial cells, atherosclerosis, COPD, tumor
Introduction
Neutrophils: The Good, The Bad, and The Ugly
The human body is equipped with a dynamic immune system that can sense and respond appropriately to microbial antigens, foreign material, and tissue injury. The immune system can be broadly categorized into two responses: 1) the innate immune response, which acts first to non-specifically attack all foreign bodies or pathogens and 2) the adaptive immune response, which is responsible for the pathogen-specific, long-lasting protective immunity. A primary component of the innate immune system are the polymorphonuclear neutrophils, which are short lived cells that act as the first responders to infection [1]. In case of an acute inflammatory response to a foreign body (i.e., bacteria), the neutrophils from the blood vessels migrate to the site of infection within minutes [1]. This migration of neutrophils across the blood vessel into the extravascular compartment is mediated through temporally sequenced mechanical, chemical, and molecular processes known together as the Leukocyte Adhesion Cascade [2,3]. The process begins with the initial capture of neutrophils from the free blood stream to the endothelial cell (EC) surface, mediated by the interaction of surface molecules, known as selectins, on both cell types. This is followed by neutrophil rolling, in which the cell-cell interaction leads to integrin up-regulation in neutrophils. These integrins are neutrophil-expressed adhesion molecules that bind to surface adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) expressed on ECs. Subsequently, the neutrophils crawl along the vascular lumen and through the vessel wall to reach the extravascular space in a process known as transmigration [4]. The last step of the cascade is the migration of neutrophils through the extracellular matrix (ECM) to reach the source of infection. Migration through the ECM entails neutrophil identification of, adhesion to, and proteolytic destruction of proteins that comprise the tissue matrix. As a defense mechanism, these cells undergo oxidative, non-oxidative, and other intracellular processes within the matrix in order to destroy foreign objects, microbes, proteins, and cell debris [5].
While primarily mediators of tissue homeostasis, there are an increasing number of studies indicating that neutrophil recruitment and transmigration can also lead to host-tissue injury. The innate immunity system is very sensitive and slight changes in the microenvironment can trigger neutrophil recruitment and response [5]. Neutrophil-induced tissue injury is highly regulated by the infiltrated tissue, which includes cytokine, chemokines, and the provisional ECM, remodeled through increased vascular permeability and other cellular infiltrates. The major components of the microenvironment that influence neutrophil recruitment and response at the site of inflammation can be categorized into the following:
The site of inflammation
The pathogen or foreign body has molecular patterns that are recognized by a variety of cells present in the tissue. These cells, in turn, release pro-inflammatory chemokines and cytokines. These chemical factors activate the neutrophils and the ECs to elicit an inflammatory response. For example, lipopolysaccharides can act as pathogen-associated molecular patterns (PAMPs), which increase expression of integrin CD18 and CD11b in neutrophils and induce reactive oxygen species (ROS) production. Certain bacterial lipoproteins, including those from Treponema denticola, also can induce ROS production by neutrophils and exocytosis of specific granules [6]. Another prominent example is the recruitment of neutrophils to the site of inflammation by Mycobacterium tuberculosis, the bacteria that causes tuberculosis. Originally recruited to kill the bacteria, there is evidence suggesting that activated neutrophils cause damage to the host lung tissue and contribute to the development of pathology [6,7].
Endothelial cell layer
The EC layer responds to inflammatory cues by differential expression of surface proteins that abet the recruitment of neutrophils and other immune cells. ECs also release nitric oxide (NO), cytokines, and chemokines to act as signals that modulate inflammation.
Neutrophils
Activated neutrophils release a variety of factors, including proteases (i.e., matrix metalloproteases, neutrophil elastase, collagenase), reactive oxygen species, cytokines, and chemokines. Proteases, cytokines, and chemokines aid in neutrophil recruitment and migration to the site of inflammation, and once there, reactive oxygen species promote pathogen death and phagocytosis. Neutrophil and EC-derived factors during inflammation constitute the majority of the literature in this area of research.
The extracellular matrix
An understudied area of research is the effect of the ECM on inflammation. There is increasing evidence suggesting that matrix proteins such as collagen, laminin, fibronectin, and fibrinogen play a crucial role in neutrophil recruitment. For this reason, matrix proteins should be included in experimental models designed to understand inflammatory diseases.
This review will focus on the role of the ECM in inflammatory diseases, as relates to modulation of neutrophil-EC interactions.
The Extracellular Matrix and Neutrophil-Endothelial Interaction
The majority of the literature representing research of neutrophil-mediated inflammation revolves around elucidating the protein expression profiles of activated neutrophils and ECs and examining the mechanisms by which inflammatory events are regulated by changes in adhesion molecule expression. There is extensive literature covering many aspects of neutrophil-EC interactions focused on the effects of ROS during inflammation and varying surface receptor expression by both cell types [3,8]. The most commonly implicated receptors are integrins, CD11b and CD18 on neutrophils and their ligands ICAM-1, VCAM-1, E-selectin, L-selectin, and P-selectin on ECs [3,4]. Many of these neutrophil integrins are altered in their expression, as an effect of neutrophil transmigration across the endothelium. In fact, CD11b is doubled in its expression, providing more adhesion molecules available for binding ECM ligands. Gonzalez et al. have demonstrated changes in neutrophil adhesion, motility, and velocity as an effect of altered integrin expression post-transendothelial migration [9]. The phenotypically altered neutrophils’ activity within the ECM is, therefore, substantially different from neutrophil response in the vasculature.
Another important aspect of the neutrophil-EC interaction is the effect of the ECM. The question was probably first raised in 1998 when Ketteritz et al. tested and confirmed the hypothesis that neutrophil-matrix interactions influence neutrophil apoptosis [10]. Luscinskas et al. continued this work to demonstrate that the interaction between fibrinogen (an ECM protein) and the CD11b integrin interaction was primarily responsible for signaling events that prolonged the life of the neutrophil [11]. Beyond signaling survival events, others have demonstrated the importance of matrix proteins in migratory events, both within the vasculature and in the extravascular space. Wang et al. have shown that neutrophils prefer transmigrating through regions with low ECM protein localization [12]. Other independent studies have confirmed the presence of these “low expression regions” that act as the preferential sites of neutrophil transmigration through the endothelium [13,14]. The past decade has seen multiple studies examining the interaction between matrix proteins and neutrophil activity [15-18]. In particular, polymer-based hydrogels have been identified as ideal research models to study the effect of matrix proteins. Specific bioactive peptide sequences derived from ECM proteins can be easily immobilized onto polymer chains of (poly)ethylene glycol (PEG), and the resultant bioactive polymer networks can be used to isolate the effects of matrix proteins on neutrophils [19].
ECM proteins can affect activated neutrophils at multiple steps of the leukocyte adhesion cascade. Matrix proteins can regulate neutrophil apoptosis through control of tumor necrosis factor-alpha (TNF-α), indirectly regulating local inflammation [10]. There is also evidence for ECM proteins and activated ECs increasing the lifespan of neutrophils [20]. Ginis et al. were able to demonstrate protection against neutrophil apoptosis as a result of adhesion to matrix proteins fibronectin and laminin and activated EC-coated substrates [20]. Moreover, studies indicate that proteolysis plays a role in neutrophil movement through the basement membranes, an integral part of the inflammation model as well [21].
Extracellular Matrix Proteins and Disease Models
Matrix proteins have been found to have substantial roles in the pathogenesis of an increasing number of inflammatory disease models. Prominently, host-tissue injury occurs as a neutrophil response to signals from ECM proteins in the immediate microenvironment. Activated neutrophils can release a host of matrix modifying proteins, including neutrophil elastase, myeloperoxidase, and defensins, contributing to the formation of atherosclerotic plaques [22-24]. Tumorigenesis, regulation of tumor growth, and tumor cell invasiveness can also be modulated by neutrophil contributions. Because neutrophils can be both anti- or pro-tumorigenic, methods of regulating their tumor cytotoxic abilities are currently being developed [25,26]. Finally, chronic obstructive pulmonary disease (COPD) is yet another inflammation-related disease in which neutrophil-released cytokines, elastase, and oxygen radicals are essential to the pathogenesis of the disease and thereby targets for its treatment [27]. Thus, neutrophil recruitment, transmigration across the EC layer, and migration through the ECM to reach the site of inflammation is key to multiple disease models [1,3,5,8,28].
While neutrophil-EC interactions have been investigated in various disease models, the role of matrix protein regulation of neutrophil activity has been underappreciated and may provide multiple therapeutic opportunities for inflammation related disorders including sepsis and arthritis [29-32]. This review will examine the current literature describing the effects of matrix proteins on neutrophil-EC interaction and its relevance to three different disease models: 1) atherosclerosis, 2) COPD, and 3) tumor growth and progression. We hope to summarize the results so far and identify opportunities for further research. Figure 1 illustrates important neutrophil-ECM interactions across these disease models.
Figure 1.

Neutrophil response to ECM proteins regulates their activity in multiple disease states. a. In the arterial development and progression of atherosclerosis, the activated neutrophil and inflamed EC produce and express proteins that contribute to the formation of the lipid plaque. Accumulation of matrix proteins, including fibrin and collagen, contribute to the immobilization and stabilization of the lipid plaque. Neutrophils and ECs subsequently release proteases and oxidants that destabilize the architecture of the plaque, inducing plaque rupture and immobilization. b. In the post-capillary venule of the lung, neutrophil and EC activation can result in the production of oxidants that damage the surrounding tissue matrix. As neutrophils enter into the remodeled matrix, they continue the process of protease release and continue to remodel the microenvironment, facilitating localized inflammation and irritation, symptomatic of COPD. c. Inflammation in the post-capillary venule contributes to the progression of tumor metastasis. Microvascular EC are activated by tumor-released chemokines to induce neutrophil adhesion and tissue infiltration. Neutrophils release oxidants and proteases that remodel the microenvironment, aiding in tumor cell recruitment to pre-metastatic tissues.
Detailed Review of the Role of Neutrophil and the ECM in Specific Disease Models
Atherosclerosis
Our understanding of atherosclerosis as a disease has evolved over the years. It is characterized by the accumulation of fatty substances that can build up to form plaques along the inner walls of the arterial lumen. The disease starts with fatty streaks on the blood vessels that give rise to fibrous or complex plaques. Plaque formation starts with the transportation and retention of lipoproteins in the arterial wall. The lipoproteins get trapped in a mesh of fibrils and fibers secreted by the arterial wall. The association of these proteins with the ECM forms the plaque, which can be oxidized by neutrophil- and EC-secreted factors [33]. Not only are the plaques composed of fatty lipids, but also platelets, foam cells, fibrinogen, and other ECM proteins. When these plaques rupture, they cause thrombosis. Atherosclerosis is a chronic inflammatory condition that converts itself into a clinically significant disease upon the initiation of plaque formation. In contrast to other inflammatory diseases, the observed inflammation and formation of lipid plaques in atherosclerosis occurs within the structure of the large vessel, rather than smaller post-capillary venule. The location of these plaques would necessitate two important features in the atherosclerotic inflammatory model: First, as a disease that is established in the vasculature, there is little contribution from the extravascular ECM proteins typically associated with neutrophil inflammatory activity in the tissue. Rather, matrix protein found in the blood plasma (i.e., fibrin) contribute to the accumulation of coagulated lipids, cells, and proteins that make up the plaque [34]. Second, the accumulation of platelets and blood matrix proteins on the wall and throughout the core of the plaque will contribute substantially to propagating the pro-inflammatory neutrophil response upon encountering the plaque. Blood flow and the structure of the plaque itself provide an opportunity for direct contact of neutrophils with the fibrotic plaque, allowing the neutrophil to promote continued plaque growth and ultimately destabilization. Assessing the role of neutrophils in the progression of atherosclerosis and destabilization of the atherosclerotic plaque could lead to identification of cellular and protein targets for therapeutic applications.
The role of neutrophils in atherosclerosis
Neutrophils play a distinct role in the formation of atherosclerotic plaques and atherosclerotic progression. Through production and degradation of plaque protein components, neutrophils are key to the development of atherosclerosis and resulting ischemic reperfusion injury, effectively resulting in myocardial infarction and stroke (Figure 2). In the development of atherosclerotic plaques, activated neutrophils contribute early on through the release of the protein Defensin at the site of EC damage [23,35]. Defensin forms stable complexes with low density lipids that effectively results in further immobilization of the complexes. These complexes bind blood plasma proteins, leading to lipid accumulation and plaque formation [36].
Figure 2.
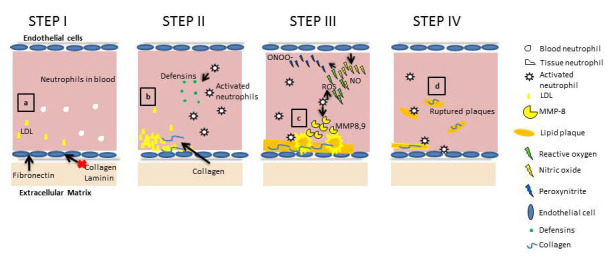
Schematic representation of important regulatory molecules and events during inflammation in atherosclerosis. a. Matrix proteins differentially regulate the activation of EC in response to changes in shear stress applied by blood flow. Fibronectin promotes localized inflammation, while collagen and laminin inhibit flow-induced atherosclerotic inflammation. b. LDL deposits become immobilized by collagen proteins to form atherosclerotic plaques. Meanwhile, platelets, fibrin, and Defensins released by neutrophils contribute to the structural integrity of the developing plaque. c. Subsequently, cytokine-activated neutrophils and ECs release oxidants and proteases that contribute to the destabilization and rupture of lipid plaques. d. Ruptured plaque fracture into the blood stream, entering the heart or brain to induce heart attack or stroke.
Atherosclerotic human arteries with plaques express matrix metalloproteases (MMPs), which are a family of homologus zinc-dependent endopeptidases enzymes capable of inducing plaque rupture by lysis of ECM proteins [37,38,39]. While atherosclerosis has been associated with vascular remodeling since the 1990s, only recently has a direct correlation been made between high neutrophil numbers, high MMP content, and destabilization of plaques [40]. However, the role of neutrophil-released MMPs in atherosclerosis is complex. For example, MMP-9 released by neutrophils during atherosclerosis is regulated by neutrophil gelatinase-associated lipocalin [41]. Gelatinase-associated lipocalin is a high-molecular weight glycoprotein, which acts as a scavenger of bacterial products, thus modulating the inflammatory environment [41]. The differential roles of distinct neutrophil-released MMPs in remodeling and degradation remain unresolved. Generally, MMPS degrade the proteins that make up the plaque, reducing its stability under flow within the vessel, allowing it to disassociate from the vessel wall [42].
While MMPs have been associated with plaque destabilization, the effects of oxidants on degradation of the ECM is a topic of increasing interest. Peroxynitrite, formed in the reaction between endothelial-released nitric oxide and neutrophil-released superoxide ions, has been shown to induce major structural and functional changes to the heparin sulfate proteoglycan perlecan, which leads to weakening of the ECM, thus, increasing the risk for heart attack and stroke [43]. Wang et al. have shown that peroxynitrite can regulate the activity of MMPs, suggesting that ROS may contribute to ECM degradation through MMP activation [44]. Peroxynitrite can oxidize tropoelastin, a monomer of elastin, and has been shown to interfere with matrix elastin fiber assembly and tropoelastin-cell binding, resulting in progression of atherosclerosis [45]. Additionally, as previously mentioned, ROS are intricately involved in the regulation of the interaction of neutrophils and EC adhesion molecules, with evidence suggesting both pro- and anti-inflammatory roles in the literature [46-49]. Another common neutrophil-released enzyme is neutrophil elastase, which can degrade elastin, damaging the matrix. On the other hand, neutrophil-mediated damage can be contained by the production of α1-antitrypsin, an anti-inflammatory agent produced during exercise. It is thought that regular exercise can increase α1-antitrypsin levels, effectively containing the progression of atherosclerosis [50].
The role of matrix proteins in atherosclerosis
Recent reports indicate that matrix proteins may modulate the development and progression of atherosclerosis [48]. This is due to ECM protein-mediated modification or stabilization of lipids deposited on the arterial wall, as well as ECM-mediated regulation of cellular contributors to plaque formation (Figure 2). Atherosclerosis can be induced in rats by feeding them diets with high low density lipid (LDL) content and is modified by extracellular components. In 1994, Nievelstein-Post et al. showed that the LDLs can associate with proteoglycan filaments, joining collagen fibers, which subsequently modify the structure of the lipid [49]. Collagen is of particular interest when it comes to atherosclerosis, because collagen content increases with plaque formation and can make up to 60 percent of the protein content of plaques [51,52]. Again, cellular interaction with collagen is integral to adhesion and transmigration of neutrophils, suggesting that it may play a role in neutrophil response to the collagen abundant plaque. Collagen content of the ECM can be degraded by neutrophil-released factors, including MMP-8 [52,53]. Multiple isotypes of this protein also bind to LDL and contribute to the structural integrity of the plaque [53,54]. Recent findings have led to the hypothesis that the behavior of vascular cells is dictated, in part, by the ECM [42]. Specifically, many matrix proteins appear to have differentially protective effects on EC, while others are pro-inflammatory. Induction of atherosclerosis by disturbed blood flow, for example, can be modified by matrix proteins. When ECs are plated on fibronectin or fibrinogen, they activate the nuclear factor kappa B (NF-κB) pathway in response to flow, but not when they are plated on collagen or laminin. Ligation of specific integrins on collagen prevents flow-induced NF-κB activation [48]. NF-κB is a protein that acts as a transcription factor and hence regulates the expression of multiple proteins with varied effects, including inflammation. Activation of NF-κB upregulates expressions of genes for E-selectin, ICAM-1, and VCAM-1, all of which are modulators of neutrophil-endothelial interactions and are also subject to alteration by ROS [46-48].
Therapeutic opportunities
As our understanding of various inflammatory contributors to atherosclerosis is growing, more and more therapeutic targets are revealing themselves. To inhibit the initial development of plaques, defensin is a plausible target. Modification of MMPs is another route currently proposed as a therapeutic approach [42]. Most recently, biomedical engineers in the field of vascular graft development have created functional blood vessels from synthetic matrices, in which immunological cells are known to contribute to the formation of the complex vascular structure. Roh et al. utilized biodegradable scaffolds composed of a synthetic (poly)glycolic acid mesh and a copolymer sealant solution to seed bone marrow mononuclear cells prior to implantation into the mouse inferior vena cava. After 24 weeks in vivo, the synthetic polymer base had transformed into a functional blood vessel, resembling native mouse vessel with layers of endothelial cells, smooth muscle cells, and collagen fibrils [55]. This work is a clear advancement in the development of vascular biology, creating a model by which the contribution of immunological cells can be elucidated in development of healthy vasculature, but also in the development of aberrant immunological responses, including atherosclerosis.
Additionally, many of the molecules and proteins that alter the ECM can be regulated by the ECM itself. With regard to neutrophils, the key may be to modulate the secretion of neutrophil ROS and MMPs, among other important proteins (i.e., defensins, neutrophil elastase) to promote the stability of atherosclerotic plaques. The matrix composition of the plaque itself dictates many of the processes that regulate neutrophil behavior. For example, collagen and laminin can prevent NF-κB activation to lower peroxynitrite levels, subsequently general inflammation in the local area [48]. Strategies to increase expression of collagen or laminin and decrease expression of fibronectin by the EC layer lining the plaques might prove efficient in decreasing neutrophil attachment to plaques. Another avenue of ongoing research is the development of drugs targeted to inhibit MMPs localized to arterial plaques, thus inhibiting plaque rupture. For example, MMP-1, MMP-13, and MMP-8 are three specific MMPs that are overexpressed in human arethroma [53]. It will be beneficial to develop anti-inflammatory drugs that could be coupled to antibodies specific to one or more of the mentioned MMPs. Silencing MMP-1 or MMP-8 might increase the stability of the atherosclerotic plaques and prevent rupture.
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease is a high-risk lung injury characterized by deterioration of small airways, emphysema, and, in some cases, bronchitis. It is often associated with inflammation and structural remodeling [27]. Some of the key concepts regarding the role of neutrophils in COPD, the effect of matrix proteins, and therapeutic opportunities have been summarized below.
The role of neutrophils in COPD
During the inflammatory response, neutrophils release oxygen radicals, elastase, and cytokines, which contribute to the pathogenesis of COPD [27]. To reach the site of inflammation, neutrophils transmigrate through the endothelial layer of small, post-capillary vessels. To initiate this process, neutrophils become activated by chemoattractants, migrate toward increasing concentrations of the chemoattractant and respond by upregulating surface receptors that attach to leukocyte adhesion molecules, including platelet endothelial cell adhesion molecule (PECAM-1) and ICAM-1 expressed by ECs (Figure 3) [3,27]. ICAM-1 and PECAM-1 serve as adhesion molecules, providing an anchor for firm neutrophil adhesion to and subsequent migration across the EC. In order to access the extravascular matrix, neutrophils have been observed to migrate directly through the EC cell body, though their preferred course of migration occurs at bi-and tri-cellular junctions between ECs. Besides an upregulation of adhesion molecules, the site of transmigration is also marked by low expression of matrix proteins (i.e., laminin) [12]. Soon after migrating across the EC layer, neutrophils interact with matrix proteins through β1 and β3 integrins found on the surface of the neutrophil. Matrix metalloproteases (including MMP-8, MMP-9) and neutrophil elastase are released by neutrophils to play a key role in degrading the ECM and provide physical pathways for matrix migration of more neutrophils during airway inflammation [56-60].
Figure 3.
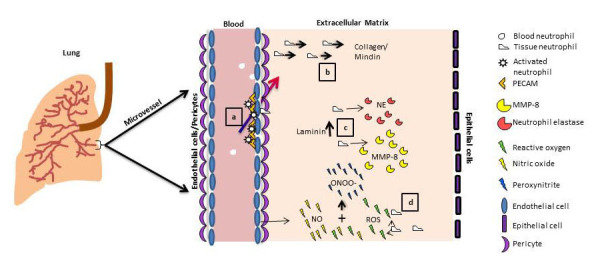
Schematic representation of important regulatory molecules and events during inflammation in COPD. a. Adhesion molecules, including PECAM, regulate neutrophil attachment to EC, encouraging neutrophil entry into the ECM. b. Neutrophils are recruited by matrix proteins, including collagen and mindin, to infiltrate the extravascular space. c. Attachment to matrix protein Laminin induces neutrophil elastase release. d. Activated neutrophils and EC continue to release both pro- (ROS) and anti-inflammatory (NO) factors. These contribute to the remodeling of the ECM and recruitment of additional leukocytes, all leading to emphysema symptomatic of COPD. (Lung image adaped with permission from http://www.buzzle.com/articles/restrictive-lung-disease.html.)
In COPD, endothelial cell mediated NO production is impaired, and ICAM-1 expression is up-regulated [61,62]. NO levels are increased in the exhaled breath of COPD patients during exacerbations in COPD symptoms [63]. NO combines with superoxide anion, or ROS, to form peroxynitrite [64]. Due to the combination of elevated NO and ROS release, patients with COPD also have higher levels of peroxynitrite in the breath condensate and sputum. The ability of endogenous antioxidants present in the lungs to inhibit the activity of peroxynitrite is also reduced, increasing the damage induced by peroxynitrite and its components [65].
In general, the role of NO and peroxynitrite in inflammation has been a matter of debate [44]. Therefore, it is relevant to cite some conflicting reports on how peroxynitrite affects neutrophil-EC interaction [46,47,66,67]. There have been studies indicating that peroxynitrite decreases adhesion of neutrophils to the endothelial surface and may contribute to decreasing the functional interaction between the two cell types [47,66]. On the contrary, other groups have confirmed that peroxynitrite increases adhesion molecule expression and helps to increase the strength of the neutrophil-EC interaction [46,67,68]. Since peroxynitrite levels have been shown to be elevated in the sputum and exhaled air of COPD patients, indicating that the lungs are experiencing more peroxynitrite generation, it is important to understand the role of these species in pulmonary diseases. Again, the role of peroxynitrite, either upstream or downstream of cell-cell and cell-matrix interactions, will be key to the development of therapeutics against extensive tissue damage in COPD. Identification of regulatory proteins governing inflammatory events may also shed light on these interactions.
The role of matrix proteins in COPD
In contrast to the large vessels affected by atherosclerosis, the lung vasculature is closely associated with an epithelial cell layer. The small blood vessels associated within the alveoli lie in close proximity to the epithelium of lung tissue, sharing a basement membrane and ECM (Figure 3). Studies have confirmed that these matrix proteins are active contributors to the process of transmigration through the endothelium of the lung microvasculature. Collagen and collagen-like polypeptides have been shown to serve as chemoattractants for neutrophils, contributing to enhanced inflammation [69,70]. Weathington et al. identified acetylated PRO-GLY-PRO (ac-PGP), a peptide segment derived from collagen, to have chemotactic activity, recruiting neutrophils for an inflammatory response both in vitro and in vivo [58]. It has been suggested that ac-PGP is a collagen-derived molecular mimic of interleukin-8 (IL-8), which activates the neutrophils and causes an upregulation of inflammation, a potential contributor in COPD. The cleavage of ECM proteins that leads to the formation of ac-PGP is mediated by metalloproteases. This fits well into the currently described role of neutrophils, shown to release neutrophil elastase, which contributes to activation of MMP-8, resulting in degradation of matrix collagen [56,63].
Besides collagen fragments, matrix proteins fibronectin, laminin, and elastin expressed in the lung have been shown to influence neutrophil-EC interactions. Particularly, MMP-12-mediated degradation of elastin could result in chemotactic elastin fragments that recruit inflammatory cells, aiding in progression of emphysema [71]. Matrix protein mindin serves as a ligand for the neutrophil integrin and has been shown to be essential for neutrophil recruitment to the site of inflammation [72]. Laminin, a fairly abundant matrix protein, has also been shown to induce neutrophil MMP release and chemotaxis specifically through interactions with the α5 domain [73]. Thus, the complex modulatory effect exerted by the matrix proteins, particularly collagen and laminin, can regulate the level of neutrophil-mediated inflammation in the lung.
Therapeutic opportunities
In summary, chronic obstructive pulmonary disease is widespread, with millions of patients being diagnosed every year. It has emerged to be the third leading cause of death in the United States, 12 years earlier than predicted [74]. Investigation into the effect of matrix proteins, including collagen, laminin, mindin, and their bioactive domains will be valuable areas of research. For example, development of drugs to shield mindin or decrease its expression in the basement membrane may contain neutrophil infiltration and neutrophil-induced inflammation in COPD. Drugs to block specific MMP cleavage sites of abundant matrix proteins, including collagen, could be a promising alternative. Elucidation of individual roles for inflammatory modulators, including elevated peroxynitrite levels in lungs of COPD patients, regulation of integrin receptor molecules, and other signaling factors, will provide a better insight to the causes and therapeutics for COPD.
Revelations of matrix protein components with protective effects against inflammation will be useful in creation of cell-therapeutic strategies. Inducing increased protective matrix proteins production by ECs or epithelial cells lining the basement membrane could significantly alter the lung microenvironment. Biomedical engineers currently are taking advantage of both synthetic and natural matrices to investigate the innate properties of the lung matrix components. Most notably, Miller et al. have demonstrated the use of natural matrix proteins for use in adhesion molecule mediated tissue development. In this work, naturally derived polymers and acellular whole tissue matrices are used as the basis for tissue-engineered bronchioles of the lung. Petersen et al. have produced a human bronchiole model composed of lung fibroblasts embedded in a collagen matrix surrounded by airway smooth muscle cells and bronchial epithelial cells [75-76]. This model can be used to examine airway remodeling events associated with chronic respiratory disease, including COPD. This is but one of the many examples of natural and synthetic ECM mimetics currently used and under development for investigation of inflammation-mediated diseases. Neutrophil elastase is another major target to treat COPD. The major challenge in targeting neutrophil elastase has been the simultaneous accomplishment of potency and selectivity of the drug [60]. Drugs to locally inhibit neutrophil-released factors that degrade the matrix (i.e., MMP-9 and MMP-8) are also attractive options for further research.
Tumor Growth and Progression
The processes of tumor progression and metastasis bear numerous similarities when compared to the process of neutrophil infiltration into tissue during inflammation. Both tumor infiltration into blood vessels and neutrophil infiltration into the matrix involves crossing cell and protein barriers. Both processes necessitate release of proteases, which degrade the matrix, to make way for infiltration. Neutrophil-released neutrophil elastase, MMPs, and cytokines matrix damage can contribute to tumor invasiveness and metastasis [26]. Thus, tumor progression and growth of many cancer types have been related to inflammation mediated factors. In addition, inflammation has been identified as a precursory event to seeding of metastatic tumor cells in unaffected tissue (Figure 4). Tumor cell transmigration, occurring as tumors cells move into the blood vessels, has been shown to be promoted by neutrophils [77]. The role of neutrophil-released inflammatory molecules as a pro-tumorigenic factor and the effect of matrix proteins in the development of cancer are new avenues of research for cancer treatment and therapy.
Figure 4.
Schematic representation of important regulatory molecules and events during inflammation in tumor growth and progression. a. Tumor-secreted GMCSF supports neutrophil recruitment to the tumor site. Simultaneously, matrix proteins, collagen and laminin, are deposited in the microenvironment immediately surrounding the tumor. b. Reactive oxygen species released by invading neutrophils is initially cytotoxic to tumor cells, and the release neutrophil proteases contribute to tumor matrix remodeling. c. Neutrophils, conditioned by VEGF and bFGF (growth factors abundant during tumor angiogenesis), continue to release factors that remodel the matrix and support tumor cell migration into the blood stream. Meanwhile, matrix proteins (i.e., matricellular glycoprotein SPARC) themselves contribute to regulation of tumor growth. d. Tumor-secreted chemokines promote preparatory neutrophil-mediated inflammation in a distant pre-metastatic tissue (lung), allowing for EC activation and matrix remodeling, for subsequent tumor cell invasion and seeding.
The role of neutrophils in tumor progression
Neutrophils are cytotoxic to tumor cells through the release of large amounts of neutrophil elastase and ROS. However, neutrophils can contribute tumor progression by preparing the microenvironment for tumor invasion. Granulocyte-macrophage colony stimulating factor (GM-CSF), present in tumor growth medium, leads to increased expression of neutrophil CD11 and CD18, enabling neutrophil-induced tumor transmigration into the extravascular space [77]. Inflammation in the pre-metastic tissue space is considered an indicator of a pro-metastatic environment. Likewise, tumors have been shown to recruit neutrophils from the blood into the extravascular space through secretion of chemokines by both direct release of interleukins and signaling production of excess IL-8 from vascular cells [78,79]. Neutrophils bind to tumor-secreted chemoattractants, become activated, adhere to the vascular endothelium, and transmigrate through the EC layer and into the ECM. This process is accompanied by neutrophil release of MMPs and ROS, remodeling the ECM (Figure 4) [78]. Figure 4 uses metastasis of breast cancer to the lung as an example to summarize the important events and regulatory molecules in tumor development, which are common to many cancer types.
Neutrophils can facilitate tumor invasiveness and growth by damaging the vasculature through release of multiple proteases and ROS, recruiting tumor cells through the release of cytokines, or by binding directly to the tumor cells to facilitate tumor cell-transmigration (Figure 4) [77,80]. Evidence of these functions has been supported by elevated numbers of neutrophils found in several tumors, including adenocarcinoma, melanoma, and myxofibrosarcoma [81]. Among the various factors released by neutrophils are neutrophil elastase, MMP-9, MMP-8, and cytokines. Neutrophil elastase can effect a broad range of substrates, including many of the ECM proteins [82]. Neutrophil elastase also acts in a dose dependent manner, inducing tumor cell proliferation at low concentrations and cell death at very high concentrations. Houghton et al. have summarized the effects of neutrophil elastase, MMP-8, and MMP-9 in tumor growth very well [26]. MMP-8, MMP-2, and MMP-9 are all released by neutrophils during inflammation and help degrade matrix components for efficient neutrophil infiltration into tissue. These proteases also have been found to contribute to tumor invasiveness by remodeling the tissue matrix [83-85]. Additionally, neutrophils and tumor cells can act together to enhance tumor cell recruitment. GM-CSF released by tumor cells serves as a chemoattractant and stimulant for neutrophils and their release of Oncostatin M. Neutrophil-secreted Oncastatin M promotes tumor progression by enhancing angiogenesis and metastasis. In this manner, both metastatic tumor cells and neutrophils work together to increase tumor progression and invasiveness. Thus, inhibiting Oncostatin M and GM-CSF may be potential avenues to reduce tumor growth and progression [86].
Neutrophils are found in elevated numbers in tumors, and hence, ROS concentrations in tumors and their immediate microenvironment are also high. Neutrophils increase the NO content of murine tumor models and have been shown to be mutagenic, contributing to the burden of genetic abnormalities associated with tumor progression [87]. Altered iNOS and NO production also have been observed in oral cavity cancer, leading to speculations that these molecules might regulate tumor-neutrophil relationships [88]. Peroxynitrite results in nitration of various molecular species, which may have relevance in cancer progression. Peroxynitrite has been shown to produce nitrated genistein, a molecule that alters the tumor-inflammatory environment [89]; however, further research is required to elucidate the effects of altered genistein on tumor invasiveness. Nevertheless, reports indicate that ROS and their intermediates might lead to decreased melanoma cell viability [90]. Again, the role of ROS is concentration dependent, having been shown to be support tumor progression at modest concentrations and cytotoxic to tumor cells and, hence, anti-tumorigenic at high concentrations [26,91].
The role of matrix proteins in tumor progression
The ECM has a variety of proteins and other structural components that can be exploited by neoplastic cells to create a tumorigenic environment. Matrix proteins including syndecan-1, collagen-IV, and laminin have been shown to be overexpressed in tumors and have been used as markers for tumor detection [92]. Vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) are ECM-bound proteins with tumorigenic properties, specifically triggering angiogenesis in the tumorigenic environment. Both VEGF and bFGF stimulate EC production of multiple proteolytic enzymes that assist in EC migration and proliferation (Figure 4). Many studies have demonstrated that upregulation of these factors effectively leads to tumor formation [93,94].
Besides the aforementioned biochemical signals, there is also evidence that the provisional ECM and its component proteins, including fibrinogen, contribute to regulating neutrophil recruitment and tumor metastasis (Figure 4) [95-97]. Multiple types of tumors have been found to contain fibrinogen, and fibrinogen degradation products have been shown to have angiogenic and chemotactic properties [95]. SPARC (secreted protein acidic and rich in cysteine) is a glycoprotein protein found in the ECM and has been shown to have anti-tumor properties. In xenograft tumors, SPARC acts to ablate VEGF, reducing the formation of vasculature throughout the tumor. However, the role of SPARC as a tumor inhibitor is controversial. While SPARC expression has been identified as a marker of aggressive tumors, it has also been shown to have anti-angiogenic properties [94,98].
Therapeutic opportunities
Tumor growth and progression are complex processes involving a multitude of factors. Neutrophil transmigration through the endothelial layer (which is influenced by the ECM) and the ECM itself are both factors that can contribute to the invasiveness of tumor cells. One consistent aspect of recent studies is that many of the effects of inflammatory molecules involved in tumor progression are concentration dependent [26,91]. This information suggests that individual molecules may have dual roles with respect to tumor progression. In general, high concentrations of inflammatory molecules have pro-host effects, while low or modest concentrations have pro-tumor effects. Another pattern observed in the literature is that activated neutrophils create a positive-feedback loop. ECM proteins can regulate activation of neutrophils and abet neutrophil transmigration. Developing strategies to control the release of inflammatory molecules by neutrophils or strategies to interrupt the communication between neutrophils and tumors cells may be key to containing tumor metastasis. In all, identifying matrix proteins and their concentration ranges, which may prevent excessive neutrophil transmigration dictated by tumor-induced chemoattractants, may be a method to curb tumor progression. The role of ROS is also to be studied in light of the matrix composition and how that affects tumor cell viability. To reiterate, one has to be mindful while studying inflammatory contributors to tumor growth and development, as they are only a singular subset of the many factors playing a role in tumorigenesis and tumor progression.
Table 1 summarizes the key molecular species associated with neutrophils, ECs and the ECM that are central to the disease models discussed and possible therapeutic targets.
Table 1. Current anti-inflammatory therapeutic targets.
| Disease/Condition | Neutrophil-released factors | Matrix proteins | Targeted anti-inflammatory therapeutic approaches |
| COPD | 1. MMP-8 | 1.Collagen | 1. Corticosteroids : Non-specific treatment for exacerbations act against IL-8, TNF-α, MMPs [99] |
| 2. Nitric Oxide | 2.Laminin | 2. ICS/LABA – Corticosteroid + β2 Antagonist – increases translocation of glucocorticoid from cell cytosol to the nucleus and hence anti-inflammatory effects [99,100] | |
| 3. Peroxynitrite | 3. Antagonists to Integrin α4β1, selectins, granulocyte adhesion – aimed at decreasing interaction between leukocytes and ECs [99] (Clinical Trials) | ||
| 4. Neutrophil Elastase | 4. Adenosine A2a receptor Agonists: Decreases neutrophil-EC adhesion and release of ROS [99] (Clinical Trial) | ||
| 5. Phosphodiesterase -4 inhibitors: Increase cAMP concentrations in neutrophils, T-cells, leads to anti-inflammatory effects [101-103] | |||
| Atherosclerosis | 1. MMP-8, MMP-9 | 1.Fibrinogen | 1. Statins: Decrease VCAM, ICAM on ECs, inhibit MMPs, lowers LDL [104,105] |
| 2. Peroxynitrite | 2.Collagen | 2. Phospholipase inhibitors: Lowers LDL levels and decreases recruitment of inflammatory cells to plaques [104] | |
| 3. Defensins | 3.Laminin | 3. TNF-α Blockade: inhibits expression of TNFα, IL-8, IL-6, MMP-1, MMP-3 [105] | |
| 4. Mutant MCP-1 gene transfection -Targeting MCP-1, which is involved in developing lesions and inflammation [106] (Animal studies) | |||
| Tumor growth and progression | 1. MMP-2, MMP-8, MMP-9 | 1.Collagen | 1. NSAIDS: Non-steroid anti-inflammatory drugs and reduce inflammation. Shown to reduce cancer occurrence in a variety of cancers [107,108] |
| 2. Oncastatin M | 2. Laminin | 2. Specific COX-2 inhibitors: reduces side effects when given in combination with NSAIDs [108] | |
| 3. Neutrophil Elastase | 3. SPARC | 3. Corticosteroids: reduced side-effects of chemotherapy and also has anti-inflammation and anti-cancer properties [109] | |
| 4. Peroxynitrite | 4. VEGF | ||
| 5. Nitric Oxide | |||
Challenges for Biomedical Engineering
In conclusion, there is increasing evidence that the ECM should be exploited as a therapeutic target against inflammatory diseases. This field of study warrants more focus to elucidate the effect of the abundant matrix proteins (collagen, laminin, fibrinogen, and fibronectin) on neutrophil recruitment and regulation of neutrophil-released factors. Neutrophil-released factors ROS and peroxynitrite remain among the least understood signals in disease models.
The challenge ahead in neutrophil-mediated inflammation research is two-fold. First, it is important to characterize the concentration dependent roles of the neutrophil and EC-derived factors during inflammation for specific disease models. It would then be relevant to explore the options to deliver drugs specifically to inhibit the pro-inflammatory signals or to enhance the effect of protective or anti-inflammatory signals. It also would be beneficial to generate smart materials that can sense the concentrations of these factors in the microenvironment and release anti-inflammatory drugs accordingly. Second, it is imperative to identify matrix proteins that inherently reduce inflammation. These matrix proteins could prove to be ideal for controlling inflammation in disease models. Investigating ways of increasing the specific “protective matrix protein” content of the provisional matrix in diseased tissues would be worthwhile. Protective protein matrix mimetics are being explored as cell-directed therapeutic devices. Further exploration of the molecular pathways responsible for pro- and anti-inflammatory effects of specific proteins and ways to manipulate these effects in vivo will lead to useful therapeutics for inflammatory diseases.
We also recommend some caution while considering these strategies. Different tissue types have strikingly different matrix compositions and, subsequently, support distinct cellular behavior. This matrix variability could pose challenges regarding molecular targets being both disease-specific and tissue-specific. Another important consideration is that the ultimate aim is not to completely inhibit pro-inflammatory signals but reduce them to normal levels in diseased tissues. A corollary to the above is that treatment for inflammatory diseases should be specifically targeted, since systemic effects can backfire by damaging the immune system. More specifically, complete ablation of inflammation could lead to devastating infection. Last, other cell types including macrophages, mast cells, and monocytes are also involved during inflammation and the role of these cells, and their signaling factors should be taken into account for further therapeutic opportunities.
The strategies proposed can be applied to multiple disease models. Here we have outlined only three of many diseases and disorders in which neutrophil function can be controlled by various ECM proteins. Therapeutic targets based on matrix proteins have the potential to target a wide variety of disease ranging from simple allergies to deadly cancers.
Acknowledgments
The authors would like to acknowledge Dr. Chantal Ayers-Sander for critical reading and editing suggestions.
Abbreviations
- EC
endothelial cells
- ECM
extracellular matrix
- COPD
chronic obstructive pulmonary disease
- LDL
low density lipoprotein
- MMP
matrix metalloproteases
- ICAM
intracellular adhesion molecule
- VCAM
vascular cell adhesion molecule
- NF-κB
nuclear factor kappa B
- NO
nitric oxide
- ROS
reactive oxygen species
- PAMPs
pathogen-associated molecular patterns
- PEG
(poly)ethylene glycol
- TNF-α
tumor necrosis factor-alpha
- ac-PGP
acetylated PRO-GLY-PRO
- IL-8
interleukin-8
- GM-CSF
granulocyte-macrophage colony stimulating factor
- VEGF
vascular endothelial growth factor
- bFGF
basic fibroblast growth factor
- SPARC
secreted protein acidic and rich in cysteine
References
- Schmidt EP, Lee WL, Zemans RL, Yamashita C, Downey GP. On, around, and through: neutrophil-endothelial interactions in innate immunity. Physiology. 2011;26(5):334–347. doi: 10.1152/physiol.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seely AJ, Pascual JL, Christou NV. Science review: Cell membrane expression (connectivity) regulates neutrophil delivery, function and clearance. Crit Care. 2003;7(4):291–307. doi: 10.1186/cc1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Simon AL, Eniola-Adefeso O. Engineering Biomaterials for Regenerative Medicine: Host Response to Biomaterials. Cambridge: Springer; 2012. pp. 143–158. [Google Scholar]
- Hickey MJ, Kubes P. Intravascular immunity: the host-pathogen encounter in blood vessels. Nat Rev Immunol. 2009;9(5):364–375. doi: 10.1038/nri2532. [DOI] [PubMed] [Google Scholar]
- Neufert C, Pai RK, Noss EH, Berger M, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein promotes neutrophil activation. J Immunol. 2001;167(3):1542–1549. doi: 10.4049/jimmunol.167.3.1542. [DOI] [PubMed] [Google Scholar]
- Eruslanov E, Lyadova I, Kondratieva T, Majorov K, Scheglov I, Orlova M. et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73(3):1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. 2009;120(20):2012–2024. doi: 10.1161/CIRCULATIONAHA.108.771170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AL, El-Bjeirami W, West JL, McIntire LV, Smith CW. Transendothelial migration enhances integrin-dependent human neutrophil chemokinesis. J Leukoc Biol. 2007;81(3):686–695. doi: 10.1189/jlb.0906553. [DOI] [PubMed] [Google Scholar]
- Kettritz R, Xu YX, Kerren T, Quass P, Klein JB, Luft FC. et al. Extracellular matrix regulates apoptosis in human neutrophils. Kidney Int. 1999;55(2):562–571. doi: 10.1046/j.1523-1755.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- Luscinskas FW, Brock AF, Arnaout MA, Gimbrone MA Jr.. Endothelial-leukocyte adhesion molecule-1-dependent and leukocyte (CD11/CD18)-dependent mechanisms contribute to polymorphonuclear leukocyte adhesion to cytokine-activated human vascular endothelium. J Immunol. 1989;142(7):2257–2263. [PubMed] [Google Scholar]
- Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M. et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203(6):1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduval P, Sillat T, Virtanen I, Dabagh M, Konttinen YT. Immigration check for neutrophils in RA lining: laminin alpha5 low expression regions act as exit points. Scand J Rheumatol. 2010;39(2):132–140. doi: 10.3109/03009740903198980. [DOI] [PubMed] [Google Scholar]
- Voisin MB, Probstl DL, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am J Pathol. 2010;176(1):482–495. doi: 10.2353/ajpath.2010.090510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgquist JD, Quinn MT, Swain SD. Adhesion to extracellular matrix proteins modulates bovine neutrophil responses to inflammatory mediators. J Leukoc Biol. 2002;71(5):764–774. [PubMed] [Google Scholar]
- Anceriz N, Vandal K, Tessier PA. S100A9 mediates neutrophil adhesion to fibronectin through activation of beta2 integrins. Biochem Biophys Res Commun. 2007;354(1):84–89. doi: 10.1016/j.bbrc.2006.12.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella C, Piazzolla G, Spaccavento F, Vella F, Pace L, Antonaci S. Regulatory role of extracellular matrix proteins in neutrophil respiratory burst during aging. Mech Ageing Dev. 2000;119(1-2):69–82. doi: 10.1016/s0047-6374(00)00171-8. [DOI] [PubMed] [Google Scholar]
- Gonzalez AL, West JL, McIntire LV, Smith CW. ECM interactions with neutrophil integrins regulate cell activity: An engineering approach to studying cell motility in inflammation. FASEB J. 2006;20(5):A1080. [Google Scholar]
- Gonzalez AL, Gobin AS, West JL, McIntire LV, Smith CW. Integrin interactions with immobilized peptides in polyethylene glycol diacrylate hydrogels. Tissue Eng. 2004;10(11-12):1775–1786. doi: 10.1089/ten.2004.10.1775. [DOI] [PubMed] [Google Scholar]
- Ginis I, Faller DV. Protection from apoptosis in human neutrophils is determined by the surface of adhesion. Am J Physiol. 1997;272(1 Pt 1):C295–C309. doi: 10.1152/ajpcell.1997.272.1.C295. [DOI] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol. 2010;17(1):9–17. doi: 10.1097/MOH.0b013e3283333930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlaan PA, Reardon CA. Thematic review series: the immune system and atherogenesis. The unusual suspects:an overview of the minor leukocyte populations in atherosclerosis. J Lipid Res. 2005;46(5):829–838. doi: 10.1194/jlr.R500003-JLR200. [DOI] [PubMed] [Google Scholar]
- Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Defensins and cathelicidins: neutrophil peptides with roles in inflammation, hyperlipidemia and atherosclerosis. J Cell Mol Med. 2005;9(1):3–10. doi: 10.1111/j.1582-4934.2005.tb00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen PA, Sallenave JM. Human neutrophil elastase: mediator and therapeutic target in atherosclerosis. Int J Biochem Cell Biol. 2008;40(6-7):1095–1100. doi: 10.1016/j.biocel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71(7):2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- Houghton AM. The paradox of tumor-associated neutrophils: fueling tumor growth with cytotoxic substances. Cell Cycle. 2010;9(9):1732–1737. doi: 10.4161/cc.9.9.11297. [DOI] [PubMed] [Google Scholar]
- Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- Kumar SD, Krishnamurthy K, Manikandan J, Pakeerappa PN, Pushparaj PN. Deciphering the key molecular and cellular events in neutrophil transmigration during acute inflammation. Bioinformation. 2011;6(3):111–114. doi: 10.6026/97320630006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves-Filho JC, de Freitas A, Spiller F, Souto FO, Cunha FQ. The role of neutrophils in severe sepsis. Shock. 2008;30(Suppl 1):3–9. doi: 10.1097/SHK.0b013e3181818466. [DOI] [PubMed] [Google Scholar]
- Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67(5):1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- Davies EV, Hallett MB. Cytosolic Ca2+ signalling in inflammatory neutrophils: implications for rheumatoid arthritis (Review) Int J Mol Med. 1998;1(2):485–490. doi: 10.3892/ijmm.1.2.485. [DOI] [PubMed] [Google Scholar]
- Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF. et al. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203(4):837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA. et al. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91(9):2488–2496. doi: 10.1161/01.cir.91.9.2488. [DOI] [PubMed] [Google Scholar]
- Bini A, Kudryk B, Numano F, Wissler R. Fibrinogen in human atherosclerosis. Atherosclerosis III: Recent Advances in Atherosclerosis Research. Ann NY Acad Sci. 1995;748:461–473. doi: 10.1111/j.1749-6632.1994.tb17342.x. [DOI] [PubMed] [Google Scholar]
- Bdeir K, Cane W, Canziani G, Chaiken I, Weisel J, Koschinsky ML. et al. Defensin promotes the binding of lipoprotein(a) to vascular matrix. Blood. 1999;94(6):2007–2019. [PubMed] [Google Scholar]
- Chavakis T, Cines DB, Rhee JS, Liang OD, Schubert U, Hammes HP. et al. Regulation of neovascularization by human neutrophil peptides (alpha-defensins): a link between inflammation and angiogenesis. Faseb J. 2004;18(11):1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A. et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4(2):197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94(6):2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henney AM, Wakeley PR, Davies MJ, Foster K, Hembry R, Murphy G. et al. Localization of stromelysin gene expression in atherosclerotic plaques by in situ hybridization. Proc Natl Acad Sci USA. 1991;88(18):8154–8158. doi: 10.1073/pnas.88.18.8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G. et al. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. 2010;30(9):1842–1848. doi: 10.1161/ATVBAHA.110.209296. [DOI] [PubMed] [Google Scholar]
- Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J. et al. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26(1):136–142. doi: 10.1161/01.ATV.0000193567.88685.f4. [DOI] [PubMed] [Google Scholar]
- Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85(1):1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- Kennett EC, Rees MD, Malle E, Hammer A, Whitelock JM, Davies MJ. Peroxynitrite modifies the structure and function of the extracellular matrix proteoglycan perlecan by reaction with both the protein core and the heparan sulfate chains. Free Radic Biol Med. 2010;49(2):282–293. doi: 10.1016/j.freeradbiomed.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sawicki G, Schulz R. Peroxynitrite-induced myocardial injury is mediated through matrix metalloproteinase-2. Cardiovasc Res. 2002;53(1):165–174. doi: 10.1016/s0008-6363(01)00445-x. [DOI] [PubMed] [Google Scholar]
- Akhtar K, Broekelmann TJ, Song H, Turk J, Brett TJ, Mecham RP. et al. Oxidative modifications of the C-terminal domain of tropoelastin prevent cell binding. J Biol Chem. 2011;286(15):13574–13582. doi: 10.1074/jbc.M110.192088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn HY, Krotz F, Zahler S, Gloe T, Keller M, Theisen K. et al. Crucial role of local peroxynitrite formation in neutrophil-induced endothelial cell activation. Cardiovasc Res. 2003;57(3):804–815. doi: 10.1016/s0008-6363(02)00786-1. [DOI] [PubMed] [Google Scholar]
- Lefer DJ, Scalia R, Campbell B, Nossuli T, Hayward R, Salamon M. et al. Peroxynitrite inhibits leukocyte-endothelial cell interactions and protects against ischemia-reperfusion injury in rats. J Clin Invest. 1997;99(4):684–691. doi: 10.1172/JCI119212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169(1):191–202. doi: 10.1083/jcb.200410073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievelstein-Post P, Mottino G, Fogelman A, Frank J. An ultrastructural study of lipoprotein accumulation in cardiac valves of the rabbit. Aterioscler Thromb. 1994;14(7):1151–1161. doi: 10.1161/01.atv.14.7.1151. [DOI] [PubMed] [Google Scholar]
- Semple SJ, McKune AJ. Alpha1–Antitrypsin: Anti-inflammatory roles in exercise and atherosclerosis. African Journal of Biochemistry Research. 2011;5(5):143–147. [Google Scholar]
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr.. et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92(5):1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- Plenz GA, Deng MC, Robenek H, Volker W. Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis. 2003;166(1):1–11. doi: 10.1016/s0021-9150(01)00766-3. [DOI] [PubMed] [Google Scholar]
- Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB. et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104(16):1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- Katsuda S, Kaji T. Atherosclerosis and extracellular matrix. J Atheroscler Thromb. 2003;10(5):267–274. doi: 10.5551/jat.10.267. [DOI] [PubMed] [Google Scholar]
- Roh JD, Sawh-Martinez R, Brennan MP, Jay SM, Devine L, Rao DA. et al. Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling. Proc Natl Acad Sci USA. 2010;107(10):4669–4674. doi: 10.1073/pnas.0911465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, Vandivier RW. The matrix degrades, neutrophils invade. Nat Med. 2006;12(3):280–281. doi: 10.1038/nm0306-280. [DOI] [PubMed] [Google Scholar]
- Senior RM, Hinek A, Griffin GL, Pipoly DJ, Crouch EC, Mecham RP. Neutrophils Show Chemotaxis to Type-Iv Collagen and Its 7s Domain and Contain a 67-Kd Type-Iv Collagen Binding-Protein with Lectin Properties. Am J Respir Cell Mol Biol. 1989;1(6):479–487. doi: 10.1165/ajrcmb/1.6.479. [DOI] [PubMed] [Google Scholar]
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS. et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12(3):317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- Vlahos R, Wark PAB, Anderson GP, Bozinovski S. Glucocorticosteroids Differentially Regulate MMP-9 and Neutrophil Elastase in COPD. PLoS ONE. 2012;7(3):e33277. doi: 10.1371/journal.pone.0033277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas SD, Costa E, Guedes RC, Moreira R. Targeting COPD: Advances on low-molecular-weight inhibitors of human neutrophil elastase. Med Res Rev. 2011 June 16; doi: 10.1002/med.20247. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Peinado VI, Barbera JA, Ramirez J, Gomez FP, Roca J, Jover L. et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. 1998;274(6 Pt 1):L908–L913. doi: 10.1152/ajplung.1998.274.6.L908. [DOI] [PubMed] [Google Scholar]
- Cella G, Saetta M, Baraldo S, Turato G, Papi A, Casoni G. et al. Endothelial cell activity in chronic obstructive pulmonary disease without severe pulmonary hypertension. Clin Appl Thromb Hemost. 2005;11(4):435–440. doi: 10.1177/107602960501100410. [DOI] [PubMed] [Google Scholar]
- Maziak W, Loukides S, Culpitt S, Sullivan P, Kharitonov SA, Barnes PJ. Exhaled nitric oxide in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 Pt 1):998–1002. doi: 10.1164/ajrccm.157.3.97-05009. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515–548. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Shiraishi S, Hirata K, Yoshikawa J. Imbalance between levels of nitrogen oxides and peroxynitrite inhibitory activity in chronic obstructive pulmonary disease. Thorax. 2003;58(2):106–109. doi: 10.1136/thorax.58.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xu B, Quilley J, Wong PY. Peroxynitrite attenuates hepatic ischemia-reperfusion injury. Am J Physiol Cell Physiol. 2000;279(6):C1970–C1977. doi: 10.1152/ajpcell.2000.279.6.C1970. [DOI] [PubMed] [Google Scholar]
- Zhao S, Zhang Y, Gu Y, Lewis DF, Wang Y. Heme oxygenase-1 mediates up-regulation of adhesion molecule expression induced by peroxynitrite in endothelial cells. J Soc Gynecol Investig. 2004;11(7):465–471. doi: 10.1016/j.jsgi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6(8):662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Gane J, Stockley R. Mechanisms of neutrophil transmigration across the vascular endothelium in COPD. Thorax. 2012;67(6):553–561. doi: 10.1136/thoraxjnl-2011-200088. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Kimura T, Sakakibara S, Riley DJ, Berg RA. Chemotactic activity of collagen-like polypeptides for human peripheral blood neutrophils. J Leukoc Biol. 1986;39(3):255–266. doi: 10.1002/jlb.39.3.255. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA. et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, Li H, He YW. The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood. 2005;106(12):3854–3859. doi: 10.1182/blood-2005-04-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair-Kirk TL, Atkinson JJ, Broekelmann TJ, Doi M, Tryggvason K, Miner JH. et al. A site on laminin alpha 5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J Immunol. 2003;171(1):398–406. doi: 10.4049/jimmunol.171.1.398. [DOI] [PubMed] [Google Scholar]
- The COPD foundation : About COPD [Internet] [cited 2012 Jan 4]. Available from: http://www.copdfoundation.org/PatientsCaregivers/AboutCOPD/tabid/81/language/en-US/Default.aspx .
- Miller C, George S, Niklason L. Developing a tissue-engineered model of the human bronchiole. Journal of Tissue Engineering and Regenerative Medicine. 2010;4(8):619–627. doi: 10.1002/term.277. [DOI] [PubMed] [Google Scholar]
- Petersen TH, Calle EA, Zhao L, Lee EJ, Gui L, Raredon MB. et al. Tissue-engineered lungs for in vivo implantation. Science. 2010;329(5991):538–541. doi: 10.1126/science.1189345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu QD, Wang JH, Bouchier-Hayes D, Redmond HP. Neutrophil-induced transmigration of tumour cells treated with tumour-conditioned medium is facilitated by granulocyte-macrophage colony-stimulating factor. Eur J Surg. 2000;166(5):361–366. doi: 10.1080/110241500750008899. [DOI] [PubMed] [Google Scholar]
- De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res. 2004;10(15):4895–4900. doi: 10.1158/1078-0432.CCR-03-0760. [DOI] [PubMed] [Google Scholar]
- Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R. et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25(14):2105–2112. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- Orr FW, Warner DJ. Effects of systemic complement activation and neutrophil-mediated pulmonary injury on the retention and metastasis of circulating cancer cells in mouse lungs. Lab Invest. 1990;62(3):331–338. [PubMed] [Google Scholar]
- Tazzyman S, Barry ST, Ashton S, Wood P, Blakey D, Lewis CE. et al. Inhibition of neutrophil infiltration into A549 lung tumors in vitro and in vivo using a CXCR2-specific antagonist is associated with reduced tumor growth. Int J Cancer. 2011;129(4):847–858. doi: 10.1002/ijc.25987. [DOI] [PubMed] [Google Scholar]
- Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med. 2001;164(5):896–904. doi: 10.1164/ajrccm.164.5.2103040. [DOI] [PubMed] [Google Scholar]
- Vayrynen JP, Vornanen J, Tervahartiala T, Sorsa T, Bloigu R, Salo T. et al. Serum MMP-8 levels increase in colorectal cancer and correlate with disease course and inflammatory properties of primary tumors. Int J Cancer. 2011 Sept 14; doi: 10.1002/ijc.26435. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP. et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179(3):1455–1470. doi: 10.1016/j.ajpath.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson V, de la Ballina LR, Munaut C, Wielockx B, Jost M, Maillard C. et al. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. Faseb J. 2005;19(2):234–236. doi: 10.1096/fj.04-2140fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res. 2005;65(19):8896–8904. doi: 10.1158/0008-5472.CAN-05-1734. [DOI] [PubMed] [Google Scholar]
- Sandhu JK, Privora HF, Wenckebach G, Birnboim HC. Neutrophils, nitric oxide synthase, and mutations in the mutatect murine tumor model. Am J Pathol. 2000;156(2):509–518. doi: 10.1016/S0002-9440(10)64755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska E, Puzewska W, Marcinczyk M, Grabowska Z, Jablonski J. iNOS expression and NO production by neutrophils in cancer patients. Arch Immunol Ther Exp (Warsz) 2005;53(2):175–179. [PubMed] [Google Scholar]
- D’Alessandro T, Prasain J, Benton MR, Botting N, Moore R, Darley-Usmar V. et al. Polyphenols, inflammatory response, and cancer prevention: chlorination of isoflavones by human neutrophils. J Nutr. 2003;133(11 Suppl 1):3773S–3777S. doi: 10.1093/jn/133.11.3773S. [DOI] [PubMed] [Google Scholar]
- Dissemond J, Weimann TK, Schneider LA, Schneeberger A, Scharffetter-Kochanek K, Goos M. et al. Activated neutrophils exert antitumor activity against human melanoma cells: reactive oxygen species-induced mechanisms and their modulation by granulocyte-macrophage-colony-stimulating factor. J Invest Dermatol. 2003;121(4):936–938. doi: 10.1046/j.1523-1747.2003.12475.x. [DOI] [PubMed] [Google Scholar]
- Haqqani AS, Sandhu JK, Birnboim HC. Expression of interleukin-8 promotes neutrophil infiltration and genetic instability in mutatect tumors. Neoplasia. 2000;2(6):561–568. doi: 10.1038/sj.neo.7900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salani R, Neuberger I, Kurman RJ, Bristow RE, Chang HW, Wang TL. et al. Expression of extracellular matrix proteins in ovarian serous tumors. Int J Gynecol Pathol. 2007;26(2):141–146. doi: 10.1097/01.pgp.0000229994.02815.f9. [DOI] [PubMed] [Google Scholar]
- Gerwins P, Skoldenberg E, Claesson-Welsh L. Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Crit Rev Oncol Hematol. 2000;34(3):185–194. doi: 10.1016/s1040-8428(00)00062-7. [DOI] [PubMed] [Google Scholar]
- Campbell NE, Kellenberger L, Greenaway J, Moorehead RA, Linnerth-Petrik NM, Petrik K. Extracellular matrix proteins and tumor angiogenesis. [Epub 2010 Jun 29];J Oncol. doi: 10.1155/2010/586905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loike JD, el Khoury J, Cao L, Richards CP, Rascoff H, Mandeville JT. et al. Fibrin regulates neutrophil migration in response to interleukin 8, leukotriene B4, tumor necrosis factor, and formyl-methionyl-leucyl-phenylalanine. J Exp Med. 1995;181(5):1763–1772. doi: 10.1084/jem.181.5.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL. et al. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood. 2000;96(10):3302–3309. [PubMed] [Google Scholar]
- Brabek J, Mierke CT, Rosel D, Vesely P, Fabry B. The role of the tissue microenvironment in the regulation of cancer cell motility and invasion. Cell Commun Signal. 2010;8:22. doi: 10.1186/1478-811X-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podhajcer OL, Benedetti LG, Girotti MR, Prada F, Salvatierra E, Llera AS. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008;27(4):691–705. doi: 10.1007/s10555-008-9146-7. [DOI] [PubMed] [Google Scholar]
- Nicola A, Hanania MC. Antiinflammatory Therapy in COPD: American college of chest physicians [Internet] [cited 2012 Jan 4]. Available from: http://www.chestnet.org/accp/pccsu/antiinflammatory-therapy-copd?page=0,3PCCSU .
- Cazzola M, Hanania NA. The role of combination therapy with corticosteroids and long-acting beta2-agonists in the prevention of exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(4):345–354. doi: 10.2147/copd.2006.1.4.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeh KM, Glaab T. Is there a role for antiinflammatory treatment in COPD? COPD. 2009;6(5):395–403. doi: 10.1080/15412550903143901. [DOI] [PubMed] [Google Scholar]
- Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(6):912–919. doi: 10.1183/09031936.03.00027003. [DOI] [PubMed] [Google Scholar]
- Rabe KF, Bateman ED, O’Donnell D, Witte S, Bredenbroker D, Bethke TD. Roflumilast--an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet. 2005;366(9485):563–571. doi: 10.1016/S0140-6736(05)67100-0. [DOI] [PubMed] [Google Scholar]
- Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov. 2011;10(5):365–376. doi: 10.1038/nrd3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg R, Hansson GK. Treating inflammation in atherosclerotic cardiovascular disease: emerging therapies. Eur Heart J. 2009;30(23):2838–2844. doi: 10.1093/eurheartj/ehp477. [DOI] [PubMed] [Google Scholar]
- Kitamoto S, Egashira K, Takeshita A. Stress and vascular responses: anti-inflammatory therapeutic strategy against atherosclerosis and restenosis after coronary intervention. J Pharmacol Sci. 2003;91(3):192–196. doi: 10.1254/jphs.91.192. [DOI] [PubMed] [Google Scholar]
- Ricchi P, Zarrilli R, Di Palma A, Acquaviva AM. Nonsteroidal anti-inflammatory drugs in colorectal cancer: from prevention to therapy. Br J Cancer. 2003;88(6):803–807. doi: 10.1038/sj.bjc.6600829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayburn ER, Ezell SJ, Zhang R. Anti-Inflammatory Agents for Cancer Therapy. Mol Cell Pharmacol. 2009;1(1):29–43. doi: 10.4255/mcpharmacol.09.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo A, Cengel KA. Anti-inflammatory therapy for pancreatic cancer: a sorely needed advance in therapeutics. Cancer Biol Ther. 2008;7(7):1051–1052. doi: 10.4161/cbt.7.7.6581. [DOI] [PubMed] [Google Scholar]