

Abstract
Novel antibiotics are needed to overcome the challenge of continually evolving bacterial resistance. This has led to a renewed interest in mechanistic studies of once popular antibiotics like chloramphenicol (CAM). Chloramphenicol acetyltransferases (CATs) are enzymes that covalently modify CAM, rendering it inactive against its target, the ribosome, and thereby causing resistance to CAM. Of the three major types of CAT (CATI-III), the CAM-specific CATIII has been studied extensively. Much less is known about another clinically important type, CATI. In addition to inactivating CAM and unlike CATIII, CATI confers resistance to a structurally distinct antibiotic, fusidic acid. The origin of the broader substrate specificity of CATI has not been fully elucidated. To understand the substrate binding features of CATI, its crystal structures in the unbound (apo) and CAM-bound forms were determined. The analysis of these and previously determined CATI-FA and CATIII-CAM structures revealed interactions responsible for CATI binding to its substrates and clarified the broader substrate preference of CATI compared to that of CATIII.
Keywords: antibacterial agent, antibiotic resistance, chloramphenicol acetyltransferase, fusidic acid, specificity, substrate recognition
Introduction
Chloramphenicol (CAM) [Fig. 1(A)] is a potent broad-spectrum antibacterial agent. Since its isolation from Streptomyces venezuelae in 1948,1 CAM was one of the primary agents used to treat many infections in the decades that followed. To date, despite its relatively high toxicity,2 CAM is used in many countries because of its affordability and its broad spectrum of activity. In the Western world, CAM is used in treatment of ophthalmic infections and as a last resort in cases of life-threatening brain infections, such as those caused by Neisseria meningitidis, which do not respond to other agents. CAM's ability to cross the blood-brain barrier makes it a potent therapeutic against brain infections. Because of the emergence of pathogens resistant to multiple drugs, CAM is now being reconsidered as a wider-spectrum therapeutic.3
Figure 1.
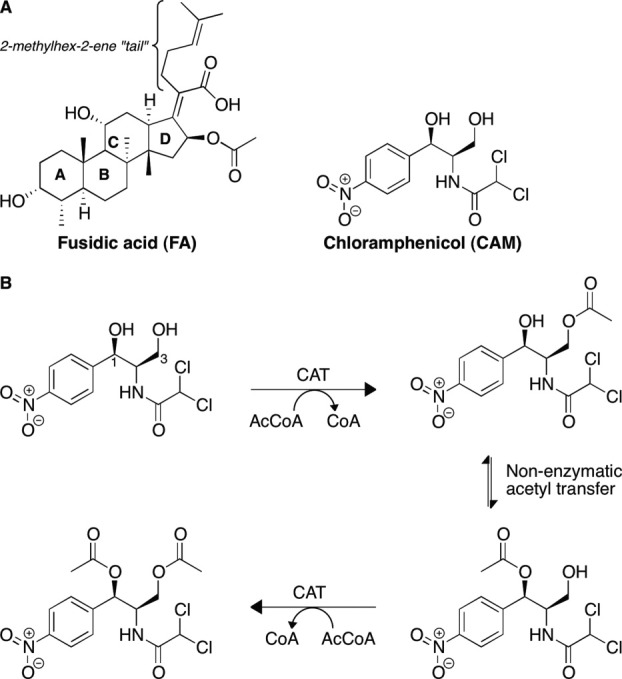
(A) Chemical structures of CAM and FA. (B) Acetylation of CAM by CATs.
CAM inhibits protein biosynthesis by binding to the 50S subunit of the bacterial ribosome. Recent crystal structures of the 50S subunit of the Escherichia coli and Thermus thermophilus ribosome in complex with CAM revealed that CAM binds to the A-site of the 50S subunit and occupies the binding site for the amino-acyl moiety of the A-site tRNA.4, 5 The 3-hydroxyl of CAM is buried in the interface with the ribosome through direct hydrogen bonding, potassium ion-mediated electrostatic interactions, as well as through van der Waals interactions with the RNA phosphosugar backbone.4, 5 The 1-hydroxyl of CAM forms hydrogen bonds with RNA bases. Therefore, any modification of the 1-hydroxyl or the 3-hydroxyl of CAM is predicted to be disruptive of CAM-ribosome binding.5 Bacterial resistance to CAM is caused by the chromosomally or plasmid-encoded enzyme chloramphenicol acetyltransferase (CAT) that catalyzes the transfer of an acetyl group from acetyl-coenzyme A (AcCoA) to the 3-hydroxyl group of CAM [Fig. 1(B)].6 A subsequent slow, non-enzymatic transfer of this acetyl group to the neighboring 1-hydroxyl group allows for a second CAT-catalyzed acetyl transfer from AcCoA onto the 3-hydroxyl group of the same CAM molecule, resulting in a di-acetylated CAM.7, 8 However, a single acetylation of CAM is sufficient to abolish its affinity for the ribosome9 as explained by the above-mentioned structural observations.4, 5
CAT proteins are historically divided into three types: CATI, CATII, and CATIII, with all three types capable of catalyzing the acetyl transfer to CAM to generate 3-O-acetyl-CAM. Genomic analysis of different CAT sequences indicates that the boundaries between these CAT types are not sharp. Members of the CATI family are present in many important pathogens such as E. coli, Shigella flexneri, Serratia marcescens, and Salmonella enterica. CATI family enzymes display high sequence conservation among themselves (e.g. S. flexneri and S. marcescens CATI proteins are 98% and 99% identical to E. coli CATI, respectively); however, they display only a modest sequence identity to CATII (∼46%) and CATIII (32–47%) (Fig. 2). The CATII family is not easily distinguishable from CATIII and has been defined historically only through its extreme susceptibility to thiol-modifying agents compared with that of CATI and CATIII.11 There are no obvious additional Cys residues or other sequence features in CATII distinguishing it from the CATIII variants. A slight variation in the pKa of the Cys31 (in CATIII nomenclature), the only Cys in vicinity of the substrate or the cosubstrate binding sites, was suggested to be responsible for the difference in reactivity with thiol-modifying agents,12 although there is no evidence confirming this idea.
Figure 2.

Sequence alignment10 of CATI, CATII, and CATIII enzymes from various bacteria (EC, Escherichia coli; YP, Yersinia pestis; KP, Klebsiella pneumoniae; SM, Serratia marcescens; SF Shigella flexneri; PM, Proteus mirabilis; HI, Haemophilus influenzae; SE, Salmonella enterica; VS, Vibrio sp.; BC, Bacillus cereus; BA, Bacillus anthracis; SP, Streptococcus pneumoniae; EF, Enterococcus faecium; LM, Listeria monocytogenes; SA, Staphylococcus aureus; CD, Clostridium difficile; CB, Clostridium botulinum; CT, Clostridium tetani). Important residues in the active site that are either conserved or non-conserved (vary) between CATI and CATIII are indicated by red and blue circles, respectively. Residues involved in CAM and FA binding are marked by orange and yellow circles, respectively.
The sequence differences between CATI and CATIII include several substitutions in the binding site (Fig. 2), potentially resulting in positional differences of CAM bound to these two proteins. A major consequence of this divergence is reflected in different substrate selectivities of CATI and CATIII. In addition to binding and modifying CAM,6 CATI, unlike CATIII, binds a much bulkier antibiotic, fusidic acid (FA).13 FA [Fig. 1(A)] is a steroidal antibacterial agent that is used topically or systemically, usually against infections caused by Gram-positive pathogens. CATI does not modify FA; rather, it sequesters it through binding by its CAM binding site. This type of mechanism of resistance through sequestration is not uncommon and has been observed for other antibiotics such as bleomycin and thiocoraline.14–18 Kinetic studies have shown that FA competes with CAM for binding to CATI, but not the other CAT types.13 Various bile salts and some triphenylmethane dyes also exhibit similar competitive binding to CATI, but not to CATII/III.13,19–21
CATI plays an important role in antibiotic resistance of many pathogenic bacteria. In addition, CATI has been used as a biochemical and proteomic tool in a number of systems22–26 and as a common CAM-resistance marker encoded in laboratory plasmids. Despite its importance in drug resistance and biotechnology, CATI27 has been much less investigated than CATIII. Structural and biochemical studies of CATIII28–34 have been mostly used to understand general features of CATI proteins. Despite this progress, differences in the substrate selectivity between CATI and CATIII remain unclear in absence of analysis of CATI-CAM and CATI-FA structures.
Herein we report crystal structures of CATI alone (apo) and in complex with CAM, which explain how CATI binds CAM despite differences in its binding site residues from those in CATIII. Analysis of these structures along with that of the structure of CATI in complex with FA (deposited in the Protein Data Bank (PDB) by Roidis and Kokkinidis; accession code: 1Q2335) provides an explanation of the differences in substrate preference among CAT types.
Results
Overall structure of CATI
E. coli CATI protein was initially co-crystallized with CAM in the P1 space group (Table I). Molecular replacement using either a monomer or trimer of apo-CATI (from the structure of a serendipitous complex of the nitric oxide synthase oxygenase domain with CATI; PDB code: 1NOC37) as a search model did not yield a solution. This complication likely arose due to the presence of several copies of the protein molecules within a very large unit cell. Further crystallization trials yielded crystals of CATI alone in the P21 space group with a smaller asymmetric unit. These crystals grew under conditions similar to those of the CATI-CAM crystals. Molecular replacement with a CATI trimer from the 1NOC entry as a search model, yielded an apo-CATI structure with three CATI trimers in the asymmetric unit (Table I). This three-trimer structure was then successfully used as a molecular replacement search model to determine the structure of the CATI-CAM complex in the P1 crystal form. The asymmetric unit of the P1 crystal form contained six CATI-CAM trimers. The crystal structure of the apo-CATI and that of the CATI-CAM complex were refined to 3.2 Å and 2.9 Å resolution, respectively (Table I).
Table I.
X-ray Diffraction Data Collection and Refinement Statistics for apo-CATI and CATI-CAM Structures
| Apo-CATI | CATI-CAM | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P1 |
| Number of trimers per asymmetric unit | 3 | 6 |
| Unit cell dimensions | ||
| a, b, c (Å) | 115.2, 102.7, 114.1 | 107.5, 114.5, 114.5 |
| α, β, γ (°) | 90, 119.9, 90 | 119.9, 97.8, 98.7 |
| Resolution (Å) | 50.0–3.2 (3.3–3.2)a | 50.0–2.9 (3.0–2.9)a |
| I/σ | 9.3 (2.1) | 14.3 (2.3) |
| Completeness (%) | 98.4 (86.1) | 87.3 (85.0) |
| Redundancy | 4.3 (3.5) | 1.7 (1.7) |
| Rmerge | 0.15 (0.476) | 0.06 (0.38) |
| Number of unique reflections | 35,818 | 82,522 |
| Structure refinement statistics | ||
| Resolution (Å) | 40.0–3.2 | 35.0–2.9 |
| R (%) | 23.8 | 24.0 |
| Rfree (%) | 30.1 | 30.9 |
| Bond length deviation (RMSD) from ideal (Å) | 0.009 | 0.006 |
| Bond angle deviation (RMSD) from ideal (°) | 1.08 | 0.907 |
| Ramachadran plot statisticsb | ||
| % of residues in most allowed regions | 84.9 | 88.7 |
| % of residues in additional allowed regions | 12.9 | 10.5 |
| % of residues in generously allowed regions | 2.3 | 0.8 |
| % of residues in disallowed regions | 0 (0 residues) | 0 (0 residues) |
Numbers in parentheses indicate the values in the highest-resolution shell.
Indicates Procheck statistics.36
The structure of CATI protein in the apo form reported here is very similar to the structure of apo-CATI (PDB code: 1NOC) used for the molecular replacement (Cα RMSD = 0.7 Å) and to another previously deposited structure of apo-CATI (PDB code: 1PD5; Cα RMSD = 0.7 Å). Furthermore, the structures of apo-CATI are highly similar to the structure of CATI in complex with CAM (Cα RMSD ∼ 0.4 Å), suggesting that no major protein conformational changes occur upon CAM binding. Analogously, no major conformational differences were observed for CATIII in the apo and the CAM-bound forms.28 The overall fold and the oligomeric organization of CATI [Fig. 3(A)] resemble those of the previously characterized CATIII28 variant. Three identical monomers of CATI form a trimer with a 3-fold rotational symmetry. The overall trimeric scaffold is formed by three 7-stranded β-sheets, each of which is formed by six strands (β6, β5, β7, β9, β10, and β2) from one monomer and one strand (β8) from another monomer [Fig. 3(B)]. In each monomer, this β-sheet is flanked on the outside by five α-helices and a small three-stranded β-sheet. In the trimeric core, the aliphatic parts of buried Asp157 side-chains (in strand β8) of the three monomers come together to form intimate hydrophobic contacts with each other, while their carboxyl groups are engaged in intricate, asymmetric network of hydrogen bonding interactions with the side-chains of Ser155 and Asn159. The hydrophobic interactions between the Asp157 residues are likely critical for complex stability as this residue is either an Asp or an Asn in most CATI/CATIII proteins. Ser155 could however be substituted by a Gly (Fig. 2). The side-chains of Asp157 residues are distorted so that the carboxyl groups form hydrogen bonds with their own backbone amide NH moieties justifying a weaker conservation of the Ser155. As the side-chains in these β-strands are generally buried away from the solvent, their identities are well conserved among CAT homologs.
Figure 3.

(A) Overall fold and trimeric organization of CAM-bound form. The strands of the β-sheets comprising the central scaffold are marked. The CAM molecule and its molecular surface are shown in blue. (B) A close-up view of the active site at the interface of two monomers in CATI-CAM structure. Substrate binding residues of the binding monomer and catalytic monomer are colored in green and orange, respectively. A few highly conserved residues involved in catalysis are marked in red. (C) A representative view of one of the CATI active sites. The violet mesh clearly defining the CAM molecule (blue sticks) is Fo-Fc omit electron density generated without the CAM in the model and contoured at 3σ. (D) A zoomed in view of the active site shown in panel C, depicted in a slightly different orientation. (E) A schematic view of residues of the CATI active site and their interactions with CAM. Hydrogen bonds and hydrophobic contacts are marked by black dashed lines and the grey hashed lines, respectively. The color coding is consistent with that of panel B. An interactive view is available in the electronic version of the article.PRO2036 Figure 3
Chloramphenicol interactions in the active site
Upon trimerization, the active site is formed at the interface of two β-sheets predominantly with residues from strands β6, β5, β7, β9, and β8 of one monomer (termed as the binding monomer) and strands β2 and β10 of the other (the catalytic monomer). Each trimer contains three identical substrate binding sites [Fig. 3(A)]. The nature of this conserved trimeric assembly strongly suggests that CATI monomers either require trimeric assembly for proper folding or, if folded, CATI would be catalytically active only in the context of a trimer. Indeed, monomeric mutants of the CATIII, whose overall fold is highly similar to that of CATI, were shown to be catalytically inactive.38 Below, we discuss features of the active site of CATI and highlight its differences from that of CATIII that specify the distinct substrate recognition properties of these two proteins.
In the structure of CATI-CAM complex, all three active site pockets of the CATI trimer are occupied with CAM molecules [Fig. 3(A)], whose positions are clearly defined in the electron density map [Fig. 3(C,D)]. One of the two monomers forming a binding site (called here the binding monomer) provides the majority of the residues (Cys91, Phe102, Ser104, Phe134, Phe144, Ser146, Leu158, and Val170) involved in binding of the CAM while the other one (called here the catalytic monomer) provides His193, which has been demonstrated to be one of the primary conserved catalytic residues7, 39, 40 [Fig. 3(B)]. A few other residues from the catalytic monomer (Phe25 and Cys31) also provide an important CAM-binding surface in the binding pocket. The disposition of the conserved catalytic residues [e.g. His193, Ser146, and Asp197; highlighted in red in Fig. 3(B)] in the CATI-CAM structure is highly similar to that observed previously in CATIII-CAM complex.41 The position of His193, the likely general base, relative to the bound CAM is identical to its counterpart in CATIII (His195). The Nε2 atom of His193 is located 2.7 Å away from the 3-hydroxyl of CAM [Fig. 3(E)]. The side-chain of His193 is in a distorted conformation (His193 χ1 = −150.2° and χ2 = −41.0° with CAM bound and χ1 = −142.0° and χ2 = −32.6° with FA bound). This conformation likely ensures that the imidazole ring is aligned appropriately for abstracting the 3-hydroxyl proton of CAM, promoting a nucleophilic attack by the oxygen on the acetyl group carbonyl of AcCoA,29, 30, 34 similarly to the proposed mechanism of the CATIII variant.40 The imidazole ring of His193 is positioned at a proper distance (approximately 3.6 Å) for a face-to-face π-π stacking contact with Phe25. This overall structural arrangement of the catalytic monomer for proper positioning of His193 at the subunit interface is stabilized by several interactions that include a chain of hydrogen bonds between His193, Asp197, Arg18, and the backbone carbonyl oxygen of Ala194 [Fig. 3(E)]. The conserved Ser146 (another catalytically important residue) of CATI is positioned similarly in the active site of all three CATI structures, and likely stabilizes the transition state oxyanion by donating a hydrogen bond (possibly water-mediated in CATI), as proposed for Ser148 of CATIII.42
The sequence alignment of CATI and CATIII from E. coli demonstrates that of the 20 amino acid residues lining the CAM binding site, 9 are different between the two types (Fig. 2, blue circles). These differences [Ala24 (CATIII, Phe24), Phe25 (CATIII, Tyr25), Val28 (CATIII, Arg28) Ala29 (CATIII, Leu29), Cys91 (CATIII, Gln92), Tyr133 (CATIII, Leu134), Phe144 (CATIII, Asn146), Phe166 (CATIII, Tyr168), and Val170 (CATIII, Ile172)] are significant as they include changes in the size and hydrophobicity of the residues. Remarkably, despite these differences, CAM binding affinities for CATI and CATIII appear to be very similar.43 Furthermore, the superposition of CATI-CAM and CATIII-CAM structures demonstrates that the orientations of the CAM molecule in the active sites of the two proteins are nearly identical. The p-NO2 group of CAM is solvent exposed when bound in the CATI active site and the aromatic ring rests on the hydrophobic surface provided primarily by Leu158 and Val170, as observed in the CATIII-CAM structure. The side-chains of Leu158, Val160, and Phe166 that line the very bottom of the substrate binding pocket [Fig. 4(C)] are positioned through interactions of the trimeric assembly and show only minor alterations between CATI and CATIII. The dichloroacetyl moiety of CAM closely interacts with Phe134 (Phe135 in CATIII), likely indicating a strong hydrophobic interaction. A major difference between the CATI-CAM and CATIII-CAM structures is that the residue analogous to Tyr133 of CATI is nonpolar (Leu134) in CATIII. Tyr133 forms a strong hydrogen bond (2.9 Å) with the carbonyl group of CAM. Interestingly, this interaction occurs in place of the interaction of that between the hydroxyl of Tyr25 in CATIII (Phe25 in CATI) and the carbonyl group of CAM, located at an O-O distance of 2.8 Å from each other.
Figure 4.

Close-up views of interactions of FA with active-site residues of (A) CATI and (B) CATIII (quadruple mutant). Schematic views of interactions of FA with (C) CATI and (D) CATIII (quadruple mutant). The color coding is consistent with that of Figure 3 (FA is shown in blue). An interactive view is available in the electronic version of the article.PRO2036 Figure 4
Fusidic acid interactions in the active site
In the CATI-FA complex [Fig. 4(A)], FA occupies the same binding site as CAM, which explains its observed behavior as a competitive inhibitor of CAM acetylation.13 The differences between the active site residues of CATI (as described above) and those of CATIII, while having little effect on CAM binding,41, 44 create a unique surface suitable for binding to FA in CATI. In particular, the placement of Ala24 and Ala29 of CATI shapes the substrate binding cavity such that the ring D and the 2-methylhex-2-ene “tail” of FA can be accommodated. The hydrophobic steroid ring system of FA makes numerous hydrophobic contacts with active site residues, including Thr93, Phe102, Phe134, Phe144, Ser146, Phe156, Leu158, Val160, Phe166, and Val170 of the binding subunit, as well as Ala24, Phe25, Val28, and Ala29 of the catalytic subunit [Fig. 4(A,C)]. The hydroxyl moiety of ring A of FA closely aligns with the 3-hydroxyl of CAM and forms a very strong hydrogen bond with the Nε2 atom of His193 at a distance of 2.9 Å [Fig. 4(C)]. The hydroxyl group of Tyr133 points inward towards the binding pocket forming a hydrogen bond with the hydroxyl moiety of ring C, the atoms being separated by a distance of about 2.6 Å [Fig. 4(C)], similarly to the interaction between the 3-hydroxyl and Tyr133 in the CATI-CAM structure [Fig. 3(E)]. Residues from both the binding monomer (Phe134) and the catalytic monomer (Ala24 and Val28) form a hydrophobic zone near the entrance of the binding pocket in CATI that cradle the “tail” section of FA and dictate its conformation [Fig. 4(A,C)].
Valuable insight can be gained by comparing this structure with the previously reported structures of CATIII in complex with CAM29 and a quadruple mutant of CATIII in complex with FA.43 In the quadruple mutant of CATIII, four catalytic pocket residues were mutated (Gln92Cys/Asn146Phe/Tyr168Phe/Ile172Val) to mimic those of CATI. This comparison [Fig. 4(B)] indicates a disruption of the FA tail-interacting hydrophobic zone in the quadruple mutant of CATIII, in particular due to the Ala24Phe and Val28Arg substitutions. The carboxylic acid and acetoxy moieties of ring D are highly solvent exposed when bound to both CATI and the CATIII mutant. The acetoxy group of FA makes a hydrophobic contact with Phe166 in CATI (Phe168 in CATIII) [Fig. 4(C,D)]. Despite the same general protein backbone scaffold of CATI and the mutant CATIII structures, there are several differences in the FA-protein contacts for the two enzymes. Most strikingly, several bulkier residues of CATIII: Phe24 (Ala24 in CATI), Tyr25 (Phe25 in CATI), Arg28 (Val28 in CATI), and Leu29 (Ala29 in CATI) prevent the FA molecule from binding in a position similar to that in CATI. A required shift of the FA molecule must not be accommodated due to structural rigidity of wild-type CATIII resulting in the lack of binding to FA. The mutations of the CATIII quadruple mutant apparently relax this rigidity and surprisingly accommodate the FA molecule in a very different position from that seen in the CATI-FA structure. The hydrophobic “tail” of FA now adopts a very different conformation and gets buried in the disordered loop region (residues 138–141) of the CATIII mutant. This disorder is very likely due to both the Asn146Phe and the Gln92Cys substitutions in the CATIII mutant, which cause displacement of the His144 and Thr140 side-chains, respectively, thereby distorting the local backbone. This displacement allows the FA-tail to occupy its altered position in the CATIII mutant.43 Notably, Tyr25 in the CATIII quadruple mutant structure (positionally analogous to Phe25 of CATI) forms hydrogen bond with FA, at a distance of 2.8 Å to the hydroxyl on the A-ring [Fig. 4(D)], and stabilizes the altered FA orientation. Phe168 and Val172 residues in the CATIII quadruple mutant make direct hydrophobic contacts with the FA molecule, which explains the contribution of these substitutions to the change in binding affinity to FA.43
We observe no major differences in the backbone conformations near the active site of CATI in the structures of apo-CATI (PDB code: 3U9B), CATI bound to CAM (PDB code: 3U9F), and CATI bound to FA (PDB code: 1Q2335). This strongly suggests that CATI has evolved to bind multiple ligands, even as large as an FA molecule, without any major protein conformational changes in its backbone.
Discussion
Chloramphenicol acetyltransferase (CAT) is found in many pathogenic bacteria and is often the cause of resistance against chloramphenicol (CAM), once a widely used antibiotic. Of many known CATs, the type-I appears to be the most prevalent. Recent studies have found CATI in many pathogenic bacteria. CATI has a preference for binding to a variety of substrates; not only does it inactivate CAM but it also binds and sequesters other antibiotics such as FA. A clear understanding of the mechanism of substrate binding by CATI is important to address the intriguing question of how CAT proteins from different classes with similar overall structures display different substrate selectivity profiles. In comparison to CATIII that has been studied almost exclusively, there are only few mechanistic studies that have been performed on CATI.
The general fold and the trimeric organization of CAT proteins have been observed in enzymes of primary metabolic pathways in bacteria and eukaryotes, such as pyruvate dehydrogenases45, 46 and α-keto acid dehydrogenases.47, 48 Therefore, CAT appears to be a product of an ancient gene duplication event, which underwent subsequent specialization through evolution to serve a protective role against toxic compounds such as CAM. The general catalytic mechanism proposed for CAT proteins is based on studies of many such proteins. The residue primarily responsible for catalysis of CATI appears to be His193 (His195 in CATIII).28 This role was proposed based on a previous study in which a mutant CATIII (His195Tyr) was shown to be devoid of catalytic activity.49 Another conserved residue, Ser146, likely stabilizes the oxyanion formed upon an attack on the AcCoA carbonyl carbon by the 3-hydroxyl of CAM. Mutagenesis studies with CATIII confirmed that Ser148 (Ser146 in CATI) is crucial for efficient catalysis.42
The CATI protein structure is similar in the apo form and in the CAM- and the FA-bound states, indicating that no major changes in the backbone conformations or in positions of the side-chains occur upon ligand binding. It is quite remarkable that such nearly rigid scaffold is evolutionarily conserved and yet CATI can bind chemically diverse substrates. Analysis of the aligned sequences shows that several residues of CATI are different than corresponding residues in CATIII. Our investigation of the CATI structures indicates that many of these differences are in residues lining the substrate binding pocket (Fig. 2, blue circles). The most striking differences are concentrated around a small patch of residues (Ala24-Cys31, contributed by the catalytic monomer) that enable the FA molecule to be accommodated only in the pocket of CATI. The bulkier residues of CATIII in this patch would push the FA towards the opposite side of the pocket and consequently disrupt the structure. Interestingly, the flexibility (apparently resulting in the reduced rigidity and increased disorder of the backbone) of the quadruple mutant of CATIII helps it accommodate the pushed out FA in a different conformation. The “tail” of FA now finds a different hydrophobic pocket to rest in and in turn provides stability to the FA in this altered binding pocket. The mutant CATIII shows a 200-fold higher affinity to FA than the wild-type CATIII. However, the quadruple mutant of CATIII binds FA with a much (4-fold) weaker affinity43 than CATI. In addition, the Km for CAM acetylation by the CATIII quadruple mutant was somewhat compromised (with respect to either CATI or CATIII) and the value of kcat was between those for CATIII and CATI. With the direct structural evidence, it is now clear how the tail of FA nests in a hydrophobic pocket and renders CATI more energetically favorable to bind to FA. In CATIII, a similarly positioned FA “tail” would be sterically blocked by Phe24 and Arg28, and it is not surprising that CATIII does not show affinity towards FA.
Our understanding of CAM's mechanism of action as well as the mechanisms of resistance to it were largely based on biochemical and structural information available on CAM binding to CATIII and to the bacterial ribosome.4 The present structural study augments this knowledge by filling in the gap in our understanding of the recognition of both CAM and FA by CATI. CAM has been largely removed from the clinic in the Western world due to its safety concerns, even though cases of extreme toxicity are exceedingly rare. CAM has remained a popular drug in underdeveloped areas due to its low cost and effectiveness against a variety of pathogens. However, as with other antibiotics, development of resistance against CAM is a major obstacle to its power to save lives. The detailed picture of the CATI structure is expected to aid in design of inhibitors of CAT enzymes that could re-sensitize CAM-resistant strains. In addition, structure-guided design of CAM analogs could lead to new antibiotics of this class that would be less toxic and more refractory to inactivation by CAT.
Materials and Methods
Expression and purification of CATI
CATI was expressed in BL21 (DE3)/RIL cells (Stratagene), which harbor a plasmid containing a constitutively expressed CAM resistance gene camR encoding untagged CATI protein. The cells were grown in LB medium (200 rpm, 37°C) containing CAM (25 μg/mL) until the culture reached an attenuance of 0.4 at 600 nm. The cells were harvested after an additional 3 h growth. Pelleted cells (centrifugation at 5,000 g, 10 min, 4°C) were resuspended in the lysis buffer [MES pH 6.5 (40 mM), NaCl (200 mM), glycerol (5%), β-mercaptoethanol (2 mM), and EDTA (0.1 mM)] and lysed by sonication. The lysate was clarified by centrifugation at 35,000 × g for 45 min at 4°C. We took advantage of the thermostability of CAT proteins50 in purifying CATI without an affinity tag. The clarified lysate was heated (75°C, 20 min) and subsequently centrifuged (35,000 × g, 45 min, 4°C) to remove unfolded precipitated proteins. The CATI in the soluble fraction was further purified by size-exclusion chromatography on an S-200 column (GE Healthcare) equilibrated with buffer [Tris pH 8.0 (40 mM) and NaCl (100 mM)]. The fractions containing pure CATI, as determined by SDS-PAGE, were concentrated to 5 mg/mL using an Amicon Ultra centrifugal filter device (Millipore) and used for crystallization.
Crystallization of CATI alone and in complex with CAM
Crystals of CATI alone and a complex of CATI with CAM (CATI-CAM) were grown by vapor diffusion in hanging drops containing 1 μL of protein and 1 μL of the reservoir solution [HEPES (100 mM) pH 7.5 (pH of 1 M stock of HEPES acid was adjusted by adding NaOH), PEG 4000 (20% w/v), isopropanol (10% v/v)] or 1 μL of the reservoir solution containing CAM (1 mM), respectively. Irregularly shaped crystals, 40–60 μm in each of the three dimensions were formed in 7–10 days when incubated at 22°C against the respective reservoir solutions. The crystals were gradually transferred into the reservoir solution containing glycerol (15% v/v) and flash frozen in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected at 100 K at the ×25 beamline of the National Synchrotron Light Source at the Brookhaven National Laboratory. The data were processed with HKL2000.51 The crystals of apo-CATI and CATI-CAM complex were in the P21 and P1 space groups, respectively. The structures of both apo-CATI and CATI-CAM complex were determined by molecular replacement with MOLREP52 as described in Results. The locations of the CAM molecules in the active sites of CATI were clearly identified and positioned in the omit Fo-Fc density and then refined. The structures were iteratively manually built and refined using programs Coot53 and REFMAC,54 respectively. The data collection and refinement statistics are given in Table I. The structures of apo-CATI and CATI-CAM complex were deposited in the Protein Data Bank with accession codes 3U9B and 3U9F, respectively.
Glossary
Abbreviations
- AcCoA
acetyl-coenzyme A
- CAM
chloramphenicol
- CAT
chloramphenicol acetyltransferase
- CoA
coenzyme A
- FA
fusidic acid.
References
- 1.Carter HE, Gottlieb D, Anderson HW. Chloromycetin and streptothricin. Science. 1948;107:113. doi: 10.1126/science.107.2770.113-b. [DOI] [PubMed] [Google Scholar]
- 2.Skolimowski IM, Knight RC, Edwards D. Molecular basis of chloramphenicol and thiamphenicol toxicity to DNA in vitro. J Antimicrob Chemother. 1983;12:535–542. doi: 10.1093/jac/12.6.535. [DOI] [PubMed] [Google Scholar]
- 3.Nitzan O, Suponitzky U, Kennes Y, Chazan B, Raul R, Colodner R. Is chloramphenicol making a comeback? Isr Med Assoc. 2010;12:371–374. [PubMed] [Google Scholar]
- 4.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci USA. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci USA. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw WV. The enzymatic acetylation of chloramphenicol by extracts of R factor-resistant Escherichia coli. J Biol Chem. 1967;242:687–693. [PubMed] [Google Scholar]
- 7.Kleanthous C, Shaw WV. Analysis of the mechanism of chloramphenicol acetyltransferase by steady-state kinetics. Evidence for a ternary-complex mechanism. Biochem J. 1984;223:211–220. doi: 10.1042/bj2230211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thibault G, Guitard M, Daigneault R. A study of the enzymatic inactivation of chloramphenicol by highly purified chloramphenicol acetyltransferase. Biochim Biophys Acta. 1980;614:339–342. doi: 10.1016/0005-2744(80)90223-5. [DOI] [PubMed] [Google Scholar]
- 9.Shaw WV, Unowsky J. Mechanism of R factor-mediated chloramphenicol resistance. J Bacteriol. 1968;95:1976–1978. doi: 10.1128/jb.95.5.1976-1978.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray IA, Martinez-Suarez JV, Close TJ, Shaw WV. Nucleotide sequences of genes encoding the type II chloramphenicol acetyltransferases of Escherichia coli and Haemophilus influenzae, which are sensitive to inhibition by thiol-reactive reagents. Biochem J. 1990;272:505–510. doi: 10.1042/bj2720505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewendon A, Shaw WV. Elimination of a reactive thiol group from the active site of chloramphenicol acetyltransferase. Biochem J. 1990;272:499–504. doi: 10.1042/bj2720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett AD, Shaw WV. Resistance to fusidic acid in Escherichia coli mediated by the type I variant of chloramphenicol acetyltransferase. A plasmid-encoded mechanism involving antibiotic binding. Biochem J. 1983;215:29–38. doi: 10.1042/bj2150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gatignol A, Durand H, Tiraby G. Bleomycin resistance conferred by a drug-binding protein. FEBS Lett. 1988;230:171–175. doi: 10.1016/0014-5793(88)80665-3. [DOI] [PubMed] [Google Scholar]
- 15.Dumas P, Bergdoll M, Cagnon C, Masson JM. Crystal structure and site-directed mutagenesis of a bleomycin resistance protein and their significance for drug sequestering. EMBO J. 1994;13:2483–2492. doi: 10.1002/j.1460-2075.1994.tb06535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawano Y, Kumagai T, Muta K, Matoba Y, Davies J, Sugiyama M. The 1.5 Å crystal structure of a bleomycin resistance determinant from bleomycin-producing Streptomyces verticillus. J Mol Biol. 2000;295:915–925. doi: 10.1006/jmbi.1999.3404. [DOI] [PubMed] [Google Scholar]
- 17.Maruyama M, Kumagai T, Matoba Y, Hayashida M, Fujii T, Hata Y, Sugiyama M. Crystal structures of the transposon Tn5-carried bleomycin resistance determinant uncomplexed and complexed with bleomycin. J Biol Chem. 2001;276:9992–9999. doi: 10.1074/jbc.M009874200. [DOI] [PubMed] [Google Scholar]
- 18.Biswas T, Zolova OE, Lombo F, de la Calle F, Salas JA, Tsodikov OV, Garneau-Tsodikova S. A new scaffold of an old protein fold ensures binding to the bisintercalator thiocoraline. J Mol Biol. 2010;397:495–507. doi: 10.1016/j.jmb.2010.01.053. [DOI] [PubMed] [Google Scholar]
- 19.Proctor GN, McKell J, Rownd RH. Chloramphenicol acetyltransferase may confer resistance to fusidic acid by sequestering the drug. J Bacteriol. 1983;155:937–939. doi: 10.1128/jb.155.2.937-939.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka H, Izaki K, Takahashi H. Some properties of chloramphenicol acetyltransferase, with particular reference to the mechanism of inhibition by basic triphenylmethane dyes. J Biochem. 1974;76:1009–1019. [PubMed] [Google Scholar]
- 21.Volker TA, Iida S, Bickle TA. A single gene coding for resistance to both fusidic acid and chloramphenicol. J Mol Biol. 1982;154:417–425. doi: 10.1016/s0022-2836(82)80004-1. [DOI] [PubMed] [Google Scholar]
- 22.Russ WP, Engelman DM. TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc Natl Acad Sci USA. 1999;96:863–868. doi: 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, He L, He N, Deng Y, Shi Z, Wang H, Li S, Liu H, Wang Z, Wang D. Polymerase chain reaction coupling with magnetic nanoparticles-based biotin-avidin system for amplification of chemiluminescent detection signals of nucleic acid. J Nanosci Nanotechnol. 2011;11:1074–1078. doi: 10.1166/jnn.2011.3061. [DOI] [PubMed] [Google Scholar]
- 24.King DA, Hall BE, Iwamoto MA, Win KZ, Chang JF, Ellenberger T. Domain structure and protein interactions of the silent information regulator Sir3 revealed by screening a nested deletion library of protein fragments. J Biol Chem. 2006;281:20107–20119. doi: 10.1074/jbc.M512588200. [DOI] [PubMed] [Google Scholar]
- 25.Speck J, Stebel SC, Arndt KM, Muller KM. Nucleotide exchange and excision technology DNA shuffling and directed evolution. Methods Mol Biol. 2011;687:333–344. doi: 10.1007/978-1-60761-944-4_24. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Ruf S, Bock R. Chloramphenicol acetyltransferase as selectable marker for plastid transformation. Plant Mol Biol. 2011;76:443–451. doi: 10.1007/s11103-010-9678-4. [DOI] [PubMed] [Google Scholar]
- 27.Van der Schueren J, Robben J, Goossens K, Heremans K, Volckaert G. Identification of local carboxy-terminal hydrophobic interactions essential for folding or stability of chloramphenicol acetyltransferase. J Mol Biol. 1996;256:878–888. doi: 10.1006/jmbi.1996.0134. [DOI] [PubMed] [Google Scholar]
- 28.Leslie AG. Refined crystal structure of type III chloramphenicol acetyltransferase at 1.75 Å resolution. J Mol Biol. 1990;213:167–186. doi: 10.1016/S0022-2836(05)80129-9. [DOI] [PubMed] [Google Scholar]
- 29.Leslie AG, Moody PC, Shaw WV. Structure of chloramphenicol acetyltransferase at 1.75-Å resolution. Proc Natl Acad Sci USA. 1988;85:4133–4137. doi: 10.1073/pnas.85.12.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Day PJ, Shaw WV, Gibbs MR, Leslie AG. Acetyl coenzyme A binding by chloramphenicol acetyltransferase: long-range electrostatic determinants of coenzyme A recognition. Biochemistry. 1992;31:4198–4205. doi: 10.1021/bi00132a007. [DOI] [PubMed] [Google Scholar]
- 31.Barsukov IL, Lian LY, Ellis J, Sze KH, Shaw WV, Roberts GC. The conformation of coenzyme A bound to chloramphenicol acetyltransferase determined by transferred NOE experiments. J Mol Biol. 1996;262:543–558. doi: 10.1006/jmbi.1996.0534. [DOI] [PubMed] [Google Scholar]
- 32.Ellis J, Bagshaw CR, Shaw WV. Kinetic mechanism of chloramphenicol acetyltransferase: the role of ternary complex interconversion in rate determination. Biochemistry. 1995;34:16852–16859. doi: 10.1021/bi00051a036. [DOI] [PubMed] [Google Scholar]
- 33.Gibbs MR, Moody PC, Leslie AG. Crystal structure of the aspartic acid-199-asparagine mutant of chloramphenicol acetyltransferase to 2.35-Å resolution: structural consequences of disruption of a buried salt bridge. Biochemistry. 1990;29:11261–11265. doi: 10.1021/bi00503a015. [DOI] [PubMed] [Google Scholar]
- 34.Day PJ, Shaw WV. Acetyl coenzyme A binding by chloramphenicol acetyltransferase. Hydrophobic determinants of recognition and catalysis. J Biol Chem. 1992;267:5122–5127. [PubMed] [Google Scholar]
- 35.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck—a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 37.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 38.Shaw WV, Bentley DW, Sands L. Mechanism of chloramphenicol resistance in Staphylococcus epidermidis. J Bacteriol. 1970;104:1095–1105. doi: 10.1128/jb.104.3.1095-1105.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleanthous C, Cullis PM, Shaw WV. 3-(Bromoacetyl)chloramphenicol, an active site directed inhibitor for chloramphenicol acetyltransferase. Biochemistry. 1985;34:5307–5313. doi: 10.1021/bi00341a006. [DOI] [PubMed] [Google Scholar]
- 40.Murray IA, Lewendon A, Shaw WV. Stabilization of the imidazole ring of His-195 at the active site of chloramphenicol acetyltransferase. J Biol Chem. 1991;266:11695–11698. [PubMed] [Google Scholar]
- 41.Lewendon A, Murray IA, Kleanthous C, Cullis PM, Shaw WV. Substitutions in the active site of chloramphenicol acetyltransferase: role of a conserved aspartate. Biochemistry. 1988;27:7385–7390. doi: 10.1021/bi00419a032. [DOI] [PubMed] [Google Scholar]
- 42.Lewendon A, Murray IA, Shaw WV, Gibbs MR, Leslie AG. Evidence for transition-state stabilization by serine-148 in the catalytic mechanism of chloramphenicol acetyltransferase. Biochemistry. 1990;29:2075–2080. doi: 10.1021/bi00460a016. [DOI] [PubMed] [Google Scholar]
- 43.Murray IA, Cann PA, Day PJ, Derrick JP, Sutcliffe MJ, Shaw WV, Leslie AG. Steroid recognition by chloramphenicol acetyltransferase: engineering and structural analysis of a high affinity fusidic acid binding site. J Mol Biol. 1995;254:993–1005. doi: 10.1006/jmbi.1995.0671. [DOI] [PubMed] [Google Scholar]
- 44.Murray IA, Lewendon A, Williams JA, Cullis PM, Lashford AG, Shaw WV. A novel substrate for assays of gene expression using chloramphenicol acetyltransferase. Nucleic Acids Res. 1991;19:6648. doi: 10.1093/nar/19.23.6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 46.de Kok A, Hengeveld AF, Martin A, Westphal AH. The pyruvate dehydrogenase multi-enzyme complex from gram-negative bacteria. Biochim Biophys Acta. 1998;1385:353–366. doi: 10.1016/s0167-4838(98)00079-x. [DOI] [PubMed] [Google Scholar]
- 47.Meng M, Chuang DT. Site-directed mutagenesis and functional analysis of the active-site residues of the E2 component of bovine branched-chain alpha-keto acid dehydrogenase complex. Biochemistry. 1994;33:12879–12885. doi: 10.1021/bi00209a020. [DOI] [PubMed] [Google Scholar]
- 48.Langley D, Guest JR. Biochemical genetics of the alpha-keto acid dehydrogenase complexes of Escherichia coli K12: genetic characterization and regulatory properties of deletion mutants. J Gen Microbiol. 1978;106:103–117. doi: 10.1099/00221287-106-1-103. [DOI] [PubMed] [Google Scholar]
- 49.Burns DK, Crowl RM. Protein structure, folding and design 2. In: Oxender DL, editor. UCLA Symposium of molecular and cellular biology. New York: A.R. Liss & Co; 1987. p. 69. [Google Scholar]
- 50.Shaw WV, Brodsky RF. Characterization of chloramphenicol acetyltransferase from chloramphenicol-resistant Staphylococcus aureus. J Bacteriol. 1968;95:28–36. doi: 10.1128/jb.95.1.28-36.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter JRMS, editor. Methods in enzymology, macromolecular crystallography, part A. Vol. 276. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 52.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]