

Abstract
Methyl CpG binding protein 2 (MeCP2) is a canonical intrinsically disordered protein (IDP), that is, it lacks stable secondary structure throughout its entire polypeptide chain. Because IDPs often have the propensity to become locally ordered, we tested whether full-length MeCP2 and its constituent domains would gain secondary structure in 2,2,2-trifluoroethanol (TFE), a cosolvent that stabilizes intramolecular hydrogen bonding in proteins. The α-helix, β-strand/turn, and unstructured content were determined as a function of TFE concentration by deconvolution of circular dichroism data. Results indicate that approximately two-thirds of the unstructured residues present in full-length MeCP2 were converted to α-helix in 70% TFE without a change in β-strand/turn. Thus, much of the MeCP2 polypeptide chain undergoes coil-to-helix transitions under conditions that favor intrachain hydrogen bond formation. The unstructured residues of the N-terminal (NTD) and C-terminal (CTD) domains were partially converted to α-helix in 70% TFE. In contrast, the central transcription regulation domain (TRD) became almost completely α-helical in 70% TFE. Unlike the NTD, CTD, and TRD, the unstructured content of the methyl DNA binding domain and the intervening domain did not change with increasing TFE concentration. These results indicate that the coil-to-helix transitions that occur in full-length MeCP2 are localized to the NTD, CTD, and TRD, with the TRD showing the greatest tendency for helix formation. The potential relationships between intrinsic disorder, coil-to-helix transitions, and MeCP2 structure and function are discussed.
Keywords: intrinsically disordered; natively unstructured; disorder-to-order transition; circular dichroism; domain; 2,2,2-trifluoroethanol
Introduction
Methyl CpG binding protein 2 (MeCP2) is a 53-kDa nuclear protein named for its ability to selectively recognize methylated DNA.1 MeCP2 is organized into five biochemically distinct domains (Fig. 1): the N-terminal domain (NTD; residues 1–78), the methyl DNA binding domain (MBD; residues 79–167), the intervening domain (ID; residues 168–205), the transcription repression domain (TRD; residues 206–309), and the C-terminal domain (CTD; residues 310–486). There is a great deal of interest in elucidating the structure/function relationships that apply to MeCP2 because missense and nonsense mutations in its sequence cause the severe neurodevelopmental disease, Rett Syndrome (RTT).3–6 Of note, pathogenic RTT mutations are found in each MeCP2 domain, rather than located in one or even a few locations (http://mecp2.chw.edu.au/mecp2/). This broad mutational spectrum indicates that all MeCP2 domains in some manner contribute to the function of the full-length protein.
Figure 1.
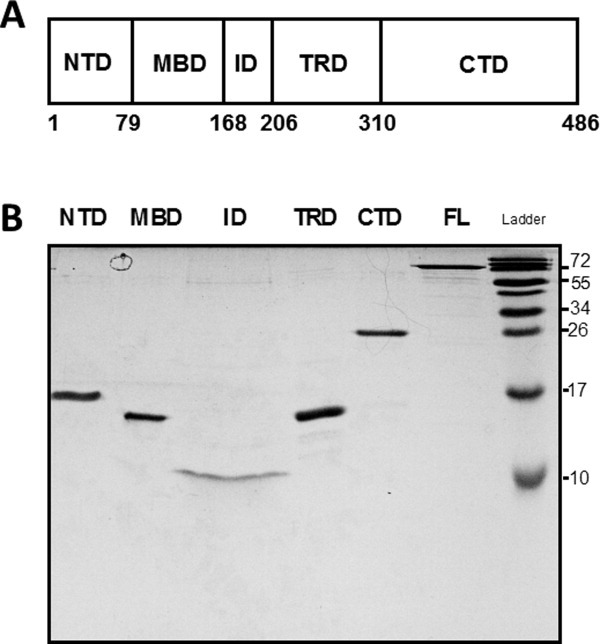
(a) Schematic illustration of the domain organization of MeCP2. NTD, N-terminal domain; MBD, methyl DNA binding domain; ID, intervening domain, TRD, transcriptional repression domain, CTD, C-terminal domain. Residue numbers of the domain boundaries are shown at the bottom. (b) SDS-PAGE of MeCP2 and its isolated domains. Full-length (FL) MeCP2 and its domains were electrophoresed on a 20% SDS-polyacrylamide gel for 120 min at 100 V. Because of different sample concentrations a larger volume of the ID was loaded relative the other domains to achieve approximately comparable band intensities, leading to horizontal band spreading. Note that full-length MeCP2 and all isolated domains exhibit anomalously slow migration during SDS-PAGE, consistent with their intrinsically disordered nature.2
Under native conditions, full-length MeCP2 is ∼60–70% unstructured as shown by circular dichroism (CD)7, 8 and has a frictional coefficient consistent with a random coil as demonstrated by analytical ultracentrifugation.8 Recent hydrogen/deuterium exchange (H/DX) experiments indicate that the entire MeCP2 polypeptide chain exhibits very rapid H/DX except for its MBD, and even the MBD has faster H/DX than a typical globular protein.9 These studies demonstrate that MeCP2 is a canonical intrinsically disordered protein (IDP), that is, it lacks stable secondary structure throughout its protein sequence.2,10–15 In many cases IDPs can locally acquire secondary structure.2, 12 For example, IDPs often become more ordered after forming complexes with other macromolecules.2,16–18 In this regard, MeCP2 binds to many different proteins, as well as DNA, RNA, and chromatin.19–28 To determine if MeCP2 can be induced to acquire secondary structure, we have used CD to measure the fraction of the full-length protein and its constituent domains that are α-helical, β-strand/turn, and unstructured as a function of 2,2,2-trifluoroethanol (TFE) concentration. TFE is a cosolvent that stabilizes intramolecular hydrogen bonds in proteins and concomitantly protein secondary structure.29, 30 Compared to native conditions, MeCP2 became substantially more α-helical and correspondingly less unstructured in TFE, while its β-strand/turn content remained constant. This indicates that TFE induced extensive coil-to-helix transitions in the full-length protein. The NTD and CTD each became more α-helical in TFE at the expense of unstructured content, although these domains remained partially unstructured even at high TFE concentrations. In contrast, essentially all of the unstructured residues of the TRD underwent coil-to helix transitions in TFE such that this domain became almost completely α-helical. The sequences of the MBD and ID did not undergo coil-to-helix transitions. These results demonstrate that coil-to-helix transitions are localized to the NTD, TRD, and CTD of full-length MeCP2. We discuss the ramifications of this work for MeCP2 structure and function and propose that the capacity of MeCP2 to undergo coil-to helix transitions may be related to its ability to interact with many different macromolecules.
Results
Histone H5 becomes more α-helical in TFE
Our experimental approach was to deconvolute CD spectra to determine the fraction of MeCP2 and its domains that were α-helical, β-strand/turn, and unstructured as a function of TFE concentration. As a control we first examined the effect of TFE on histone H5, which has been previously studied by CD.31 Representative CD spectra for H5 are shown in Figure 2(A). Visual inspection of the curves showed a qualitative trend toward α-helix formation with increasing TFE, as indicated by the disappearance of the trough at ∼200–205 nm and an increase in the peaks at ∼195 and 220 nm, respectively. To quantitate these results, the CD curves were analyzed with CDPro32 and the % calculated secondary structure plotted against TFE concentration [Fig. 2(B)]. Results indicate that there was an interconversion of unstructured content to α-helical content with increasing TFE concentration, while the amount of β-strand/turn stayed essentially constant. In 0% TFE, H5 was 68% unstructured, 9% α-helix, and 23% β-strand/turn, while in 50 and 70% TFE H5 was ∼30% unstructured, ∼40% α-helix, and ∼30% β-strand/turn. The H5 data reproduce the results of earlier studies,31 validating the experimental results obtained below with MeCP2 and its domains.
Figure 2.
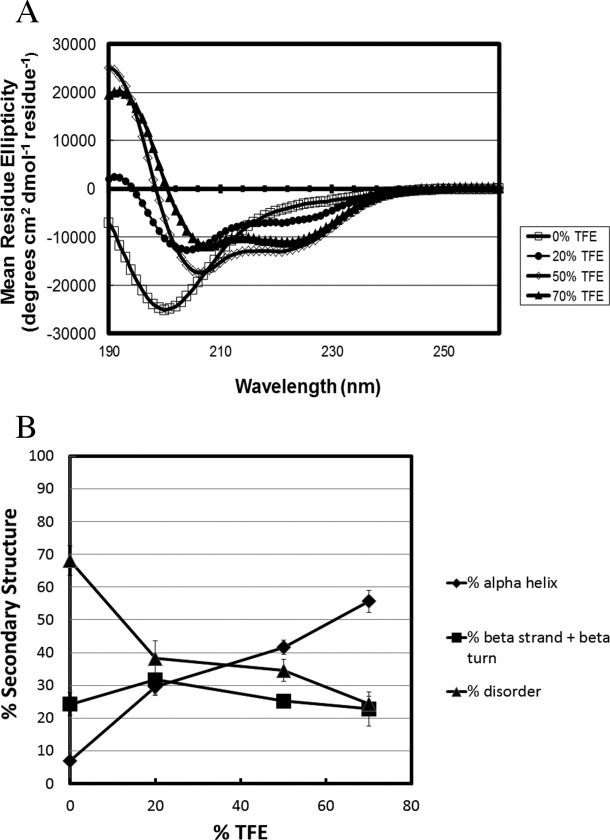
Chicken histone H5 gains α-helical content in a dose dependent manner with increased TFE concentration. (a) Circular dichroism spectra of chicken histone H5 measured in 0% (□), 20% (•), 50% (⋄), and 70% (▴) TFE. (b) Percent secondary structure was obtained after deconvolution of the CD spectra from panel (a) using CDpro software (see Materials and Methods). The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Effect of TFE on the secondary structure of full-length MeCP2
The CD spectra for full-length MeCP2 in TFE are shown in Figure 3(A), and a plot of % secondary structure against TFE concentration is shown in Figure 3(B). As with H5, MeCP2 showed an increase in α-helix with increasing TFE concentration. For full-length MeCP2, the maximum changes in secondary structure content occurred in 70% TFE. In 0% TFE MeCP2 was 68% unstructured and 7% α-helix. This changed to 24% unstructured and 55% α-helix in 70% TFE. The β-stand/turn component was ∼25% in all TFE concentrations. Thus, for full-length MeCP2, about two-thirds of the unstructured residues present under native conditions were converted to α-helix in 70% TFE while the amount of β-strand/turn remained essentially constant. This indicates that TFE induces coil-to-helix transitions in full-length MeCP2.
Figure 3.
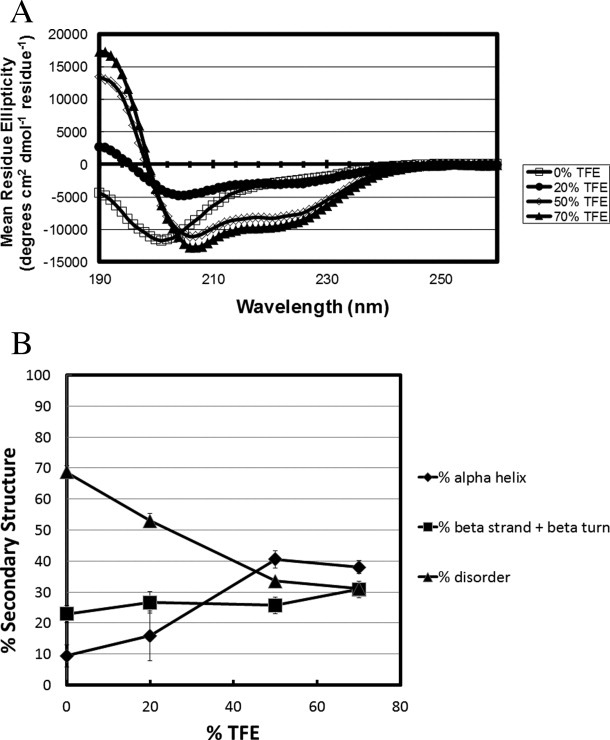
Full-length human MeCP2 gains α-helical content with increasing TFE concentration. (a) Circular dichroism spectra of MeCP2 measured in 0% (□), 20% (•), 50% (⋄), and 70% (▴) TFE. (b) Percent secondary structure was obtained after deconvolution of the CD spectra from panel (a) using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Effect of TFE on the secondary structure of isolated MeCP2 domains
To determine how the gain in secondary structure is distributed along the length of the MeCP2 sequence, we next characterized the effects of TFE on isolated MeCP2 domains. A schematic illustration of the MeCP2 domain organization is shown in Figure 1. A plot of % secondary structure against TFE concentration for the NTD is shown in Figure 4. In 20% TFE, the % unstructured residues increased slightly while the β-strand/turn decreased correspondingly and the α-helical content remained unchanged. The α-helix content that was 5% in 0% TFE changed to 34% in 70% TFE. Under these same conditions, the unstructured content decreased from ∼65% to ∼34%. Thus, compared to 0% TFE, roughly half of the initial unstructured residues of the NTD were converted to α-helix in 70% TFE. The β-strand/turn content fluctuated slightly as a function of TFE concentration, but it was ∼30% in both 0% and 70% TFE. These data indicate that TFE-induced coil-to-helix transitions occur in the NTD.
Figure 4.
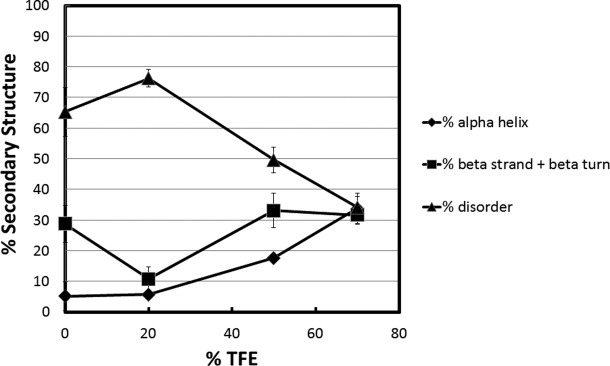
Secondary structure content as a function of TFE concentration for the MeCP2 N-terminal domain. Percent secondary structure was obtained after deconvolution of the CD spectra using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Analysis of the MBD is shown in Figure 5. In 0% TFE, the MBD was ∼32% unstructured, ∼10% α-helix, and ∼54% β-strand/turn. The structure of the isolated MBD solved by NMR33 is 31% unstructured (residues 74–93 and 162–171) and 10% α-helix (residues 135–144), with the rest of the sequence distributed between β-strand/turn and connecting loops. The crystal structure of MBD in complex with DNA34 yielded similar secondary structure content. Thus, the CD data in 0% TFE are in good agreement with the structures determined by NMR and X-ray crystallography. At 20% TFE, the values were ∼29% unstructured, ∼35% α-helix, and ∼37% β-strand/turn. This indicates that for the MBD ∼25% of the β-strand/turn present in 0% TFE was converted to α-helix in 20% TFE, while the % unstructured residues stayed constant. Unlike any of the other domains, the MBD showed no further changes in secondary structure content between 20 and 70% TFE. This result suggests that the more structured central core of the MBD has the capacity to undergo conformational change(s) in low TFE concentrations. The data in Figure 4 further indicate that TFE does not induce coil-to-helix transitions in the MBD.
Figure 5.
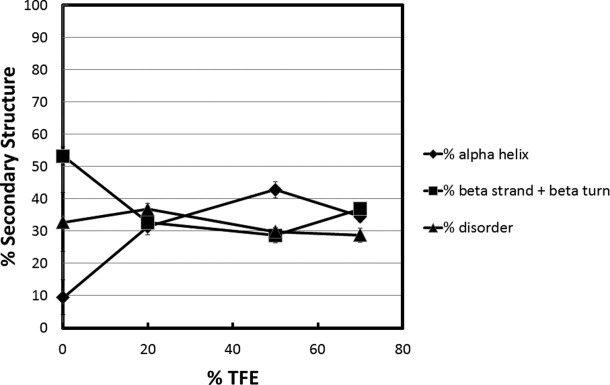
Secondary structure content as a function of TFE concentration for the MeCP2 methyl CpG binding domain. Percent secondary structure was obtained after deconvolution of the raw CD data using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Results obtained for the ID are shown in Figure 6. The data indicate that there was no significant change in secondary structure content in TFE for this domain; the % unstructured, α-helical and β-strand/turn content remained constant at ∼32%, ∼12%, and ∼55%, respectively in all TFE concentrations.
Figure 6.
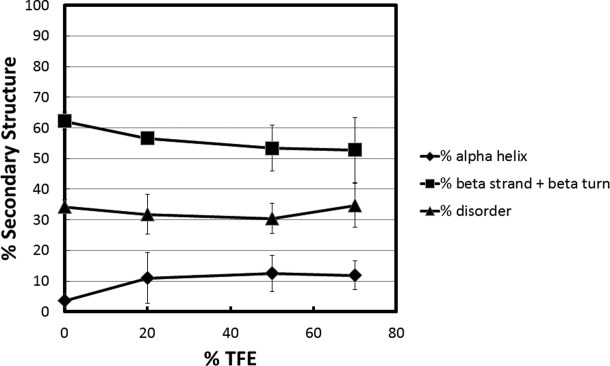
Secondary structure content versus TFE concentration for the intervening domain (ID) of MeCP2. Percent secondary structure was obtained at each % TFE after deconvolution of the raw CD data using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Characterization of the TRD is shown in Figure 7. This domain showed fundamentally different behavior compared to the others. In 0% TFE, the unstructured content was 81%, α-helix was 7%, and β-strand/turn was 12%. In 20% TFE, there was a substantial increase in α-helix (to 42%) and a smaller increase in β-strand/turn (to 28%) at the expense of unstructured content. By 70% TFE the unstructured content had decreased to 5%, α-helix increased to 89% and β-strand/turn decreased to 7%. Thus, despite having the highest degree of unstructured content under native conditions, the TRD became almost completely α-helical in 70% TFE. These data indicate that essentially the entire TRD can undergo coil-to-helix transitions.
Figure 7.
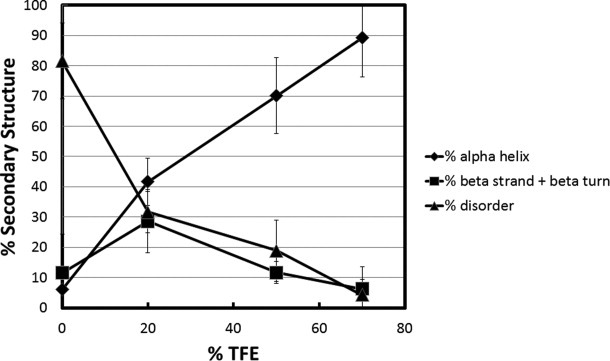
Secondary structure content versus TFE concentration for the transcription repression domain (TRD) of MeCP2. Percent secondary structure was plotted against % TFE after deconvolution of CD data from using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Plots of % secondary structure against TFE concentration for the intact CTD peptide are presented in Figure 8. In 0% TFE, the unstructured content was ∼70%, α-helix ∼5% and β-strand/turn ∼25%. In 20% TFE, there were increases in the α-helix and β-strand/turn while the unstructured content decreased proportionally. By 70% TFE the CTD was 36% unstructured, 32% α-helical and 34% β-strand/turn. This indicates that ∼50% of the unstructured CTD residues present under native conditions were converted to α-helix in 70% TFE. After its initial increase in 20% TFE, the β-strand/turn of this domain remained constant at ∼35–40% in 50% and 70% TFE. The data in Figure 8 demonstrate that coil-to-helix transitions also are localized to the CTD.
Figure 8.
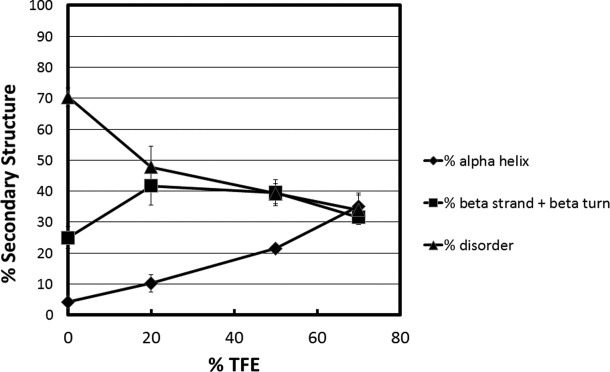
Secondary structure content versus TFE concentration for the C-terminal domain of MeCP2. Percent secondary structure is plotted against % TFE after deconvolution of CD data from using CDpro software. The % disordered (▴), % α-helix (♦), and % β strand/turn (▪) subsequently were plotted against each TFE concentration.
Discussion
In this study, we have established that TFE causes MeCP2 to become significantly more α-helical in a concentration-dependent manner. TFE is a cosolvent that stabilizes intramolecular hydrogen bonding in proteins.30 Most previous studies have examined TFE effects on model peptides and well-structured proteins.30 Depending on the system studied, TFE has been shown to stabilize α-helices, β-sheets, and β-turns.30 More recently, TFE has been used to probe disorder-to-order transitions in IDPs.35–37 Here we find that in TFE, full-length MeCP2 became substantially less unstructured (∼68% in 0% TFE vs. ∼24% in 70% TFE) and correspondingly more α-helical (∼7% in 0% TFE vs. 55% in 70% TFE), while the β-strand/turn content remained unchanged. This result indicates that roughly half of the full-length MeCP2 sequence can undergo coil-to-helix transitions under solution conditions that stabilize intrachain hydrogen bonds. By determining the behavior of the isolated MeCP2 domains in TFE, we have gained insight into how the ability to acquire secondary structure is distributed along the length of the MeCP2 polypeptide chain. In the cases of the NTD and CTD, about half of the unstructured residues present under native conditions became α-helical in 70% TFE. The overall secondary structure content of these two domains in 70% TFE was one-third α-helix, one-third β-strand/turn, and one-third unstructured. The TRD also gained α-helix in TFE at the expense of disorder. In contrast, essentially all of the unstructured residues in 0% TFE were converted to α-helix in 70% TFE, such that the TRD became almost completely α-helical under these conditions. The MBD acquired a small amount of α-helix in TFE, but at the expense of β-strand/turn rather than unstructured residues. The secondary structure content of the ID did not change, even in 70% TFE. Taken together, these results indicate that TFE-induced coil-to-helix transitions are localized to the NTD, TRD, and CTD. Importantly, Table I shows that the secondary structure content measured for full-length MeCP2 in Figure 3 is very similar to the summed content derived from Figures 4 to 8 in all TFE concentrations examined. This result suggests that the MeCP2 domains behave as autonomous structural units, and that the ability of the isolated NTD, TRD, and CTD to undergo coil-to-helix transitions accurately reflects their ability to undergo coil-to-helix transitions in the full-length protein.
Table I.
A Comparison of Measured and Summed Secondary Structure Content of MeCP2
| 0% TFE | 20% TFE | 50% TFE | 70% TFE | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %Ua | %αb | %βc | %U | %α | %β | %U | %α | %β | %U | %α | %β | |
| Measuredd | 68 | 8 | 24 | 37 | 31 | 33 | 34 | 42 | 26 | 23 | 56 | 23 |
| Summede | 65 | 6 | 28 | 45 | 20 | 34 | 34 | 34 | 33 | 27 | 43 | 30 |
% Unstructured residues.
% α-helical residues.
% β-sheet/turn residues.
Taken from Figure 2(B).
For each TFE concentration, the measured % U, %α, and %β values for the NTD, MBD, ID, TRD, and CTD were multiplied by 0.16, 0.18, 0.08, 0.2, and 0.38, respectively (the fraction of the total protein sequence corresponding to each domain). The resulting values were then summed.
Induction of coil-to-helix transitions by TFE does not appear to be due to a high helix-forming potential of the MeCP2 protein sequence. Predictions using the Agadir algorithm38, 39 indicate that only 0.6% of the NTD, 2.9% of the TRD, and 0.3% of the CTD are expected to be α-helical. The predictions are in good agreement with the experimental data obtained under native conditions; only ∼5% of the NTD, TRD, and CTD were observed to be α-helical in 0% TFE. Thus, TFE is able to convert unstructured segments of the MeCP2 polypeptide chain with very low inherent helix forming potential to α-helix. The action of TFE has been proposed to originate from preferential clustering of cosolvent around the polypeptide chain, which diminishes hydrogen bonding to bulk water and establishes a low dielectric medium that favors intrachain hydrogen bonding.40 From a functional perspective, we hypothesize that TFE/water solutions may mimic the environment found at the binding interfaces between IDPs and other macromolecules. IDPs often gain secondary structure concomitant with macromolecular interactions.2,16–18 In the case of MeCP2, the NTD interacts with the protein HP1,19 the TRD with many different co-repressor proteins22–27 and unmethylated DNA,8, 41 and the CTD with unmethylated DNA,41 chromatin,41 and RNA splicing proteins.20, 21 Moreover, some proteins interact with multiple MeCP2 domains, e.g., PU.1 binds to the NTD and TRD,28 CDKL5 binds to the TRD and CTD.26 The demonstration of inducible coil-to-helix transitions in the NTD, TRD, and CTD provides a testable working model for how MeCP2 can be extensively disordered in its native state yet bind to nucleic acids and many different proteins.
Materials and Methods
Protein expression and purification
Linker histone variant H5 was purified from native chromatin derived from mature chicken erythrocytes as described previously.31, 42 Full-length MeCP2 (isoform e2), and the NTD, MBD, ID, and TRD fragments were expressed in E. coli and purified using the Intein Mediated Purification with an Affinity Chitin-binding Tag (IMPACT) system (New England Biolabs). The CTD was expressed using a modified pET28a vector plasmid (Novagen). The MBD and TRD constructs contained an added sequence, EFLEGSSC, on their C-terminal ends as a result of previously described cloning methods.8, 41 The ID was cloned without this vestigial sequence and corresponded to the DNA sequence that codes for amino acids 168–205 from wild-type MeCP2 using the following primers (ID5′) 5′-GGA GCC CCC ATA TGC GAG AGC AGA AAC CAC CTA AGA AGC C-3′; and (ID3′) 5′-CAT AGG CTC TTC GGC ACT CTG ACG TGG CCG CCT TGG GTC TC-3′. Primers were designed to amplify DNA fragments cleavable by restriction enzymes Nde1 on the 5′ side and Sap1 on the 3′ side. The NTD construct was amplified to express wild type MeCP2 amino acids 1–78 using the following primers; (NTD5′) 5′-GAC ATA TGG TAG CTG GGA TGT TAG GGC TCA GGG AAG-3′; and (NTD3′) 5′-CAG AAT TCA GAA GCT TCC GGC ACA GCC GGG GC-3′. The NTD insert was amplified from the wild-type MeCP2 template such that it was cleaved by NdeI and EcoR1, gel-purified, and ligated into correspondingly digested and purified pTYB1 vector plasmid. The NTD also expresses with an additional non-native eight amino acids, EFLEGSSC as a result of the NdeI and EcoR1 cloning strategy.
E. coli BL21RP+ was used as host bacteria for expression by pTYB1 plasmid. A transformed bacterial colony was selected and grown in lysogeny broth to an optical density of 0.5 absorbance units (590 nm) at 37°C. Translation was induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside at 18°C for 3 h before harvest. Bacteria were centrifuged in an Avanti J-26 XPI preparative centrifuge (Beckman Coulter) in a JLA 8.100 rotor at 5000g for 10 min. Pellets were resuspended in wash buffer (25 mM Tris, pH 7.5/100 mM NaCl) and centrifuged again under the previous conditions. Bacterial pellets were resuspended in column buffer (25 mM Tris–HCl pH 8.0, 500 mM NaCl) with 0.1% Triton X-100, 0.2 mM PMSF, and Protease Inhibitor Cocktail Set II (Calbiochem) added. Resuspended bacteria were subjected to two rounds of sonication, 90 s each, with a Branson Sonifier 450. A large tip set to 50% duty cycle with a power output of 7 was used. Sonicated lysate was poured into Oakridge tubes and centrifuged at 21,000g for 25 min in a JA-17 rotor (Beckman). Clear supernatant was mixed with chitin beads (New England Biolabs) previously equilibrated in column buffer, and the mixture was incubated at 4°C overnight. Errant contaminating bacterial DNA-MeCP2 complexes were washed from the samples while bound to the chitin beads with five column volumes column buffer, followed by an equal volume column buffer at 900 mM NaCl final concentration. Chitin beads were re-equilibrated with an additional five column volumes 500 mM NaCl column buffer. Column buffer with 50 mM DTT was passed over the column such that 1 cm buffer remained between the meniscus and top of the column bed in a 10-cm Kontes FlexColumn (Fischer). The column was left for 48–72 h that allowed intein-mediated cleavage. Column buffer was used to elute protein from the chitin column. The eluant was diluted from 500 to 100 mM NaCl with column buffer containing no salt and loaded onto a HiTrap Heparin HP column (GE Healthcare). Proteins were eluted from the heparin column with a 100 mM NaCl to 1M NaCl step gradient. Specifically, a gradient consisting of 100 mM NaCl steps in 25 mM Tris, pH 7.5, 10% glycerol buffer was used. Peak fractions were loaded on a 20% SDS polyacrylamide gel to analyze for purity and/or degradation [Fig. 1(B)], then pooled and dialyzed into 10 mM Tris pH 7.5.43
The CTD was expressed using a modified pET28a vector plasmid (Novagen) with an N-terminal 6-histidine tag cleavable by PreScission protease (GE Healthcare). Modified vector was kindly supplied by Dr. Wayne Lilyestrom. The CTD fragment corresponds to residues 310–486. The CTD construct was amplified with an Nde1 site on the 5′ end and a BamH1 site on the 3′ end with a stop codon included directly 3′ to the BamH1 site. The forward primers used to amplify the CTD construct was 5′-GAC ATC CAT ATG GAG ACG GTC AGC ATC GAG G-3′. The reverse primer sequence is (MeCP2 486 3′) 5′-CTG GGA TCC CTA GCT AAC TCT CTC GGT CAC G-3′. Constructs were expressed and purified using Ni-NTA agarose beads (Qiagen).
Circular dichroism
Stock solutions of the respective proteins were prepared at ∼500 μg/mL concentration and stored in glass test tubes. Concentrations were determined using a bicinchoninic acid assay (Pierce BCA™ protein assay kit, Thermo Scientific) conducted on a 96-well microplate and measured in a Bio-Rad Model 680 microplate reader at 560 nm. Peptide concentration accuracy was verified by total amino acid analysis (Biophysics Core, Department of Biomolecular Structure, University of Colorado Denver Anschutz Medical Campus). Dilutions were prepared in glass test tubes to a final volume of 220 μL such that the final protein concentration was 0.12 mg/mL in 1 mM Na phosphate, 0.2 mM Na3EDTA with either 0, 20, 50, or 70% volumetric concentrations of 99.8% trifluoroethanol (Acros organics, Fisher). The final pH was adjusted to 7.4 when necessary. All buffers were purified through a 0.2-μm filter (Metricel® Pall Corporation), and proteins were dialyzed extensively against their respective buffers using 3500 MWCO dialysis tubing (Spectrum labs).
CD spectra were recorded from 260 to 190 nm on a Jasco-720 spectropolarimeter. Samples were transferred to a cuvette with a 1-mm path length and cooled to 20°C. Spectra were collected at a bandwidth of 1 nm with a scanning rate of 10 nm/s in continuous scanning mode and a response time of 16 s. From raw data, collected spectra were buffer-subtracted and converted from millidegrees to molar ellipticity using the following equation:
where [θ] is the mean residue ellipticity (degrees cm2/dmol/residue), θobs is the ellipticity measured in millidegrees, M is the protein mean residue molecular weight, l is the optical path length of the cuvette in cm, and C is the concentration of the protein in mg/mL. Ellipticity data were processed using Spectra Manager software version 1.53.01 (Jasco Corporation) and saved as text files. Data files were then deconvoluted using the SDP48 basis set, which contains CD spectra from five unfolded proteins and 43 native proteins. Deconvolution was performed using CDpro software including CONTINLL, SELCON3, and CDSSTR methods.32 Estimates of percent secondary structure obtained from the three methods were averaged and standard deviations calculated.
Acknowledgments
The authors thank Dr. Robert W. Woody for critical review of this manuscript, Dr. Christopher Woodcock, and Dr. Wayne Lilyestrom for kindly supplying plasmid reagents, and Christine Krause for technical assistance with experiments.
References
- 1.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 2.Tompa P. Structure and function of intrinsically disordered proteins. Chapman & Hall: CRC Press; 2009. [Google Scholar]
- 3.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 4.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 5.Huppke P, Laccone F, Krämer N, Engel W, Hanefeld F. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000;9:1369–1375. doi: 10.1093/hmg/9.9.1369. [DOI] [PubMed] [Google Scholar]
- 6.Cheadle JP, Gill H, Fleming N, Maynard J, Kerr A, Leonard H, Krawczak M, Cooper DN, Lynch S, Thomas N. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet. 2000;9:1119–1129. doi: 10.1093/hmg/9.7.1119. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, Woodcock CL. Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem. 2008;283:20523–20534. doi: 10.1074/jbc.M803021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams V, McBryant S, Wade P, Woodcock C, Hansen J. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J Biol Chem. 2007;282:15057–15064. doi: 10.1074/jbc.M700855200. [DOI] [PubMed] [Google Scholar]
- 9.Hansen JC, Wexler BB, Rogers DJ, Hite KC, Panchenko T, Ajith S, Black BE. DNA binding restricts the intrinsic conformational flexibility of methyl CpG binding protein 2 (MeCP2) J Biol Chem. 2011;286:18938–18948. doi: 10.1074/jbc.M111.234609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 11.Dunker AK, Obradovic Z. The protein trinity-linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 12.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 13.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Uversky VN. Seven lessons from one IDP structural analysis. Structure. 2010;18:1069–1071. doi: 10.1016/j.str.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 16.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 17.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal N, Hardt T, Brero A, Nowak D, Rothbauer U, Becker A, Leonhardt H, Cardoso MC. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 2007;35:5402–5408. doi: 10.1093/nar/gkm599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedford MT, Chan DC, Leder P. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 1997;16:2376–2383. doi: 10.1093/emboj/16.9.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buschdorf JP, Stratling WH. A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J Mol Med. 2004;82:135–143. doi: 10.1007/s00109-003-0497-9. [DOI] [PubMed] [Google Scholar]
- 22.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 23.Kaludov NK, Wolffe AP. MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 2000;28:1921–1928. doi: 10.1093/nar/28.9.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806–4812. doi: 10.1074/jbc.M209923200. [DOI] [PubMed] [Google Scholar]
- 25.Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T, Yasukawa T, Colmenares C, Ishii S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J Biol Chem. 2001;276:34115–34121. doi: 10.1074/jbc.M105747200. [DOI] [PubMed] [Google Scholar]
- 26.Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- 27.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki M, Yamada T, Kihara-Negishi F, Sakurai T, Oikawa T. Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene. 2003;22:8688–8698. doi: 10.1038/sj.onc.1207182. [DOI] [PubMed] [Google Scholar]
- 29.Goodman M, Listowsky I. Conformational aspects of synthetic polypeptides. VI. Hypochromic spectral studies of oligo-γ-methyl-L-glutamate peptides. J Am Chem Soc. 1962;84:3770–3771. [Google Scholar]
- 30.Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- 31.Clark D, Hill C, Martin S, Thomas J. Alpha-helix in the carboxy-terminal domains of histones H1 and H5. EMBO J. 1988;7:69–75. doi: 10.1002/j.1460-2075.1988.tb02784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 33.Wakefield RI, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN. The solution structure of the domain from MeCP2 that binds to methylated DNA. J Mol Biol. 1997;291:1055–1065. doi: 10.1006/jmbi.1999.3023. [DOI] [PubMed] [Google Scholar]
- 34.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Roque A, Iloro I, Ponte I, Arrondo JL, Suau P. DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J Biol Chem. 2005;280:32141–32147. doi: 10.1074/jbc.M505636200. [DOI] [PubMed] [Google Scholar]
- 36.Bourhis JM, Canard B, Longhi S. Structural disorder within the replicative complex of measles virus: functional implications. Virology. 2006;344:94–110. doi: 10.1016/j.virol.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Pattaramanon N, Sangha N, Gafni A. The carboxy-terminal domain of heat-shock factor 1 is largely unfolded but can be induced to collapse into a compact, partially structured state. Biochemistry. 2007;46:3405–3415. doi: 10.1021/bi061124c. [DOI] [PubMed] [Google Scholar]
- 38.Lacroix E, Viguera AR, Serrano L. Elucidating the folding problem of alpha-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 39.Munoz V, Serrano L. Development of the multiple sequence approximation within the AGADIR model of alpha-helix formation: comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 1997;41:495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 40.Roccatano D, Colombo G, Fioroni M, Mark AE. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc Natl Acad Sci USA. 2002;99:12179–12184. doi: 10.1073/pnas.182199699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghosh RP, Nikitina T, Horowitz-Scherer RA, Gierasch LM, Uversky VN, Hite K, Hansen JC, Woodcock CL. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry. 2010;49:4395–4410. doi: 10.1021/bi9019753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Ramirez M, Leuba SH, Ausio J. One-step fractionation method for isolating H1 histones from chromatin under nondenaturing conditions. Protein Expr Purif. 1990;1:40–44. doi: 10.1016/1046-5928(90)90043-x. [DOI] [PubMed] [Google Scholar]
- 43.Clark DJ, Thomas JO. Salt-dependent co-operative interaction of histone H1 with linear DNA. J Mol Biol. 1986;187:569–580. doi: 10.1016/0022-2836(86)90335-9. [DOI] [PubMed] [Google Scholar]