

Abstract
Nucleoside diphosphate kinase (NDK) is known to form homotetramers or homohexamers. To clarify the oligomer state of NDK from moderately halophilic Halomonas sp. 593 (HaNDK), the oligomeric state of HaNDK was characterized by light scattering followed by X-ray crystallography. The molecular weight of HaNDK is 33,660, and the X-ray crystal structure determination to 2.3 and 2.7 Å resolution showed a dimer form which was confirmed in the different space groups of R3 and C2 with an independent packing arrangement. This is the first structural evidence that HaNDK forms a dimeric assembly. Moreover, the inferred molecular mass of a mutant HaNDK (E134A) indicated 62.1–65.3 kDa, and the oligomerization state was investigated by X-ray crystallography to 2.3 and 2.5 Å resolution with space groups of P21 and C2. The assembly form of the E134A mutant HaNDK was identified as a Type I tetramer as found in Myxococcus NDK. The structural comparison between the wild-type and E134A mutant HaNDKs suggests that the change from dimer to tetramer is due to the removal of negative charge repulsion caused by the E134 in the wild-type HaNDK. The higher ordered association of proteins usually contributes to an increase in thermal stability and substrate affinity. The change in the assembly form by a minimum mutation may be an effective way for NDK to acquire molecular characteristics suited to various circumstances.
Keywords: nucleoside diphosphate kinase, X-ray crystallography, light scattering, oligomerization, halophilic enzyme
Introduction
Halophilic enzymes, especially those from extremely halophilic archaea and the extracellular fractions of moderately halophilic bacteria, are highly stable and functional in the presence of high salt concentrations, a condition under which most nonhalophilic enzymes aggregate and become inactive. Most halophilic enzymes are highly acidic and negatively charged under physiological conditions.1–5 Moderately halophilic bacteria optimally grow in the presence of 1–2M NaCl, and those enzymes that are secreted into the medium or the periplasm space are stable and active in the presence of salts at high concentrations.6, 7 In contrast, cytoplasmic enzymes of moderately halophilic bacteria exhibit less halophilicity than secreted enzymes.
Nucleoside diphosphate kinase (NDK) is a typical “housekeeping” enzyme that catalyzes the transfer of γ-phosphate from nucleoside triphosphates to nucleoside diphosphates. NDK is highly conserved from prokaryotes to eukaryotes; the sequences are >40% identical among different species. NDK is a member of a multifunctional protein family with broad functionality.8–10 Extensive genetic, biochemical, and crystallographic studies on NDK have been carried out. X-ray crystallographic studies of NDK of many organisms from bacteria to humans have demonstrated that all NDKs have common subunits of about 150 residues with a very similar fold based on the βαββαβ fold or “ferredoxin” fold. All known NDKs are confirmed to have an oligomeric structure11, 12 in which dimer units are assembled into more extensive multimers. Three common dimer units assemble into a homohexameric form in NDKs from eukaryotes, archaea, and gram-positive bacteria11, 13 and two common dimers assemble into a tetrameric form in gram-negative bacteria.14–16 In the case of gram-negative bacteria, X-ray structural analysis showed that there are at least two kinds of tetrameric assemblies, Type I and II tetramers, which are observed in the Myxococcus NDK (MxNDK) tetramer and E. coli NDK (EcNDK) tetramer, respectively. The Type I tetramer in MxNDK (PDB ID: 1NHK, 1NLK, and 2NCK) uses the C-terminal region as the monomeric interface, whereas the Type II tetramer in EcNDK (PDB ID: 2HUR) uses a loop comprising residue positions 93–112 for the interface. This region from residue position 93–112 is termed the “Kpn loop.”17 Kpn refers to the “Killer of prune,” and is a point mutation in the abnormal wing disc (awd) gene of Drosophila. This mutation is a substitution of serine for P97 in NDK, and causes dominant lethality in affected individuals.18–20
To investigate the mechanism for subunit assembly of NDK, we chose NDK from moderately halophilic bacteria Halomonas sp. 593 (HaNDK), because we have already shown that HaNDK is unique in having a dimer oligomeric assembly.21 From an amino acid comparison of HaNDK, Pseudomonas NDK (PaNDK) and Myxococcus NDK (MxNDK), it was hypothesized that in the C-terminal region of NDK, residue 134 (glutamic acid in HaNDK, alanine in PaNDK, arginine in MxNDK) plays an important role in the oligomeric assembly.22 We have also reported that the E134A mutation in dimeric HaNDK converted it into a tetramer, whereas the converse A134E mutation in tetrameric PaNDK converted it into a dimer.22, 23 Although it was clear that position 134 is critical for the oligomeric assembly of HaNDK, the molecular mechanism and the association scheme of dimeric and tetrameric assemblies of the wild-type and E134A mutant HaNDKs, respectively, have not been elucidated from a structural point of view.
Thus, we elucidated characteristics of the oligomeric forms of the wild-type and E134A mutant HaNDKs using high-performance size-exclusion chromatograph coupled to a multiangle laser light scattering detector (SEC-MALLS), in combination with structural analysis by X-ray crystallography. The structural information provides evidence that the biological structure of the wild-type HaNDK is a dimer, and that the single amino acid mutation E134A converted dimeric HaNDK into the Type I tetrameric assembly, due to removal of charge repulsion at E134 in HaNDK. A single point mutation is therefore shown to be capable of regulating higher-order oligomeric assembly of HaNDK.
Results
Enzymatic activities of the wild-type and E134A mutant HaNDKs
The specific enzymatic activities of the wild-type and E134A mutant HaNDK were measured and compared with that of the wild-type PaNDK tetramer. Under conditions where the enzyme specific activity of the wild-type HaNDK was set to 100% the specific activity of tetrameric PaNDK was 129% ± 7% and is consistent with previous experiments.21, 22 The specific activity of the tetrameric E134A mutant HaNDK was determined to be 119% ± 9%, which is slightly larger than that of the wild-type HaNDK dimer.
Oligomerization of the wild-type and E134A mutant HaNDKs observed by SEC-MALLS
The gel filtration chromatograms of the wild-type HaNDK, the E134A mutant HaNDK, and bovine serum albumin (BSA, standard sample) are shown in Figure 1(A,B). Under the existence of 0.2M NaCl, peaks belonging to the wild-type HaNDK, the E134A mutant HaNDK, and BSA were eluted at 9.4, 8.8, and 8.4 mL in the gel filtration [Fig. 1(A)], and the molecular mass of the wild-type and E134A mutant HaNDK were determined to be 33.7 ± 2.0 kDa and 62.1 ± 4.3 kDa (Table I). In addition, under the existence of 2.0M NaCl, peaks belonging to the wild-type HaNDK, the E134A mutant HaNDK, and BSA were eluted at 10.7, 9.9, and 9.2 mL in the gel filtration [Fig. 1(B)], and the molecular mass of the wild-type and E134A mutant HaNDK were determined to be 32.1 ± 2.2 kDa and 65.3 ± 5.2 kDa, respectively (Table I). Because the theoretical mass calculated from amino acids sequence was 15.1 kDa for the monomeric HaNDK, the results of SEC-MALLS indicate that the wild-type HaNDK and E134A mutant HaNDK form dimeric and tetrameric structures, respectively, in both 0.2 and 2.0M NaCl solutions.
Figure 1.
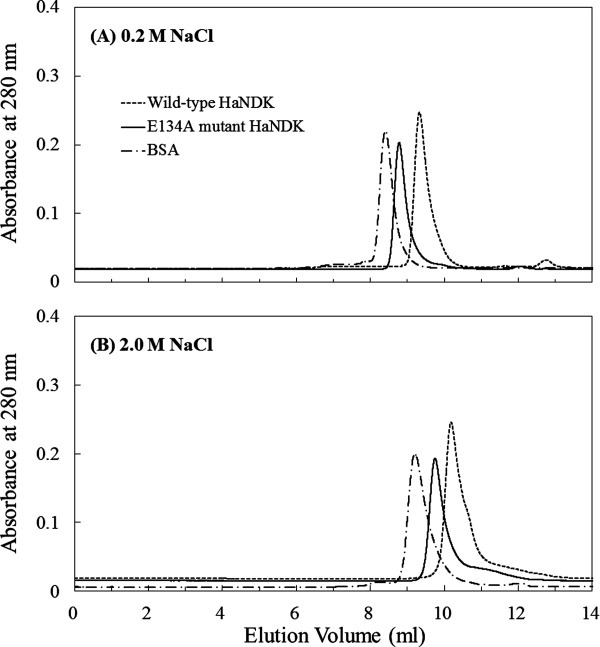
Gel filtration chromatogram of the wild-type and E134A mutant HaNDKs. Approximately 100 μg of wild-type HaNDK (1 mg mL−1) and of the E134A mutant HaNDK (1 mg mL−1) were injected onto SK-GEL G3000 SWXL column equilibrated with 50 mM Tris HCl buffer at pH 8.0, containing 2 mM MgCl2 and (A) 0.2M NaCl or (B) 2.0M NaCl. Approximately 100 μg of the bovine serum albumin (1 mg mL−1) was also analyzed as a standard sample.
Table I.
Molecular Mass of the Wild-Type and E134A Mutant HaNDKs Determined Using Light Scattering Analysis
| Theoretical mass (Da) | ||||||
|---|---|---|---|---|---|---|
| Sample | NaCl in eluting solvent (M) | Observed mass (Da) | Monomer | Dimer | Tetramer | Assignment |
| Wild-type HaNDK | 0.2 | 33,700 ± 2000 | 15,137 | 30,274 | 60,548 | Dimer |
| 2.0 | 32,100 ± 2200 | |||||
| E134A mutant HaNDK | 0.2 | 62,100 ± 4300 | 15,076 | 30,158 | 60,316 | Tetramer |
| 2.0 | 65,300 ± 5200 | |||||
| BSA (standard sample) | 0.2 | 70,800 ± 7400 | 66,776 | — | — | Momomer |
| 2.0 | 72,600 ± 2900 | |||||
The theoretical mass was calculated from amino acids sequence. Assignment of multimer was determined by comparison between observed and theoretical mass.
Crystal packing of the wild-type and E134A mutant HaNDKs
The crystal packing in the asymmetric unit of crystal Forms 1 through 4 is shown in Figure 2. Form 1 and 2 crystals (wild-type HaNDK) have two and five monomers in their asymmetric unit, respectively, which corresponds to one dimer in the asymmetric unit of the Form 1 crystal and 2.5 dimers in the Form 2 crystal. In the Form 2 crystal the monomer colored in red (Fig. 2) forms a dimer with the neighboring symmetric monomers. On the other hand, Form 3 and 4 crystals (E134A mutant HaNDK) have two and four tetramers in their asymmetric unit, respectively. The solvent content was 76.0% for Form 1, 79.8% for Form 2, 48.9% for Form 3, and 49.5% for Form 4. The Form 1 and 2 wild-type structures have higher R-factor, Rfree, and mean B values compared to Forms 3 and 4, resulting from the high mobility of the dimers in Forms 1 and 2 as a likely consequence of the high solvent contents of Forms 1 and 2. In both the Form 1 and 2 crystals, the RMSDs of Cα atoms of each monomer in the asymmetric unit is <0.6 Å. Similarly, in both the Form 3 and 4 crystals, the RMSD of Cα atoms of each monomer in their asymmetric unit is <0.5 Å. These values for RMSD indicate that there are no significant differences in the structures of HaNDK monomers located in the asymmetric unit.
Figure 2.

Crystal packing of wild-type and E134A mutant HaNDKs. (A) and (B) shows the crystal packing of the wild-type HaNDK observed in Form 1 and Form 2 crystals. (C) and (D) shows the crystal packing of the E134A mutant HaNDK observed in Form 3 and 4 crystals. Asymmetric units constructing each crystal packing are surrounded by dotted squares.
Monomer and dimer structures of the wild-type and E134A mutant HaNDKs
The monomeric structure and subunit assembly of the wild-type (Form 1) and E134A mutant HaNDKs (Form 3) are shown in Figure 3. As shown in Figure 3(A,B), the wild-type and E134A mutant HaNDKs were formed in a single α/β domain consisting of a β-sheet of four antiparallel strands and seven α-helices, in which the all β-strands (β1: T3-I10, β2: K33-Q41, β3: P71-E80, and β4: V116-G118) are surrounded by the α-helices. Such a monomeric structure of NDK is similar to the ferredoxin-like fold observed in Pseudomonas aerogenes ferredoxin.24 The monomeric structures of the wild-type and E134A mutant HaNDKs were similar to each other. The RMSDs of Cα atoms of the HaNDK monomers between Form 1 and the other forms were <0.4 Å (Form 2), 0.3 Å (Form 3), and 0.4 Å (Form 4), respectively. The overall structure of HaNDK monomer was conserved despite the E134A mutation and the change in crystal packing.
Figure 3.

Monomeric structure and subunit assembly of wild-type and E134A mutant HaNDKs. (A), the wild-type HaNDK monomer. (B), the E134A mutant HaNDK monomer. In (A) and (B), 134th residues are shown in sticks. Kpn loop is shown in dotted circle. (C), the wild-type HaNDK dimer. (D) and (E), the E134A mutant HaNDK tetramer. The tetramers (D) and (E) are called Type I and II tetramers in this article. (F), Crystal packing in the asymmetric unit of the E134A mutant HaNDK (Form 3). Eight subunits (a–h) were observed in the asymmetric unit of Form 3 crystal. There are two kinds of tetrameric assembly (Type I and II tetramers) in the asymmetric unit of the E134A mutant HaNDK. Tetrameric comformation of Type I tetramer is similar to that of Myxococcus NDK (MxNDK). On the other hand, tetrameric conformation of Type II tetramer is similar to that of E. coli NDK (EcNDK). Red circles in (F) show the dimmer–dimer interfaces in Type I tetramer including mutation site (E134A). Black circles in (F) show the Kpn loops.
The dimer structure of the wild-type HaNDK, in which two HaNDK monomers are related by a noncrystallographic twofold axis is shown in Figure 3(C). In all the crystal forms (Forms 1 through 4), monomer–monomer interfaces for intermolecular interactions were created at α2–α2 (residues 18–30) and at β2–β2 (residues 35–41) contacts. Eighteen intermolecular hydrogen bonds are observed in the monomer–monomer interfaces of the HaNDK dimer (Table II). Twelve out of 18 hydrogen bonds are observed in the α2–α2 interface, 2 hydrogen bonds are observed in the β2–β2 interface, and the other 4 hydrogen bonds are located between R141 of the C-terminal and V15 in the α1 domain and between R141 and N18 in the α2 domain. These interfaces extend the four-stranded antiparallel β-sheet in monomer form to an eight-stranded β-sheet in dimer form. Such a dimer unit of HaNDK has been commonly observed in both the wild-type and E134A mutant HaNDK, as well as other known structures of NDKs. The RMSDs of Cα atoms of HaNDK dimers between Form 1 crystal and other crystals are <0.5 Å (Form 2), 0.4 Å (Form 3), and 0.5 Å (Form 4). The dimer structure shown in Figure 3(C) was conserved in all crystal Forms 1 through 4.
Table II.
Hydrogen Bonds in the Wild-Type and E134A Mutant HaNDKs
| Domain | Residue | Atom | Distance, Å | Atom | Residue | Domain |
|---|---|---|---|---|---|---|
| Monomer–monomer interface of the wild-type HaNDK dimer (Form 1) | ||||||
| α1 | V15 | O | 3.3 | Nη1 | R141 | C-terminal |
| α2 | N18 | Oδ1 | 2.2 | |||
| O | 3.2 | Oε1 | E28 | α2 | ||
| I20 | N | 3.2 | Oε1 | |||
| 3.3 | Oε2 | |||||
| G21 | N | 2.9 | Oε1 | |||
| O | 3.4 | O | G21 | |||
| E22 | N | 3.2 | Oγ | S25 | ||
| S25 | Oγ | 3.3 | N | E22 | ||
| E28 | Oε1 | 2.7 | N | G21 | ||
| 3.3 | O | N18 | ||||
| 3.2 | N | I20 | ||||
| Oε2 | 3.2 | O | N18 | |||
| 3.1 | N | I20 | ||||
| β2 | A37 | O | 2.8 | N | M39 | β2 |
| M39 | N | 3.0 | O | A37 | ||
| C-terminal | R141 | Nη1 | 3.1 | O | V15 | α1 |
| 2.7 | Oδ1 | N18 | α2 | |||
| Dimer–dimer interface of the E134A mutant HaNDK (Type I tetramer in Form3) | ||||||
| Chain | Residue | Atom | Distance, Å | Atom | Residue | Chain |
|---|---|---|---|---|---|---|
| A | E135 | N | 2.9 | O | A130 | b |
| S136 | N | 3.1 | O | Y131 | ||
| Oγ | 2.8 | |||||
| A103 | O | 2.9 | N | E135 | ||
| Y131 | O | 3.0 | N | S136 | ||
| 3.0 | Oγ | |||||
| E | E135 | N | 2.9 | O | A130 | f |
| 3.3 | O | Y131 | ||||
| S136 | N | 2.9 | ||||
| Oγ | 3.2 | |||||
| A130 | O | 2.8 | N | E135 | ||
| Y131 | O | 3.1 | N | S136 | ||
| 2.7 | Oγ | |||||
| Dimer–dimer interface of the E134A mutant HaNDK (Type II tetramer in Form3) | ||||||
| B | K29 | Nζ | 3.2 | Oε1 | E22 | c |
| D88 | Oδ1 | 3.3 | Oε2 | E99 | ||
| f | G101 | O | 2.9 | Nη2 | R26 | g |
| 3.3 | Nη1 | |||||
Names of chains are as in Figure 3(F).
Assembly of common dimer units in the structures of the wild-type and E134A mutant HaNDKs
The Form 1 and Form 2 structures of the wild-type HaNDK were dimer units stabilized by a large number of intermolecular van der Waals contacts. This is the first observation of the biologically active dimer structure of NDK. The number of van der Waals contacts common to both of the crystal forms is 127 for the dimer interface, but is <79 for each neighbor molecule crystal contacts.
On the other hand, in the asymmetric unit of Form 3 crystal of the E134A mutant HaNDK, there are two kinds of dimer–dimer assembly created by a noncrystallographic twofold interaction [Fig. 3(D,E)]. One dimer–dimer interface comprises the 38–43rd and 130–137th residues, and another dimer–dimer interface comprises 26–30th and 85–106th residues. These two assemblies were identified as the Type I and II tetramers seen in MxNDK and EcNDK, respectively.16 This is the first time that Type I and II tetramers appeared simultaneously in the same crystal. The RMSD of Cα atoms in Type I tetramer between the E134A mutant HaNDK and MxNDK (PDB ID: 1NHK, 1NLK, and 2NCK) is <0.8 Å, and the RMSD in the Type II assembly between E134A mutant HaNDK and EcNDK tetramer (PDB ID: 2HUR) is 0.9 Å. The asymmetric unit of Form 3 crystal contains eight copies of the E134A mutant HaNDK (chains a–h), with four dimers each comprising both the Type I and II tetramers, as shown in Figure 3(F). Type I and II tetramers are repeated alternately in the asymmetric unit [Fig. 3(F)]. In the same manner, the asymmetric unit of Form 4 crystal contains alternately repeated Type I and II tetramers, although the numbers of molecules in the asymmetric units of Form 3 (8 molecules) and Form 4 (16 molecules) are different.
Conformation of active site
The NDK active site cleft consists of H117 in the very short β4 (116–118th residue), α4 (59–69th residue), and Kpn loop (93–112th residue).12, 14 H117 located on the bottom of the cleft acts as a catalytic residue of NDK. The active site structures in the wild-type and E134A mutant HaNDK are similar to other known NDKs. The Kpn loop observed in the wild-type and E134A mutant HaNDK has three intramolecular hydrogen bonds, between O of L89 and N of I103, O of M90 and N of R104, and O of G91 and Nη1 of R104, the same as EcNDK [Fig. 4(a)].16 These hydrogen bonding interactions are also observed in all crystal Forms 1 through 4.
Figure 4.
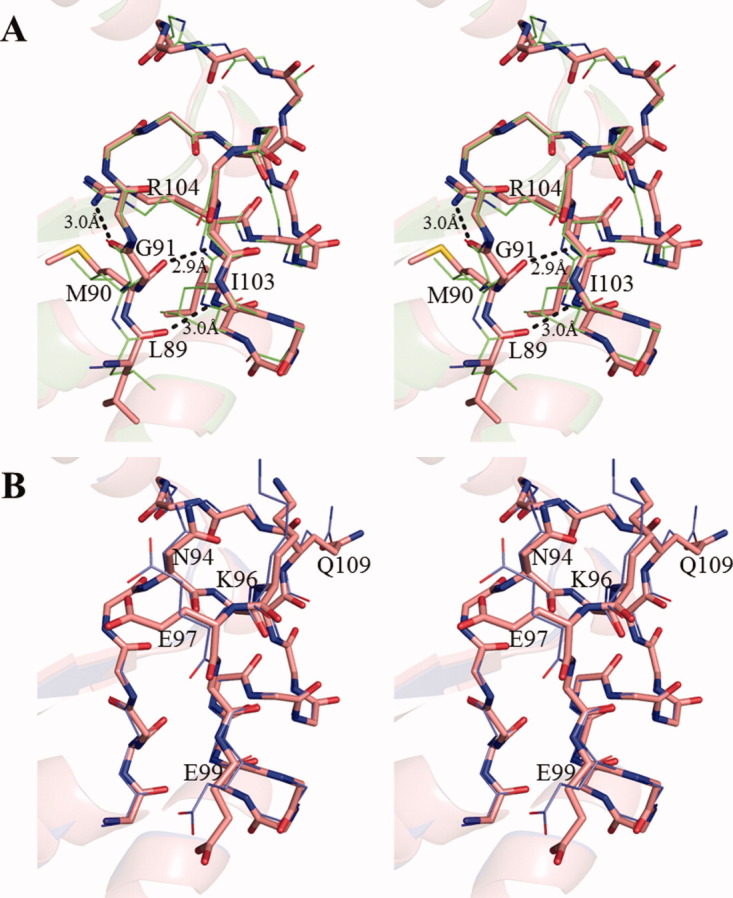
Stereo view of the Kpn loop. (A), Structural comparison of the Kpn loop of Form 1 crystal (red) and EcNDK (PDB ID: 2HUR, green). These structures are superposed based on the main chain atoms. Residues (L89, M90, Q91, I103, and R104) that form the intramolecular hydrogen bonds are shown with the side chains, and the side chains of other residues are omitted. The hydrogen bonds in Form 1 crystal are shown by black dotted line. (B), Structural comparison of Kpn loop of the wild-type HaNDK (red) and the E134A mutant HaNDK (blue). The wild-type and E134A mutant HaNDKs are superposed based on the main chain atoms. N94, K96, E97, E99, and Q109 are shown with the side chains, and the side chains of other residues are omitted.
In addition, the Kpn loop is important for the formation of Type II tetramer in the E134A mutant HaNDK, because the Kpn loop is located in the dimer–dimer interface and interacts with a neighboring molecule [Fig. 3(F)]. The RMSDs of the Kpn loop (93–112th residue) between the wild-type and E134A mutant HaNDKs are <0.6 Å for Cα atoms and 1.1 Å for all atoms. The main chain structures of the wild-type and E134A mutant HaNDKs in the region of the active site are almost the same [Fig. 4(B)], whereas the side chain structures of N94, K96, E97, E99, and Q109 are rotated. The RMSD of all the active site residues (comprising α4, Kpn loop, and β4) between the wild-type and E134A mutant HaNDKs are <0.5 Å for Cα atoms and 1.0 Å for all atoms. It is clear that the active site structures in Type I and II tetramers are similar, though the active sites in Type I and II tetramers are located near the outside and in the interior of tetrameric assembly, respectively.
Discussion
The assembly forms of both the wild type and E134A mutant HaNDKs were identified to be dimer and tetramer, respectively, by X-ray crystallographic analysis and SEC-MALLS. The crystal structure of the dimeric form of NDK was first observed in the case of HaNDK. It was also determined that the tetrameric form of the E134A mutant HaNDK was converted by a single mutation (E134A) from the dimeric wild type structure. Because two kinds of tetrameric assembly, namely Type I and II tetramers shown in Figure 3, were observed in the crystal packing of the E134A mutant HaNDK, the structural property of the dimer–dimer interface in Type I and II tetramers of the E134A mutant HaNDK and the effect of mutation at E134 on the oligomerization of HaNDK are further discussed.
The protein–protein interface of tetramer assemblies observed in the E134A mutant HaNDK
In the crystal structures of the E134A mutant HaNDK shown in Figure 3(F), dimmer–dimer interactions are created by chains ae/chains bf interface or chains cg/chains dh interface (the contact surface area: 821 Å2) in Type I tetramer and by chains bf/chains cg interface (the contact surface area: 1040 Å2) in Type II tetramer. Because the dimeric assembly of the wild-type HaNDK is converted to the tetrameric assembly by the E134A mutation and the mutation site of E134A is not located in the dimer–dimer interface of Type II tetramer but in that of Type I tetramer, it is considered that Type I tetramer is the assembly form of the E134A mutant HaNDK and Type II tetramer is formed by crystal packing.
To investigate how the tetrameric assembly of E134A mutant HaNDK was formed, we further compared the characteristics of the dimer–dimer interface affinities (van der Waals interactions, hydrophobic interactions, hydrogen bonding interactions, and intermolecular water-bridges) of Type I and of Type II tetramers in the crystal structures of the E134A mutant HaNDK determined from Form 3 and 4 crystals [shown in Fig. 2(C,D) and Table IV] by the web-based program PISA25 (URL: http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) and NCONT in the CCP4i package.26
Table IV.
Data Collection and Refinement Statistics of X-Ray Diffraction
| Form 1 (wild-type) | Form 2 (wild-type) | Form 3 (E134A mutant) | Form 4 (E134A mutant) | |
|---|---|---|---|---|
| Collected beamline | PF BL-6A | SPring-8 | SPring-8 | PF BL-6A |
| Wavelength (Å) | 0.978 | 1.0 | 1.0 | 0.978 |
| Space group | R3 | C2 | C2 | P21 |
| Unit cell parameters | a = 112.5 Å | a = 138.2 Å | a = 186.5 Å | a = 112.2 Å |
| b = 112.5 Å | b = 170.6 Å | b = 93.1 Å | b = 92.0 Å | |
| c = 126.0 Å | c = 77.6 Å | c = 68.4 Å | c = 113.6 Å | |
| α = 90.0° | α = 90.0° | α = 90.0° | α = 90.0° | |
| β = 90.0° | β = 93.5° | β = 103.6° | β = 94.7° | |
| γ = 120.0° | γ = 90.0° | γ = 90.0° | γ = 90.0° | |
| Resolutiona (Å) | 2.3 (2.38–2.30) | 2.7 (2.80–2.70) | 2.3 (2.38–.30) | 2.5 (2.59–.50) |
| No. of measured reflections | 102,607 | 139,159 | 129,988 | 171,705 |
| No. of unique reflections | 25,691 (2424) | 89,278 (8,059) | 47,185 (4,559) | 73584 (6,525) |
| Redundancy | 4.1 (2.6) | 1.6 (1.3) | 2.9 (2.3) | 2.5 (1.9) |
| Rmergeab (%) | 4.6 (33.2) | 4.7 (27.7) | 6.3 (25.1) | 10.0 (33.4) |
| Completeness of dataa (%) | 97.3 (92.1) | 91.3 (82.3) | 93.8 (91.0) | 91.9 (82.2) |
| I/sigIa (%) | 26.0 (2.4) | 17.1 (1.7) | 16.0 (2.9) | 9.07 (1.9) |
| R factorac (%) | 26.3 (33.6) | 28.7 (35.0) | 18.9 (21.3) | 23.1 (31.4) |
| Rfreea (%) | 28.9 (36.1) | 31.4 (41.5) | 23.5 (26.2) | 29.8 (34.2) |
| No. of protein atoms | 2185 | 5364 | 8894 | 16,48 |
| No. of water molecules | 57 | 44 | 432 | 279 |
| RMSD bonds (Å) | 0.01 | 0.01 | 0.02 | 0.01 |
| RMSD angles (deg) | 1.13 | 1.32 | 1.63 | 1.30 |
| Mean B value (Å) | 48.1 | 46.8 | 28.8 | 29.5 |
| Used reflection | 24,388 | 47,868 | 44,779 | 73,412 |
| PDB ID | 3VGS | 3VGT | 3VGU | 3VGV |
alues for highest-resolution shells are shown in the parentheses.
Rmerge = S |I − 〈I〉|/SI.
R-factor and Rfree = S ||Fo| − |Fc||/S|Fo|, where the free reflections (5% of total used) were held aside for Rfree throughout refinement.
The van der Waals interactions and hydrophobic interactions in dimer–dimer interfaces of the E134A mutant HaNDK were characterized. The number of van der Waals interactions in dimer–dimer interfaces observed by NCONT are 123 for Type I tetramer and 125 for Type II tetramer. The Gibbs free energy for hydrophobic interactions (ΔiG) in the dimer–dimer interface, which is calculated by PISA and does not include the effect of hydrogen bonds and salt bridges,25 are −50.6 kJ mol−1 for Type I tetramer and −51.5 kJ mol−1 for Type II tetramer. Thus, these characteristics of the contact surfaces of dimer–dimer interface in Type I and II tetramers are indistinguishable by these observations.
Subsequently, we focused on the hydrogen bonding interactions (3.4 Å or less) and intermolecular water-bridges in the dimer–dimer interface of Type I and of Type II tetramers. There are more hydrogen bonds in Type I tetramer than those of Type II tetramer as shown in Table II: there are 13 hydrogen bonds at the dimer–dimer interface of Type I tetramer, whereas there are four hydrogen bonds in Type II tetramer. These hydrogen bonds were observed as follows; the numbers of hydrogen bonds between main chain atoms at the dimer–dimer interfaces are nine for Type I tetramer (i.e., between O of A130 and N of E135, O of Y131 and N of E135, and O of Y131 and N of S136), whereas no hydrogen bonds are observed for Type II tetramer. The numbers of hydrogen bonds between main chain and side chain atoms at the dimer–dimer interfaces are four for Type I tetramer (i.e., between O of Y131 and Oγ of S136), and two for Type II tetramer (i.e., between Nη1 of R26 and O of G101, and O of K29 and Oδ2 of D106). No hydrogen bonds between side chain atoms at the dimer–dimer interfaces are observed for Type I tetramer, whereas two hydrogen bonds are observed for Type II tetramer (i.e., between Oε1 of E22 and Nζ of K29, and Oδ1 of D88 and Oε2 of E99). Average distances of observed hydrogen bonds at the dimer–dimer interfaces are 3.0 ± 0.2 Å for Type I tetramer and 3.2 ± 0.2 Å for Type II tetramer. These results indicate that the hydrogen bonding interactions at the dimer–dimer interface in Type I tetramer are stronger than that in Type II tetramer.
Moreover, to clarify the water-mediated dimer–dimer interactions, water molecules at the dimer–dimer interface in Type I and II tetramers were characterized. There are 11 water molecules in the dimer–dimer interface of Type I tetramer and 19 water molecules in that of Type II tetramer, respectively; thus, there are more water-mediated indirect interactions in the dimer–dimer association of Type II tetramer. This result suggests that the gap at the dimer–dimer interface in Type II tetramer, where water molecules can enter, is larger than that in Type I tertramer. Therefore Type II tetramer may dissociate easier than Type I tetramer.
From these observations, it is concluded that the Type I tetramer reflects the tetrameric assembly of the E134A mutant HaNDK observed in solution conditions such as the gel-filtration experiment.
Effect of mutation at E134 on the oligomerization of wild-type HaNDK
The E134A mutation converts the assembly of HaNDK from a wild-type dimer to a tetramer in solution, which was indicated from our SEC-MALLS and X-ray results. To clarify the effect of mutation at 134th residue and/or neighboring 135th residue on the oligomerization of NDK, we compared oligomeric forms of HaNDK with other NDKs. In Table III, the oligomer states of HaNDK, PaNDK, and MxNDK are summarized.14, 22 Chemical crosslink experiments revealed that HaNDK with glutamic acid at 134th residue, i.e., wild-type HaNDK, and also E135A mutant HaNDK form a dimeric assembly.22 In addition, a mutation of E134 to alanine, i.e., E134A mutant HaNDK and double mutant HaNDK (both 134th and 135th residues are alanine), convert HaNDK to a tetramer. Conversely, tetrameric PaNDK converts to a dimeric assembly with an A134E mutation.22 In both HaNDK and PaNDK, if the 134th residue is alanine, NDKs assemble into a tetramer. The SEC-MALLS and X-ray results in our study are consistent with previous chemical crosslinking experiments.22 Thus, it is clear that the mutation of 134th residue can regulate the oligomeric assembly of HaNDK and PaNDK.
Table III.
List of NDK Experiments From Gram-Negative Bacteria
| Samples | Source | Mutation | Amino acid sequence from 131th | Assembly (cross link)a | Assembly (SEC-MALTTS) | X-ray structure |
|---|---|---|---|---|---|---|
| HaNDK | Halophiles | None (wild-type) | YFFEESEICSR | 2 | 2 | 2.3 Åb |
| Halophiles | E135A | YFFEASEICSR | 2 | |||
| Halophiles | E134A | YFFAESEICSR | 4 | 4 | 2.3 Åb | |
| Halophiles | E134A E135A | YFFAASEICSR | 4 | |||
| PaNDK | non-Halophiles | None (wild-type) | YFFAATEVCERIR | 4 | 4 | |
| non-Halophiles | A135E | YFFAETEVCERIR | 4 | |||
| non-Halophiles | A134E | YFFEATEVCERIR | 2 | |||
| non-Halophiles | A134E; A135E | YFFEETEVCERIR | 2 | |||
| MxNDK | non-Halophiles | None (wild-type) | YFFRETEIHSYPYQK | 2.0 Åc |
To elucidate the reason why the dimeric wild-type HaNDK is converted to a tetramer by the E134A mutation, the tertiary structure of wild-type HaNDK was superposed in a manner based on the location of the Cα atoms of Type I HaNDK tetramer, as shown in Figure 5. This superposition shows that if the wild-type HaNDK forms a tetramer, 4 glutamic acids comprising E134 (chain a), E137 (chain a), E134 (chain b), and E137 (chain b) cluster within 3.5 Å, which should generate an electrostatic repulsive interaction. Even if the side chain of E134 is rotated to avoid steric hindrance against the side chain of E137 in the neighboring HaNDK, the side chain of E134 will then collide with the main chain atoms of Y131, F132, and F133 in the neighboring HaNDK. Therefore, we conclude that the electrostatic repulsion and steric hindrance caused by the side chain of E134 prevent dimer–dimer association of wild-type HaNDK.
Figure 5.
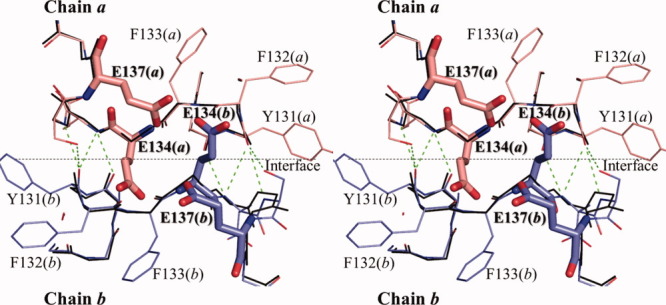
Stereo view of dimmer–dimer interface around 134th residue in Type I tetramer. Main chain structure of the E134A mutant HaNDK dimer (derived from Form 3) formed by chain a and b are shown in thin stick colored in black, in which the side chains are omitted. The interface between chain a and b of the E134A mutant HaNDK is shown by black dotted line. Two wild-type HaNDK monomers (derived from Form 1) are superposed based on Cα atoms of the E134A mutant HaNDK, which are colored in pink and blue. Bold sticks show E134 and E137 of the wild-type HaNDK. Green dotted lines show the intermolecular hydrogen bonds.
Biological meaning for changing the oligomeric form of HaNDK
Although the amino acid sequences are more than 40% identical among NDKs in different species,12 the oligomeric structure varies depending on the organism.11 For example, the hexameric assembly of NDK was widely distributed in eukaryotes, archaea, and some gram-positive bacteria.5, 11, 16 Tetrameric assemblies were observed in gram-negative bacteria (Salmonella typhymurium, Myxococcus xanthus (MxNDK) and E. coil (EcNDK)) and a gram-positive bacteria Streptomyces coelicolor.16 Only two types of tetrameric assemblies of NDK, Type I tetramer in MxNDK (PDB ID: 1NHK, 1NLK and 2NCK) and Type II tetramer in EcNDK (PDB ID: 2HUR), have been determined by X-ray crystallography. In this study, the natural dimeric structure of an NDK was determined for the first time: wild-type HaNDK from gram negative bacteria. Therefore, it is clear that the oligomeric assemblies of NDK from gram negative bacteria range at least from a dimer to Type I and II tetramers,21 although the amino acid sequences of their NDKs are highly conserved (more than 68% identity) as shown in Figure 6. Moreover, the oligomeric structure of HaNDK and of PaNDK can be converted by the single point mutation at 134th residue.22 These results suggest that a small number of amino acid changes are sufficient to convert the NDK oligomeric assembly.
Figure 6.

Amino acid sequence alignments of NDKs from bacteria. NDK_Halomonas: NDK from Halomonas sp. #593 (HaNDK). NDK_Pseudomonas: NDK from Pseudomonas aeruginosa (PaNDK). NDK_Escherichia: NDK from Escherichia coli (EcNDK). NDK_Myxococcus: NDK from Myxococcus xanthus (MxNDK). NDK_Salmonella: NDK from Salmonella typhimurium. These five bacteria belong to gram-negative species. NDK_Streptomyces: NDK from Streptomyces coelicolor. NDK_Bacillus: NDK from Bacillus subtilis. These two bacteria belong to gram-positive species. Sequence homologies highlighted in red; sequence identities are shown as white letters on red background box. The location of secondary structure is schematically shown above the primary sequence of enzyme. β-strands are shown as arrows; α-helices as coils and strict beta turns as TT letters. The figure was created using ESPript.27
The increase of number of assemblies may bring some benefit in the activity and stability of NDK. As with the avidity effect observed in antibody,28 the increment of the local concentration of the substrate binding sites caused by oligomerization increases the affinity of protein with substrates. The specific activity of the tetrameric E134A mutant HaNDK was 19% greater than that of the wild-type HaNDK dimer as mentioned above. The increase in the local concentration of nucleoside binding sites by tetramerization due to the E134A mutation causes the increase in the specific enzymatic activities of HaNDK. Moreover, the increase of the number of assemblies results in increased thermal stability of the protein,29–31 which is mainly caused by the shift in the equilibrium between folded and unfolded state accompanied by oligomerization. Previous circular dichroism measurements revealed that the melting temperature of the mutant HaNDK tetramer is higher than that of the wild-type HaNDK dimer.23 The result that a small number of mutations can affect the oligomeric state of NDK may suggest the effective way for bacterial NDK to adapt to different environmental circumstances.
Material and Methods
Expression and purification of NDK proteins
The construction of the E134A mutant of HaNDK was described previously.22 The wild-type and E134A mutant HaNDK were expressed in E. coli BL21star(DE3) and purified using ATP-agarose affinity chromatography described previously.21 For crystallization, further purification of the wild-type and E134A mutant HaNDKs was performed by ion-exchange chromatography using Hi-Trap Q resin (GE Healthcare).
Enzyme activity assay
The enzyme activity of the purified wild-type and E134A mutant HaNDKs was measured at 303 K by the enzyme-coupling method as described previously.22 The enzyme activity of tetrameric PaNDK was also measured as a control; the protein concentration was 10–20 μg mL−1. These measurements for each NDK were carried out at least seven times to improve accuracy. A reaction mixture (0.5 mL) containing 0.4 mM TDP, 2 mM ATP, 25 mM MgCl2, 3 mM phosphoenolpyruvate, 0.15 mM NADH, 0.1M KCl, 0.8 U pyruvate kinase, 2.3 U lactate dehydrogenase, 0.1M Tris HCl buffer at pH 7.5, and the enzyme sample was incubated at 303 K in a cuvette placed in a photometer chamber. The reaction was detected by the deoxdation of NADH and was followed by A340. The enzyme activity was also measured in a 96-well microtiter plate with the same reaction mixture (0.2 mL) except with a higher NADH concentration of 0.24 mM, and A340 was measured by a plate reader (Bio-Rad Benchmark Plus) at 303 K.
Molecular mass determination by high performance size-exclusion chromatography and multiangle laser light scattering (SEC-MALLS)
Light scattering analysis was performed using a miniDAWN multiangle laser light scattering detector (Wyatt Technology) equipped with a gel filtration column. Approximately 100 μg of the wild-type HaNDK (1 mg mL−1) and of the E134A mutant HaNDK (1 mg mL−1) were injected onto an SK-GEL G3000 SWXL size exclusion column equilibrated with 50 mM Tris HCl buffer at pH 8.0, 2 mM MgCl2 and 0.2M or 2.0M NaCl, and eluted at a flow rate of 0.5 mL min−1. The elution of HaNDK protein was monitored by both refractive index and UV absorbance. Approximately 100 μg of bovine serum albumin (1 mg mL−1, molecular weight: 66,776) was also analyzed as a standard sample.
Crystallization of wild-type and E134A mutant HaNDKs
Initial screening for crystallization conditions of the wild-type and E134A mutant HaNDKs proteins was performed using the sitting-drop vapor-diffusion method in a 96-well Intelliplate (Hampton Research) and Hydra II Plus One crystallization workstation (Matrix Technology) at 293 K. In both the crystallization experiments of the wild-type and the mutant HaNDKs, protein was dialyzed against 50 mM Tris HCl buffer (pH 8.0) containing 0.2M NaCl and 2 mM MgCl2. The dialyzed protein solutions (60 mg mL−1 wild-type HaNDK and 57 mg mL−1 mutant HaNDK) were stored at 277 K for several weeks. Sitting drops were prepared by mixing 0.3 μL each of the protein solution and the reservoir solution, and the resulting drop was equilibrated against 70 μL of reservoir solution. The initial search for crystallization conditions was performed using commercially available precipitant solutions including PEG/ION Screen I and II, Crystal Screen I and II (Hampton Research) and Wizard Screen I and II (Emerald Biostructures). After initial screening identified useful crystallization conditions, protein concentration and precipitant conditions were optimized to obtain diffraction-quality crystals.
In the case of wild-type HaNDK, crystals were obtained in two different morphologies (Form 1 and Form 2). Form 1 was a cubic shaped crystal grown from 0.2M ammonium acetate, 0.1M sodium citrate tribasic dehydrate at pH 5.6, and 30%(w/v) polyethylene glycol (PEG) 4000 (i.e., Crystal Screen I, solution No. 9) with 60.0 mg mL−1 protein. Form 2 was a rod-shaped crystal grown from 0.19M sodium acetate, 0.09M Tris HCl at pH 8.5, and 28%(w/v) PEG 4000 (originated from Crystal Screen I, solution No.22) with 60.0 mg mL−1 protein.
The E134A mutant HaNDK also produced crystals in two different morphologies (Form 3 and 4). Form 3 was a rod-shaped crystal grown from 0.18M calcium acetate hydrate, 90 mM sodium cacodylate trihydrate at pH 6.5, and 16%(w/v) PEG 8000 (originated from Crystal Screen I, solution No.46) and 28.5 mg mL−1 protein. Form 4 was also a rod-shaped crystal grown from 0.2M calcium acetate hydrate, 10 mM dithiothreitol (DTT), 0.1M sodium cacodylate trihydrate at pH 6.5, and 18%(w/v) PEG 8000 (originated from Crystal Screen I, solution No. 46) and 57.0 mg mL−1 protein.
Diffraction experiments and structure determination
Four datasets were collected for each HaNDK crystal (Form 1–4) at beamline BL-6A at the photon factory (PF), Tsukuba, Japan and at Structural Biology III beamline BL38B1 at the SPring-8, Hyogo, Japan. All datasets comprised single wavelength experiments and were collected at 100 K. Crystals were cryoprotected with 20% glycerol. Form 1 and 4 diffraction data were collected at PF (BL-6A) using the UGUI user interface and a Quantum 4R CCD detector (Area Detector Systems Corporation). Diffraction data for Form 2 and 3 crystals were obtained at SPring-8 (BL38B1) using the program BSS (Beamline Scheduling Software)32 managed by the web-based database application D-Cha.33 In this case, the mounting of cryocooled samples was automated by the sample changer robot SPACE,34 and diffraction data were collected using a Jupiter210 CCD detector (Rigaku, Japan). Diffraction data were integrated and scaled using the HKL2000 suite of programs.35 Form 1 (space group R3) and Form 2 (space group C2) crystals diffracted to 2.3 and 2.7 Å resolution, respectively. Form 3 (space group C2) and Form 4 (space group P21) crystals diffracted to 2.3 and 2.5 Å resolution, respectively.
Initial phase information was obtained by the molecular replacement (MR) method using the program MOLREP36 and Myxococcus xanthus nucleoside diphosphate kinase (MxNDK; PDB ID: 1NHK) as the search model. Model building and refinement including TLS (translation/libration/screw-motion) protocol was carried out using programs Coot,37 CNS 1.21,38 and REFMAC5.39 All data sets exhibited good overall completeness and refined structures exhibited acceptable values of stereochemistry and crystallographic residual (Table IV) (PDB ID: 3VGS, 3VGT, 3VGU, and 3VGV). According to RAMPAGE,40 the final model obtained from crystal Form 1 has 96.4% of the residues in the favored conformation of Ramachandran plot and no residues in the outlier region. The 91.3, 98.0, and 98.5% of Form 2, 3, and 4 crystals were in the favored conformation, respectively. The RMS deviations of atoms of NDKs for structural comparison were calculated using Lsqkab in the CCP4i package26 and the web-based program EBI-SSM (URL: http://www.ebi. ac.uk/msd-srv/ssm/).41
Acknowledgments
The authors thank Drs. N. Shimizu, M. Kawamoto, and M. Yamamoto of SPring-8 (proposal No. 2006B2647) and Profs. N. Igarashi and S. Wakatsuki of Photon Factory (proposal No. 2007G137) for X-ray data collection.
Glossary
Abbreviations and Symbols
- BSA
bovine serum albumin
- EcNDK
NDK from E. coli
- HaNDK
NDK from Halomonas sp. 593
- MxNDK
NDK from Myxococcus xanthus
- NDK
nucleoside diphosphate kinase
- PaNDK
NDK from Pseudomonas aeruginosa
- PF
the photon factory
- SEC-MALLS
high-performance size-exclusion chromatograph coupled to a multiangle laser light scattering detector.
References
- 1.Lanyi JK. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol Rev. 1974;38:272–290. doi: 10.1128/br.38.3.272-290.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madern D, Ebel C, Zaccai G. Halophilic adaptation of enzymes. Extremophiles. 2000;4:91–98. doi: 10.1007/s007920050142. [DOI] [PubMed] [Google Scholar]
- 3.Mevarech M, Frolow F, Gloss LM. Halophilic enzymes: proteins with a grain of salt. Biophys Chem. 2000;86:155–164. doi: 10.1016/s0301-4622(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 4.Ishibashi M, Tokunaga H, Hiratsuka K, Yonezawa Y, Turumaru H, Arakawa T, Tokunaga M. NaCl-activated nucleoside diphosphate kinase from extremely halophilic archaeon, Halobacterium salinarum, maintains native conformation without salt. FEBS Lett. 2001;493:134–138. doi: 10.1016/s0014-5793(01)02292-x. [DOI] [PubMed] [Google Scholar]
- 5.Yamamura A, Ichimura T, Kamekura M, Mizuki T, Usami R, Makino T, Ohtsuka J, Miyazono K, Okai M, Nagata K, Tanokura M. Molecular mechanism of distinct salt-dependent enzyme activity of two halophilic nucleoside diphosphate kinases. Biophys J. 2009;96:4692–4700. doi: 10.1016/j.bpj.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tokunaga H, Ishibashi M, Arakawa T, Tokunaga M. Highly efficient renaturation of beta-lactamase isolated from moderately halophilic bacteria. FEBS Lett. 2004;558:7–12. doi: 10.1016/S0014-5793(03)01508-4. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi R, Tokunaga H, Ishibashi M, Arakawa T, Tokunaga M. Salt-dependent thermo-reversible α-amylase: cloning and characterization of halophilic α-amylase from moderately halophilic bacterium, Kocuria varians. Appl Microbiol Biotech. 2011;89:673–684. doi: 10.1007/s00253-010-2882-y. [DOI] [PubMed] [Google Scholar]
- 8.Lascu I. The nucleoside diphosphate kinases 1973–2000. J Bioenerg Biomemb. 2000;32:213–214. [PubMed] [Google Scholar]
- 9.Kimura N. Introduction: nucleoside diphosphate kinases: genes and protein functions. J Bioenerg Biomemb. 2003;35:3–4. [Google Scholar]
- 10.Wieland T. Interaction of nucleoside diphosphate kinase B with heterotrimeric G protein βγ dimers: consequences on G protein activation and stability. Naunyn-Schmiedeberg's Arch Pharmacol. 2007;374:373–383. doi: 10.1007/s00210-006-0126-6. [DOI] [PubMed] [Google Scholar]
- 11.Lascu I, Giartosio A, Ransac S, Erent M. Quaternary structure of nucleoside diphosphate kinases. J Bioenerg Biomemb. 2000;32:227–236. doi: 10.1023/a:1005580828141. [DOI] [PubMed] [Google Scholar]
- 12.Janin J, Dumas C, Moréra S, Xu Y, Meyer P, Chiadmi M, Cherfils J. Three-dimensional structure of nucleoside diphosphate kinase. J Bioenerg Biomemb. 2000;32:215–225. doi: 10.1023/a:1005528811303. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Morera S, Mocan J, Lascu I, Janin J. X-ray structure of Mycobacterium tuberculosis nucleoside diphosphate kinase. Proteins. 2002;47:556–557. doi: 10.1002/prot.10113. [DOI] [PubMed] [Google Scholar]
- 14.Williams RL, Oren DA, Muñoz-Dorado J, Inouye S, Inouye M, Arnold E. Crystal structure of Myxococcus xanthus nucleoside diphosphate kinase and its interaction with a nucleotide substrate at 2.0 A resolution. J Mol Biol. 1993;234:1230–1247. doi: 10.1006/jmbi.1993.1673. [DOI] [PubMed] [Google Scholar]
- 15.Strelkov SV, Perisic O, Webb PA, Williams RL. The 1.9 A crystal structure of a nucleoside diphosphate kinase complex with adenosine 3′,5′-cyclic monophosphate: evidence for competitive inhibition. J Mol Biol. 1995;249:665–674. doi: 10.1006/jmbi.1995.0327. [DOI] [PubMed] [Google Scholar]
- 16.Moynié L, Giraud MF, Georgescauld F, Lascu I, Dautant A. The structure of the Escherichia coli nucleoside diphosphate kinase reveals a new quaternary architecture for this enzyme family. Proteins. 2007;67:755–765. doi: 10.1002/prot.21316. [DOI] [PubMed] [Google Scholar]
- 17.Dumas C, Lascu I, Moréra S, Glaser P, Fourme R, Wallet V, Lacombe ML, Véron M, Janin J. X-ray structure of nucleoside diphosphate kinase. EMBO J. 1992;11:3203–3208. doi: 10.1002/j.1460-2075.1992.tb05397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biggs J, Tripoulas N, Hersperger E, Dearolf C, Shearn A. Analysis of the lethal interaction between the prune and Killer of prune mutations of Drosophila. Genes Dev. 1988;2:1333–1343. doi: 10.1101/gad.2.10.1333. [DOI] [PubMed] [Google Scholar]
- 19.Lascu I, Chaffotte A, Limbourg-Bouchon B, Véron M. A Pro/Ser substitution in nucleoside diphosphate kinase of Drosophila melanogaster (mutation killer of prune) affects stability but not catalytic efficiency of the enzyme. J Biol Chem. 1992;267:12775–12781. [PubMed] [Google Scholar]
- 20.Timmons L, Xu J, Hersperger G, Deng XF, Shearn A. Point mutations in awdKpn which revert the prune/Killer of prune lethal interaction affect conserved residues that are involved in nucleoside diphosphate kinase substrate binding and catalysis. J Biol Chem. 1995;270:23021–23030. doi: 10.1074/jbc.270.39.23021. [DOI] [PubMed] [Google Scholar]
- 21.Yonezawa Y, Izutsu K, Tokunaga H, Maeda H, Arakawa T, Tokunaga M. Dimeric structure of nucleoside diphosphate kinase from moderately halophilic bacterium: contrast to the tetrameric Pseudomonas counterpart. FEMS Microbiol Lett. 2007;268:52–58. doi: 10.1111/j.1574-6968.2007.00626.x. [DOI] [PubMed] [Google Scholar]
- 22.Tokunaga H, Ishibashi M, Arisaka F, Arai S, Kuroki R, Arakawa T, Tokunaga M. Residue 134 determines the dimmer-tetramer assembly of nucleoside diphosphate kinase from moderately halophilic bacteria. FEBS Lett. 2008;582:1049–1054. doi: 10.1016/j.febslet.2008.02.054. [DOI] [PubMed] [Google Scholar]
- 23.Tokunaga H, Arakawa T, Tokunaga M. Engineering of halophilic enzymes: two acidic amino acid residues at the carboxy-terminal region confer halophilic characteristics to Halomonas and Pseudomonas nucleoside diphosphate kinases. Protein Sci. 2008;17:1603–1610. doi: 10.1110/ps.035725.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adman ET, Sieker LC, Jensen LH. Structure of a bacterial ferredoxin. J Biol Chem. 1973;248:3987–3996. [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick E. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 28.Karush F. Affinity and the immune response. Ann N Y Acad Sci. 1970;169:56–64. doi: 10.1111/j.1749-6632.1970.tb55970.x. [DOI] [PubMed] [Google Scholar]
- 29.Jaenicke R, Boèhm G. The stability of proteins in extreme environments. Curr Opin Struct Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 30.Maes D, Zeelen JP, Thanki N, Beaucamp N, Alvarez M, Thi MHD, Backmann J, Martial JA, Wyns L, Jaenicke R, Wierenga RK. The crystal structure of TIM from Thermotoga maritima: a comparative thermostability structural analysis of ten different TIM structures. Proteins. 1999;37:441–453. [PubMed] [Google Scholar]
- 31.Dams T, Jaenicke R. Stability and folding of dihydrofolate reductase from the hyperthermophilic bacterium Thermotoga maritima. Biochemistry. 1999;38:9169–9178. doi: 10.1021/bi990635e. [DOI] [PubMed] [Google Scholar]
- 32.Ueno G, Kanda H, Kumasaka T, Yamamoto M. Beamline Scheduling Software: administration software for automatic operation of the RIKEN structural genomics beamlines at SPring-8. J Synch Rad. 2005;12:380–384. doi: 10.1107/S0909049505004735. [DOI] [PubMed] [Google Scholar]
- 33.Okazaki N, Hasegawa K, Ueno G, Murakami H, Kumasaka T, Yamamoto M. Mail-in data collection at SPring-8 protein crystallography beamlines. J Synch Rad. 2008;15:288–291. doi: 10.1107/S0909049507064679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno G, Hirose R, Ida K, Kumasaka T, Yamamoto M. Sample management system for a vast amount of frozen crystals at SPring-8. J Appl Cryst. 2004;37:867–873. [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi, and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 41.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]