

Abstract
Genetic studies have established that lysis inhibition in bacteriophage T4 infections occurs when the RI antiholin inhibits the lethal hole-forming function of the T holin. The T-holin is composed of a single N-terminal transmembrane domain and a ∼20 kDa periplasmic domain. It accumulates harmlessly throughout the bacteriophage infection cycle until suddenly causing permeabilization of the inner membrane, thereby initiating lysis. The RI antiholin has a SAR domain that directs its secretion to the periplasm, where it can either be inactivated and degraded or be activated as a specific inhibitor of T. Previously, it was shown that the interaction of the soluble domains of these two proteins within the periplasm was necessary for lysis inhibition. We have purified and characterized the periplasmic domains of both T and RI. Both proteins were purified in a modified host that allows disulfide bond formation in the cytoplasm, due to the functional requirement of conserved disulfide bonds. Analytical centrifugation and circular dichroism spectroscopy showed that RI was monomeric and exhibited ∼80% alpha-helical content. In contrast, T exhibited a propensity to oligomerize and precipitate at high concentrations. Incubation of RI with T inhibits this aggregation and results in a complex of equimolar T and RI content. Although gel filtration analysis indicated a complex mass of 45 kDa, intermediate between the predicted 30 kDa heterodimer and 60 kDa heterotetramer, sedimentation velocity analysis indicated that the predominant species is the former. These results suggest that RI binding to T is necessary and sufficient for lysis inhibition.
Keywords: lysis, holin, lysis inhibition, T4
Introduction
For most bacteriophages, the host lysis event depends on the temporally regulated action of holin, an extremely diverse class of small phage-encoded membrane proteins.1, 2 The holin proteins accumulate harmlessly in the inner membrane (IM) throughout the morphogenesis period of the infection cycle. Suddenly, the holin forms lethal membrane lesions, or holes that results in a cessation of respiration and termination of macromolecular synthesis.3 This lethal event, called holin triggering, occurs at a time specified by the allelic state of the holin gene. The timing of triggering is thought to reflect the instant when the holin reaches a critical concentration within the membrane, leading to massive oligomerization and then lethal hole formation.4 Hole formation can also be prematurely triggered by artificial depolarization of the membrane. The mechanistic and structural linkages between oligomerization and triggering are not yet known. For many phages, including the well-studied lambda and T4, the holes formed are sufficiently large to allow nonspecific escape of the phage endolysin, a small cytoplasmic protein with muralytic activity, across the bilayer, resulting in the rapid destruction of the peptidoglycan.5, 6 Holins are genetically malleable, in that conservative mutations throughout the primary structure, and especially in the transmembrane domains (TMDs), can have drastic effects on the triggering time.7 Since the triggering time defines the length of the infection cycle and indirectly the average burst size of the infection, it has been proposed that the apparent universality of holin-controlled lysis reflects the requirement for phages to be able to rapidly evolve to shorter or longer infection cycles as the environment changes.7, 8
Real-time regulation of lysis timing, however, has been observed only with phage T4 and its close relatives. Specifically, lysis can be delayed indefinitely in the lysis-inhibited state (LIN) depending on the supply of free virus particles in the vicinity of the infected cell.9, 10 LIN has been the focus of numerous studies since its discovery in the 1940s,10 leading researchers to many fundamental advances, including the definition of a gene,11, 12 the nature of the genetic code,13 and the basics of recombination.14–16 At the cellular level, LIN is imposed when a T4-infected cell is superinfected at 5 min or later after the original infection. The basic physiology of this event has been established in detail. The superinfection begins normally, with irreversible adsorption to the host surface leading to massive conformational changes in the virion and penetration of the outer membrane by the central tail tube. However, the cytoplasmic membrane is not punctured, and the contents of the virion capsid, including the 170 kb DNA molecule and more than 1000 protein molecules, are instead ectopically ejected into the host periplasm. Somehow, this abortive injection leads to activation of a small (11.1 kDa) phage protein, RI, which then inhibits T (25.2 kDa), the T4 holin, blocking it from triggering and thus prolonging the infection cycle. If superinfections continue to occur at <10 min intervals, RI-mediated inhibition of T persists indefinitely, so that virions can accumulate intracellularly to levels ten-fold or more higher than the normal burst size.17
Both T and RI are synthesized as type II integral membrane proteins, with an N-terminal TMD tethering 19.2 kDa (163 aa) [Fig. 1(A)] and 8.8 kDa (75 aa) [Fig. 1(B)] periplasmic domains to the bilayer, respectively. The presence of a significant periplasmic domain makes T unique among holins, which in general consist of two or more TMDs linked by short loops.18 In addition, the TMD of RI escapes spontaneously from the membrane and is thus designated a SAR domain, for “signal-anchor-release”.19In vivo, the SAR domain confers extreme functional and proteolytic instability (t½ = 2 min) on the periplasmic form of RI.19 We have proposed a model in which the abortive superinfection, specifically the ectopic injection of the capsid contents in the periplasm, stabilizes RI and allows it to bind to the periplasmic domain of T,17 which in turn blocks oligomerization of T and thus prevents triggering. Replacement of the SAR domain with a cleavable signal sequence results in the accumulation of the periplasmic domain of RI, designated as sRI [Fig. 1(B)].17 In this situation, sRI binds to the soluble domain of T (sT) [Fig. 1(A)] and blocks lysis. Moreover, secretion of sT to the periplasm can spare full-length T from RI inhibition.17
Figure 1.

The primary structure of T4 T and RI. A: T4 T. The predicted TMD and location of the C-terminal oligohistidine tag are underlined. The region of T cloned into pET11a and purified is labeled with “sT-his” (see “Materials and Methods”). Location of the amber and cysteine-to-serine substitution mutants are indicated by arrows. B: T4 RI. The SAR domain and C-terminal oligohistidine tag are underlined. The region of RI cloned into pET11a and purified is labeled with “sRI-his” (see “Materials and Methods”). Location of cysteine-to-serine substitution mutants are indicated by arrows.
The proteolytic instability of RI and the lethality of T have been major impediments to studying the mechanism of T-hole formation and RI-mediated LIN at the biochemical or structural level. Here, we report the results of efforts to purify and characterize the soluble periplasmic domains, sRI and sT. The results are discussed in terms of a model for the RI-T interaction and its role in LIN.
Results
Cysteines are required for T lytic and RI LIN function
The choice of strategies for over-production and purification of the T4 holin and antiholin was affected by the presence of two Cys residues in both RI and T. If the Cys residues could be replaced by conservative substitutions like Ser, the potential for oxidative damage during purification would be minimized. However, sequence alignment of the holins (T) and antiholins (RI) of T4-like phages revealed that in both proteins the Cys residues were conserved both in terms of number (always two), spacing (30–31 intervening residues for T proteins; 3–5 intervening residues for RI proteins), and position in the sequence (C-terminal 20% of both proteins; Supporting Information Figs. S1 and S2). This suggested that, like many periplasmic proteins, both RI and T are likely stabilized by intramolecular disulfide bonds. To test whether these residues were essential for the lytic function of T, single and double Ser substitutions were made, generating Cys175Ser, Cys207Ser, and Cys175/207Ser. Induction of the wild type construct resulted in the characteristic, sharply defined lysis at 20 min [Fig. 2(A)]. In contrast, each of the cysteine substitution alleles exhibited an identical absolute lysis defect. Similarly, to test whether the two Cys residues in RI are essential for LIN, the Cys69Ser, Cys75Ser, and Cys69/75Ser single and double substitutions were constructed. Synchronous induction of t and the parental rI resulted in bona fide lysis inhibition [Fig. 2(B)]. Addition of 1% chloroform at 60 min after induction resulted in immediate clearing of the T and RI induced culture, indicating that the lack of lysis was not due to the lack of expression of the endolysin, coexpressed on the plasmid, but rather due to a holin defect. In contrast, all three-substitution alleles failed to support LIN, with lysis occurring at 20 min, as it does in inductions of t alone. Thus the conserved pairs of Cys residues in the periplasmic domains of both the holin T and antiholin RI are essential for lytic and LIN functions, respectively. Even-numbered complements of Cys residues in periplasmic proteins are almost always associated with disulfide bonds important for stable folding,20 so these results indicated that a useful over-production and purification strategy must retain the capacity to form and maintain disulfide bonds in both T and RI.
Figure 2.
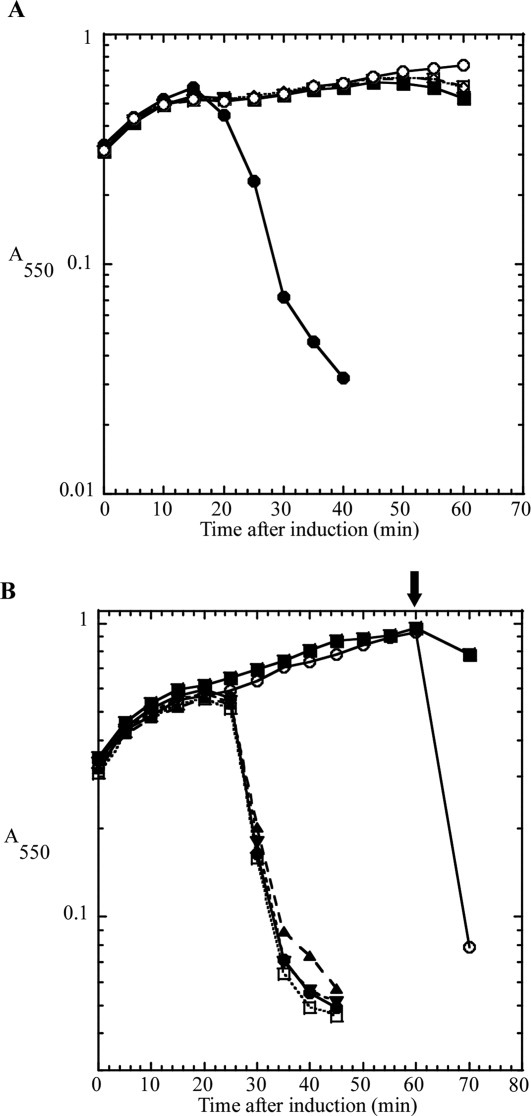
Lysis and lysis inhibition defect in cysteine to serine substitutions. A: Cysteine-to-serine substitutions in T show a severe lysis defect. CQ21 λkanΔ (SR) cells carrying the indicated plasmids were grown to A550 = 0.3 and induced by a shift to 42°C. Symbols: •, pSM-t; ▪, pSM-t C175S; ⋄, pSM-t C207S; □, pSM-t C175,207S; ○, pSM-t D19am. B: Cysteine-to-serine substitutions in RI show a LIN-defective phenotype, similar to inductions of T alone. CQ21 λkanΔ (SR) cells carrying the indicated plasmids were grown to A550 = 0.3 and induced by a shift to 42°C and addition of IPTG. Symbols: •, pSM-t only; ▪, pZA-ssPhoAϕsRI only; ○, pSM-t and pZA-ssPhoAϕsRI; ▴, pSM-t and pZA-ssPhoAϕsRI C69S; ▾, pSM-t and pZA-ssPhoAϕsRI C75S; □, pSM-t and pZA-ssPhoAϕsRI C69,75S. Arrow indicates addition of CHCl3.
Soluble sRI accumulates in a host cytoplasm optimized for disulfide bond formation
Attempts to obtain native, full length RI by overexpression were unsuccessful, and in fact, no native RI could even be detected by immunoblot (not shown). The SAR domain directs RI to the sec translocon, which itself severely constrains the maximum output from over-expression systems,21 and also confers extreme proteolytic instability on periplasmic RI.19 In view of the fact that the periplasmic domain, sRI, is fully functional in vivo when directed to the periplasm by a normal, cleavable signal sequence, a strategy focusing on sRI was pursued. Using the T7-based hyper-expression vector, pET11a,22 we constructed a plasmid encoding sRI with a C-terminal hexahistidine tag. Cultures of BL21(DE3) carrying this construct and induced at 37°C accumulated detectable levels of the 9.7 kDa sRI-his product [Fig. 3(A)]. However, none of the protein proved to be soluble. In view of the essential character of the cysteine residues, it was suspected that the sRI protein was misfolding and aggregating in the host cytoplasm. Nevertheless, there was no improvement when using Origami(DE3)™ cells, in which the cytoplasm is more conducive to disulfide bond formation as a result of gor and trxB mutations.23 In contrast, nearly 25% of total sRI was soluble when SHuffle® T7 [SHuffle(DE3)] cells were used. SHuffle(DE3) cells are similar to Origami(DE3) cells except for the ectopic, cytoplasmic presence of the disulfide bond isomerase, DsbC.24 Finally, ∼75% of the sRI protein was found in the soluble fraction when cells were incubated at 16°C overnight, following induction. sRI was purified by immobilized metal affinity chromatography (IMAC) at 4°C and yielded 4 mg L−1.
Figure 3.
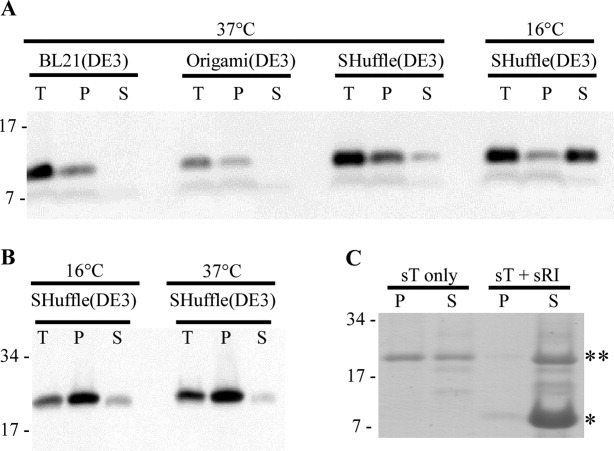
Protein solubility in various over-expression backgrounds and temperatures. Determination of protein solubility of sRI (A) and sT (B) in various over-expression backgrounds and temperatures. T: total sample, P: insoluble fraction, and S: soluble fraction. Molecular mass standards used are indicated on left. A: Western blot of sRI samples over-expressed at 37°C in BL21(DE3), Origami(DE3), and both 37 and 16°C in the SHuffle(DE3) backgrounds. B: Western blots of sT samples over-expressed at 16 and 37°C in the SHuffle(DE3) background. C: Coomassie-stained gel of samples in which sT was eluted with or without the presence of sRI. Migration of sRI, * and sT, ** are indicated.
sRI is a predominantly helical monomer in solution
In an effort to determine whether the purified sRI was properly folded, the secondary structure of the periplasmic domain of RI was assessed by circular dichroism (CD) spectroscopy. Analysis of the spectrum using the deconvolution program K2D225 indicated that sRI was ∼78% alpha-helical, slightly higher than the 65% predicted by the secondary sequence prediction program JPred3,26 and strongly indicating that, despite its small size and its ectopic expression, the purified sRI protein was quantitatively folded [Fig. 4(A,C)]. The oligomeric status of the purified sRI was assessed by gel filtration chromatography, where it eluted in a major peak corresponding to 10 kDa, consistent with monomeric status, and a minor peak at 20 kDa [Fig. 5(A)]. This conclusion was buttressed by sedimentation velocity and equilibrium experiments performed on the purified sRI. Sedimentation velocity analysis of three samples at varying concentrations revealed a coefficient of 1.4S and a frictional ratio (f/f0) of 1.3, consistent with a globular monomer of 9.7 kDa [Fig. 5(B,C)].27 Additionally, a sedimentation equilibrium analysis experiment revealed a protein of 9.2 kDa in solution, within 5% of the 9.7 kDa calculated from the sRI primary structure [Fig. 5(D)]. Taken together, these results indicate that sRI exists as a globular monomer of primarily helical character in solution.
Figure 4.
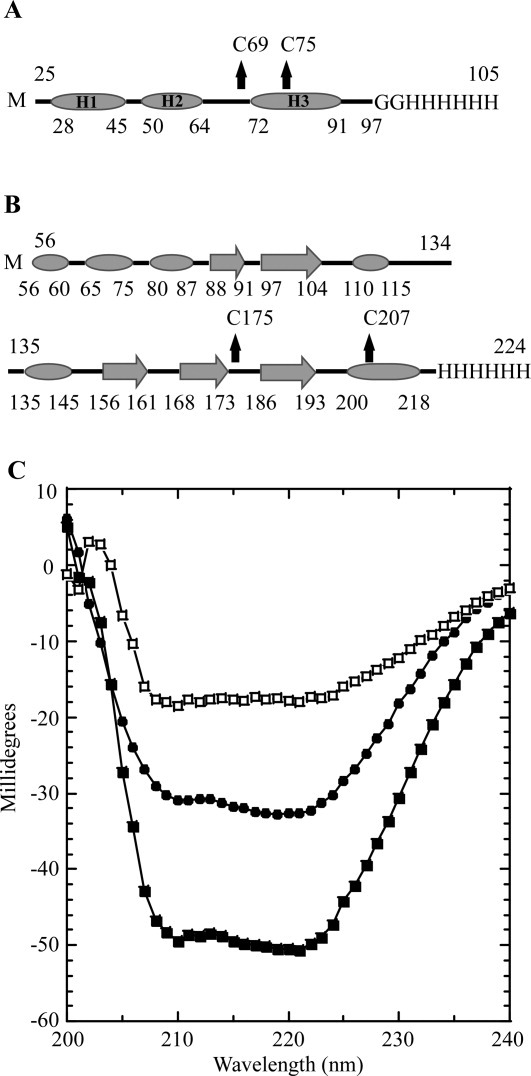
Secondary structure prediction and CD spectroscopy. A: Secondary structure prediction of sRI by JPred. α-helices are indicated by ovals and unstructured elements indicated by lines. The location of the two cysteine residues are indicated by arrows and the C-terminal oligohistidine tag is labeled. sRI is predicted to contain 65% alpha-helical content with no other structured elements predicted. B: Secondary structure prediction of sT by JPred. α-helices are indicated by ovals, β-sheets are indicated by large arrows, and unstructured elements are indicated by lines. The location of the two-cysteine residues is indicated by arrows and the C-terminal oligohistidine tag is labeled. sT is predicted to contain 30.5% alpha-helical character, and 19% beta-sheet with no other structured elements predicted. C: CD spectra of sRI (•) and sT-sRI complex (▪) at a final concentration of 1 μM each. The difference spectrum (of sRI and the sT-sRI complex) is indicated (□).
Figure 5.
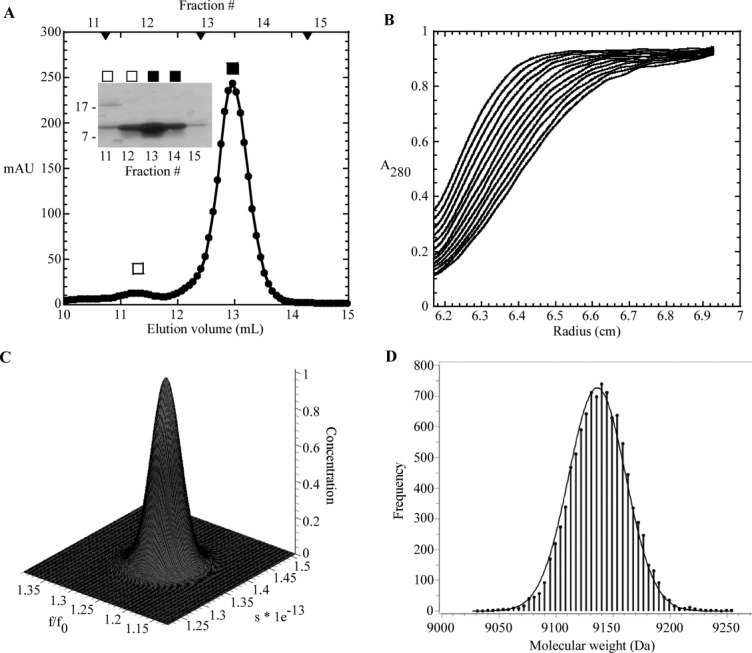
S-75 gel filtration and analytical ultracentrifugation of sRI. A: Elution profile of sRI with major monomer peak (▪) and minor dimer peak (□) indicated. S-75 standards are indicated by arrow heads, from left to right: 29, 13.7, and 6.5 kDa. Inset: Coomassie-stained gel of samples collected from each gel filtration fraction. B: Sedimentation velocity of sRI (at various concentrations, see “Materials and Methods”). The positions of the moving boundaries shown are at ∼6 min intervals by spectrophotometric scanning at 280 nm. C: Genetic algorithm/Monte Carlo analysis of the sRI sedimentation velocity data with frictional ratio and sedimentation coefficient shown. The concentration axis shows that no other species exist in the samples analyzed in the sedimentation velocity experiment. D: Measurement of the molecular mass of sRI by fitting of sedimentation equilibrium data performed at varying sRI concentrations (see “Materials and Methods”). The frequency distribution describes the statistical confidence level for each molecular weight calculation.40
sT oligomerizes at high concentrations and requires sRI to remain soluble
Previous experiments showed that the periplasmic domain of T, sT, is fully capable of interacting with RI.17 Accordingly, a pET11a construct encoding sT with a C-terminal oligohistidine tag was used for over-expression. Because sT also contains two essential cysteine residues, over-expression was carried out in the SHuffle(DE3) background with inductions at 37 and 16°C. Inductions at 16°C slightly increased the amount of soluble protein as compared to 37°C [Fig. 3(B)]. The inductions at 16°C yielded ∼1.5 mg of soluble sT per liter. However, unlike sRI, elution of sT from the IMAC resin resulted in immediate precipitation of more than 60% of the protein, presumably reflecting the tendency of holin proteins to oligomerize. Since experiments carried out in vivo had shown that sRI binds to T and inhibits hole formation, we reasoned that sT eluted into excess sRI might form stable complexes blocked from aggregation.17 This was indeed the case, since very little aggregation (<5%) was detected after elution of sT into a molar excess of sRI [Fig. 3(C)].
The sT-sRI complex is a heterodimer in solution that contains both helical and beta sheet character
Gel filtration analysis of the sT-sRI mixture was performed to gauge the molecular mass of the formed complex [Fig. 6(A,B)]. An apparent sT-sRI complex eluted in a major peak corresponding to 45.6 kDa, calculated using previously analyzed standards [Fig. 6(A)]. This mass was not consistent with the 29.7 kDa mass predicted for a heterodimer of sT-his (20 kDa) and sRI-his (9.7 kDa). N-terminal analysis by Edman degradation sequencing of the complex revealed that sT and sRI were present in a 1:1 ratio (data not shown). At this ratio, the predicted mass of the complex would be either 29.7 kDa for the heterodimer or 59.4 kDa for the sT2-sRI2 heterotetramer. To resolve this apparent discrepancy, sedimentation velocity experiments were performed, revealing the complex to have a sedimentation coefficient of 4.1S, consistent with the 29.7 kDa heterodimer state [Fig. 6(C)]. CD spectroscopy was performed to assess the secondary structure found in the complex. Using K2D2, the complex exhibited 48.5% α-helical character along with 10% β-strand, with nearly 40% unstructured elements [Fig. 4(C)]. A difference spectrum of the sT-sRI complex and sRI only CD spectra was performed to understand the secondary structure character of sT alone, assuming that secondary structures of sT and sRI are not changed significantly upon binding. Based on this analysis, sT is slightly more than 23.7% α-helical, 26.2% β-strand, and nearly 50% unstructured, similar to what is predicted by JPred3 (30% α-helical, 20% β-strand, and 50% unstructured) [Fig. 4(B,C)].
Figure 6.
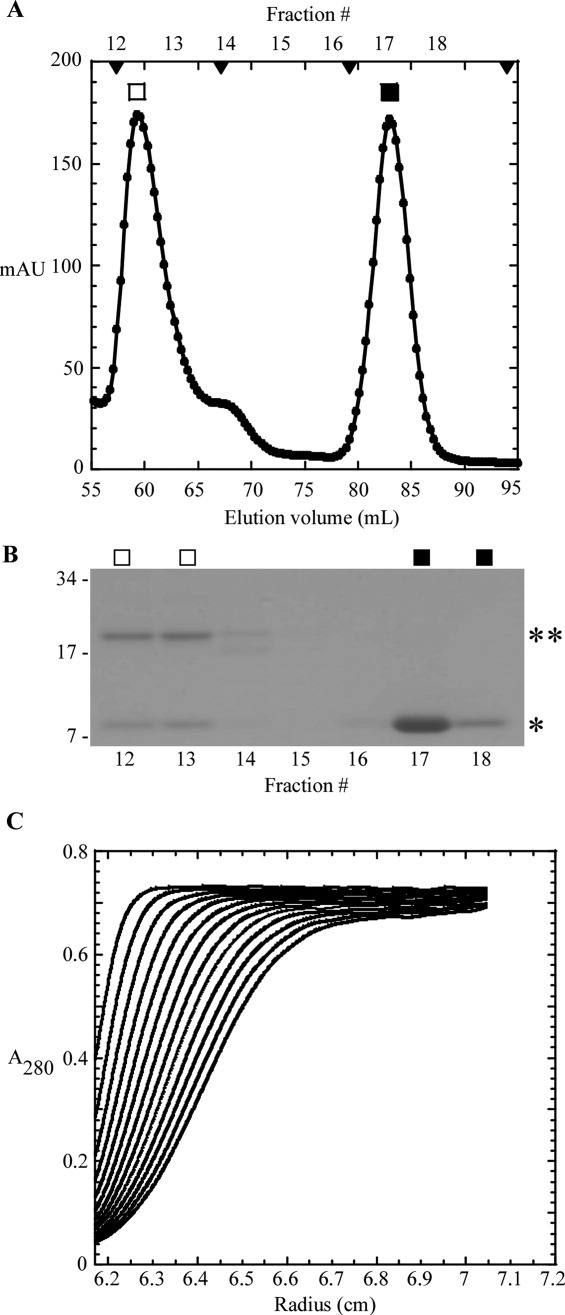
S-75 gel filtration and analytical ultracentrifugation of the sT-sRI complex. A: Elution profile of sT-sRI complex (□) and sRI (▪). S-75 standards are indicated by arrow heads, from left to right: 44, 29, 13.7, and 6.5 kDa. B: Coomassie-stained gel of samples collected from each gel filtration fraction in A. *: sRI and **: sT. C: Sedimentation velocity of sT-sRI complex (at various concentrations, see “Materials and Methods”).
The RI and T cysteines form a disulfide bond necessary for function
The state of the RI and T thiols in the purified sRI and sT-sRI complex protein was assessed with Ellman's reagent (5,5′-dithio-bis-(2-nitrobenzoic acid). The purified sRI and sT-sRI complex exhibited no reactive thiols (Supporting Information Tables I and II). These results indicate that formation of the disulfide bond between Cys69 and Cys75 of sRI and Cys175 and Cys207 of sT is essential for the proper function of sRI and sT. Taken together with the results from Figure 2(A,B), disulfide formation is essential for T and RI function.
Quantitation of T and RI during T4 infections
Useful models for holin and antiholin function during the phage infection cycle require knowledge of the stoichiometry of these proteins in vivo. The availability of purified periplasmic domains of RI and T made it possible to address this issue, using quantitative Western blotting. In both cases, antibodies previously raised against RI and T could be used for detection of the periplasmic domains. Using purified sT as a standard, quantitative blotting was performed on samples taken from wild-type T4 infections of mid-log (∼1.1 × 108 cells/mL; input MOI = 10; Fig. 7). Care was taken to sample the cultures directly into ice-cold TCA for rapid and quantitative denaturation of all cell proteins, so that the immunoblots would reflect an instantaneous snap-shot of the T and RI content. Under these infection conditions, LIN is imposed, so samples could be taken from cells at 60 min after infection, revealing, reproducibly, that ∼8000 molecules of T had accumulated at this point. A T4rI infection under the same conditions results in lysis at ∼30 min; accordingly, the level of T in these samples was found to be ∼4000 molecules, reproducibly. Since holin triggering stops macromolecular synthesis, this quantity represents the critical concentration for T. These numbers are roughly comparable to the levels of the lambda S and phage 21 S68, the only other holin proteins whose quantities have been determined during a phage infection.
Figure 7.
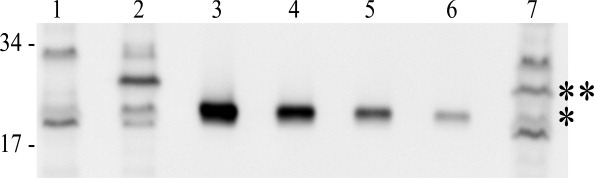
Quantitation of T in a wild type and LIN-defective phage infection. Quantitative Western blot of samples that were collected. Molecular mass standards are indicated at the left. Lane 1, B834 cells only; 2, wild-type T4 infection at 60 min; 3-6, sT standards, from left to right: 30, 15, 7.5, and 3.75 pmol; 7, T4rI infection at 30 min. *: sT and **: T, full length.
RI could not be detected in the samples from T4 wild type infections at t = 60 min. Using the sensitivity of the anti-RI antibody, we determined that there could be no more than 10,000 molecules of RI in the average LIN-inhibited cell.
Discussion
Previous work from our laboratory has shown that, in vivo, the soluble domains of T and RI interact in the periplasm and that this interaction is essential for the inhibition of lysis.17, 28 Here, we have presented results from an effort to purify and characterize these periplasmic domains, sRI and sT, as the initial steps in determining a mechanistic basis for this inhibition. Ultimately, structural information will be required, but the findings presented here have a number of useful implications.
Essential Cys residues and cytoplasmic over-expression in the SHuffle® host
The pair of Cys residues in the C-terminal region of the soluble domains of both RI and T were found to be essential for biological function. Since, in the sRI-sT complex described in this work, there are no free thiols detectable, the two Cys residues in each protein are likely to be involved in a disulfide bond. Since there is no precedent for intermolecular disulfides in native periplasmic proteins, presumably both disulfides are intramolecular linkages important for the stable fold of the protein. Whether or not either or both disulfides are involved in T or RI function or just required for the stable fold cannot be determined from our data. Nevertheless, the requirement for the disulfides in T should make the host dsb system essential for T4, although it is not for the host itself. To our knowledge, this has not been tested, despite the extensive genetics of the T4 system, including screens for host mutants that block T4 vegetative growth. It will be interesting to test this idea by constructing E. coli B mutants with various dsb mutations and then assessing plating efficiency and the kinetics of viral reproduction on isogenic hosts. If the dsb dependency is sufficiently strict, it may be possible to exploit it by looking for intragenic suppressors within t, to explore the structure-function relationship of the holin periplasmic domain.
Interestingly, cytoplasmic production of soluble sRI and sT was much better in the SHuffle® host compared with the parental BL21(DE3) or the Origami™ host. Both Origami and SHuffle strains are E. coli B derivatives carrying mutations in gor, trxB, and aphC that, together, allow disulfide bond formation in the cytoplasm.23, 24 The substantive genotypic difference between Origami and SHuffle strains is supposed to be the presence of an engineered dsbC gene encoding a DsbC disulfide bond isomerase lacking its signal sequence, thus causing DsbC activity to be redirected to the cytoplasm. sRI and sT have only a pair of Cys residues each and are thus unlikely substrates for a disulfide bond isomerase. However, there are reports that DsbC can participate in other yet uncharacterized ways in disulfide bond formation, perhaps by acting as a chaperone.29–31
sRI is monomeric, globular, and highly alpha-helical
Analysis of purified sRI by gel filtration chromatography and sedimentation equilibrium both indicate that sRI exists as a monomer in solution. In addition, CD spectroscopy indicates that sRI is nearly completely in alpha-helical conformation, even more so than suggested by predictions based on secondary structure propensity analysis of the polypeptide sequence. Moreover, sedimentation velocity analysis indicates that sRI has an axial ratio of a rather compact globular shape, suggesting that the extensive alpha helices must be bundled in some way, rather than existing in an extended conformation. Indeed, the position of the disulfide bond that we have inferred from the results discussed above suggests that sRI has a hairpin structure, with the two predicted alpha-helical domains H2 and H3 linked by a Cys69-Cys75 disulfide bond [Fig. 4(A)].
The periplasmic domain of the T holin has a propensity for oligomerization that is blocked by complexing with sRI
The periplasmic domain of the T4 holin, sT, can be produced in soluble form in the host cytoplasm. However, upon elution from an IMAC matrix, sT oligomerizes into insoluble aggregates. These insoluble aggregates can be rescued by elution from the IMAC matrix into buffer containing purified sRI. Gel filtration and ultracentrifugation experiments confirmed that a complex of sT and sRI is indeed formed. This complex is formed at a 1:1 ratio and is completely soluble as shown in Figure 3(C). These results unambiguously confirm the direct role of the periplasmic domain of T for both oligomerization, a necessary step in holin-mediated hole formation, as well as regulation by RI during LIN by the inhibition of oligomerization. This system is the first well-studied example of holin regulation by a protein that shares no sequence homology. The bacteriophage lambda holin, S105, is negatively regulated by S107, another product of the S gene protein with a translation start two codons upstream.32 Likewise, the holin of lambdoid phage 21 is also regulated by a second protein produced from an upstream translational start.33 In both of these cases, the antiholin has a polypeptide sequence nearly identical to that of the holin and thus complex formation with the holin is essentially homomeric.
The T holins have been specialized for real-time regulation by environmental cues
Unlike other holins characterized by experiment or identified by bioinformatics, which have at least two transmembrane segments, the holins of T4-like phages have only a single predicted TMD. Moreover, the holins of T4-like phages are the only holins, which have a substantial soluble domain, in contrast to other predicted holins, which consist almost entirely of TMDs connected by short loops and a short but highly hydrophilic C-terminal tail. The T4-like holins appear to have been adapted so that a signal for superinfection can be recognized and transduced to a lysis inhibition output in the periplasmic space before the IM can be compromised. Presumably, this gives the phage an advantage in preserving membrane integrity and host energetics for the purpose of continued macromolecular synthesis and virion production. Extrapolating from our results with the purified periplasmic domains it seems likely that in superinfected cells, activated RI acts as an antiholin by complexing stoichiometrically with T and thus blocking T homooligomerization. However, in the aqueous context of the periplasm, which is rich in proteolytic activity, soluble RI is highly unstable. This confers an important transitory character to lysis inhibition, so that when superinfections cease, indicating that the extracellular environment is no longer over-stocked with virions, the LIN signal dissipates, allowing lysis to occur.
Despite the regulatory advantages of having a globular soluble domain controlling the formation of homooligomeric holes, the presence of a bulky 19.2 kDa periplasmic component tethered to the single TMD puts constraints on models for the formation and structure of the T-hole. The quantification of T done here using the purified sT as a protein standard indicates that at the normal lysis time of an infected cell (t = 30 min) there are ∼4000 holin molecules (Fig. 7). Assuming that the T-hole is lined by the single TMD, this indicates a maximum of ∼4 μm in hole perimeter could be supported by T, consistent with the micron scale holes observed for the lambda holin S105, which has three TMDs.6 However, there are two conceptual issues that distinguish T-hole formation and structure from that of the S105-hole. First, our results suggest that the oligomerization of the T holin is driven, at least in part, by the oligomerization of the sT domain, while TMD interactions drive the oligomerization of S105.32 Moreover, although both the lambda antiholin, S107, and the T4 antiholin, RI, function by specifically inhibiting oligomerization of the respective holin, in the latter case the entire interaction is within the soluble phase, by the formation of the sT-sRI complex. The simplest interpretation is that the sT domain dominates in the lysis timing function of the holin, in that oligomerization of the sT domain results in creating a high local two-dimensional concentration of the hole-forming component, the single TMD. This perspective could explain why a very conservative missense change in the TMD, I39V17, confers RI-insensitivity on T, presumably by increasing the intrinsic self-affinity of the TMD, even though it is unlikely to affect RI-T complex formation. In any case, the 19.2 kDa periplasmic domain of T will likely have a diameter several-fold greater than the ∼1 nm of hole perimeter occupied by the TMD. Unless the sT domain undergoes a dramatic conformational reorganization during hole-formation, any model of the hole structure would thus require a flexible linker between the membrane and soluble domains. In addition, accommodating the sT domain along the perimeter of the hole wall will likely require multiple homomeric but heterotypic interaction surfaces for sT, any one of which if blocked by RI occupancy could disrupt hole formation. Structural analysis potentiated by our ability to generate purified sRI and sT-sRI in crystallographically useful quantities should further delineate these issues.
Materials and Methods
Bacterial growth and induction
The phages, bacterial strains, and plasmids used in this study are listed in Supporting Information (Table III). Bacterial cultures were grown in standard LB media supplemented with ampicillin (100 μg mL−1) and/or chloramphenicol (10 μg mL−1) when appropriate. Cultures were grown from overnight starter cultures of a single colony from a fresh plate. Starter cultures were diluted 300:1 and grown with aeration at 30°C for lysogenic strains and 37°C for cultures of BL21(DE3), Origami(DE3), and SHuffle(DE3) used for over-expression of proteins. The over-expression strains carrying the indicated plasmids were induced for expression by the addition of 1 mM final concentration of isopropyl β-d-thiogalactosidase (IPTG) at an A550 of 0.6, at 37 or 16°C when appropriate.
Monitoring lysis function
To assess lysis function under physiologically relevant conditions, a plasmid complementation system used with a thermo-inducible lambda lysogen [λkanΔ(SR)] and a medium-copy plasmid carrying the t holin gene and the lambda R endolysin gene under the control of the lambda late promoter.17 In this system, the induction of the prophage provides RzRz1 function and transactivates the plasmid promoter, ensuring that the necessary lysis proteins (holin, endolysin, and Rz-Rz1) are provided with physiologically relevant timing and expression level. Lysogens were thermally induced at A550 = 0.3 by aeration at 42°C for 15 min, followed by continuous growth at 37°C.
DNA manipulations and plasmid construction
Isolation of plasmid DNA, DNA amplification by polymerase chain reaction (PCR), DNA transformation, and DNA sequencing were performed as previously described.17, 19 Oligonucleotides (primers) were obtained from Integrated DNA Technologies (Coralville, IA) and were used without further purification or modification. Restriction and DNA-modifying enzymes were purchased from New England Biolabs (Ipswich, MA); all reactions using these enzymes were performed according to the manufacturer's instructions. Site-directed mutagenesis was performed using the QuikChange kit from Stratagene (La Jolla, CA) as described previously.17, 19 Primer sequences are listed in Supporting Information (Table IV). Inverse PCR was carried out through a modified version of QuikChange site-directed mutagenesis as previously described.34 The DNA sequence of all constructs was verified by automated fluorescence sequencing performed at the Laboratory for Plant Genome Technology at the Texas Agricultural Experiment Station and by Eton Bioscience (San Diego, CA).
All plasmids used and generated in the course of this study are listed in Supporting Information (Table III). The plasmid pSM-t, used for the expression of T in the lysogenic background, was constructed by Inverse PCR using plasmid pER-t as the template and primers pERtdelRzRz1For/Rev to delete the Rz/Rz1 genes. Cysteine to serine substitutions at positions 175 and 207 of the t gene in pSM-t were constructed by site-directed mutagenesis using the primer pairs T4TC175SFor/Rev and T4TC207SFor/Rev, respectively. Cysteine to serine substitutions at positions 69 and 75 in RI were achieved by site-directed mutagenesis of the template pZA-ssPhoAϕsRI using primer pairs T4RIC69SFor/Rev and T4RIC75SFor/Rev. The plasmid pET11a-sThis, used for over-expression of sT was constructed by deletion of codons 2-55 of t in the plasmid pET11a-This using inverse PCR and primers T56-218For/Rev.
Assessment of protein solubility
Induced 25 mL cultures at either 37 or 16°C (80 and 640 min inductions, respectively) were collected by centrifugation at 5000g for 15 min in a Sorvall Superspeed RC2-B centrifuge and resuspended in 3 mL of buffer (20 mM sodium phosphate, 0.1M NaCl, and pH 8.0) supplemented with Protease Inhibitor Cocktail (Sigma, St. Louis), and 100 μg mL−1 final concentrations of DNase and RNase (Sigma, St. Louis). Cells were disrupted by passage through a French pressure cell (Spectronic Instruments, Rochester, NY) at 16,000 lb/in2. Pressate of 250 μL of pressate was set aside on ice and corresponded to the total protein sample (T) while another 250 μL was centrifuged at 15,000g in a tabletop microcentrifuge at 4°C for 20 min. The supernatant was set aside and corresponded to the soluble protein fraction (S) while the pellet fraction was resuspended with an equivalent volume of buffer (250 μL) and corresponded to the insoluble or pellet protein fraction (P). Protein from each sample was precipitated with trichloroacetic acid (TCA, Sigma, St. Louis) and washed with acetone as previously described.34 Pelleted protein was air dried and resuspended in reducing protein sample buffer (0.5 mg mL−1 bromophenol blue in 0.25M Tris, pH 6.8, and 100 mM β-mercaptoethanol) so that 30 μL contained 0.3 O.D. units of culture.
Protein purification
Proteins containing oligohistidine tags were isolated through IMAC, as previously described.34 Six 2 L cultures of SHuffle(DE3) carrying the plasmid pET11a-sRIhis or pET11a-sThis were grown at 37°C to an A550 of ∼0.5, cooled on ice to 16°C, and induced overnight (12–16 h) with 1 mM IPTG, final. Cells were harvested by centrifugation in a Beckman JA-10 rotor at 8000 rpm for 15 min at 4°C. Cell pellets were resuspended in 120 mL of 20 mM sodium phosphate, 0.1M NaCl pH 8.0 (purification buffer) supplemented with Protease Inhibitor Cocktail (Sigma, St. Louis), and 100 μg mL−1 final concentrations of DNase and RNase. Cells were disrupted in a French pressure cell, as previously described,34 and passed over Talon metal affinity resin (Clontech, Mountain View, CA) and eluted with purification buffer supplemented with 0.5M imidazole. For purification of the sT-sRI complex, sT was eluted from the Talon metal affinity resin into a 500-μL bed volume containing 4 mg mL−1 of purified sRI. Elution fractions of each bed volume for purification of sRI and the sT-sRI complex were assessed for purity by SDS-PAGE and Coomassie blue staining before further analysis.
Quantitation of T and RI in T4 wild type and LIN-deficient infections
Wild type T4 (T4D) and LIN-defective T4 (T4ΔrI) bacteriophages were used to infect E. coli B834 cells to quantify the amount of T and RI produced during a T4 infection. Cultures of B834 were grown to an A550 = 0.3 (1.1 × 108 cells mL−1) and were infected with either T4D or T4ΔrI at a multiplicity of infection of 10. Ten-milliliter samples were collected after 60 or 30 min for T4D and T4ΔrI infections, respectively, and total protein precipitated with TCA and resuspended in reducing sample buffer, as described above. The protein standards used were collected from gel filtration experiments in which sT [Fig. 6(A)] (fraction 14) and sRI [Fig. 5(A)], (fraction 13) were purified. Standards were prepared by dilution, with sT standards of 7, 3.5, 1.75, and 0.875 pmol while the sRI standards were 30, 15, 7.5, and 3.75 pmol. All samples were run on SDS-PAGE gels, Western blotted, and analyzed using the software ImageJ (NIH).
SDS PAGE and Western blotting
SDS-PAGE, Western blotting, and immunodetection were performed as previously described.35 sRI and sT in over-expression experiments were detected using a mouse monoclonal antibody against the oligohistidine epitope tag at a dilution of 1:3000 (GE Healthcare, UK). sT and full length T in the T quantification experiments was detected using a rabbit polyclonal antibody against the periplasmic domain of T at a dilution of 1:1000. The anti-mouse and anti-rabbit IgG horseradish peroxidase-conjugated secondary antibodies, which were supplied with the chemiluminescence kit, were used at a 1:5000 dilution. Blots were developed with the West Femto SuperSignal chemiluminescence kit (Thermo Scientific, Rockford, IL). Chemiluminescence signal was detected using a Bio-Rad Chemidoc XRS.
Gel filtration
Gel filtration chromatography was carried out using either an analytical or prep grade Superdex 75 column calibrated with the low molecular weight gel filtration calibration kit on an AKTA FPLC (GE Healthcare, UK). Columns were equilibrated with 20 column volumes of purification buffer before sample injection. Once sample was injected, gel filtration was performed at 0.4 mL min−1 (for analytical grade) or 1 mL min−1 (for prep grade) until one column volume of buffer had eluted.
CD spectroscopy
CD spectra were obtained using an Aviv model 62DS spectropolarimeter (Lakewood, NJ). The sRI and sT-sRI complex protein were purified in 20 mM sodium phosphate and 0.1M NaCl pH 8 buffer and concentrated using an Ultra-4 3K MWCO concentrator (Amicon) to 1 μM. Protein concentrations of sRI and sT-sRI were determined by A280 using calculated extinction coefficients.36 Three-milliliter samples were loaded into 1 cm quartz cuvettes and equilibrated to 25°C for 3 min before being scanned. Scans from 250 to 195 nm with a 1 nm step size and 30 s averaging time were applied to all samples. All CD spectra were fitted by K2D225 using data from 200 to 240 nm that were converted to differential absorption units (Δε).37
Analytical ultracentrifugation
Sedimentation velocity experiments were performed on three sRI samples (0.3, 1, and 1.6 mg/mL) and three sT-sRI complex samples (0.15, 0.4, and 0.6 mg/mL) in purification buffer. Samples and buffer (380 and 400 μL, respectively) were loaded into cells assembled with 12-mm double sector Epon charcoal-filled centerpieces and sapphire windows. Samples were run in a Beckman Model XL-A analytical ultracentrifuge at 50,000 rpm (for sRI) and 37,500 rpm (for sT-sRI complex) at 25°C in an An-60Ti rotor for 8 and 5 h, respectively. Scans were performed at 280 nm, with ∼3 min elapsed between each scan. Data were analyzed and sedimentation coefficients for each sample determined using the programs SVEDBERG version 6.39 and UltraScan II version 9.9, using a genetic algorithm and Monte Carlo analysis, as previously described.38, 39 Sedimentation equilibrium experiments were performed on sRI samples (0.3, 1, and 1.6 mg/mL) in purification buffer. Samples and buffer (110 and 120 μL, respectively) were loaded into cells assembled with a six-channel Epon charcoal-filled centerpiece and sapphire windows. Samples were run in a Beckman Model XL-A analytical centrifuge at 24,000, 28,800, 33,600, 38,400, 43,200, and 48,000 for 13, 14, 13.5, 13.5, 12, and 11 h, respectively, at 25°C. Two scans were performed at 280 nm at the end of each equilibration time. Data for each sample were analyzed with UltraScan II version 9.9, as previously described.39, 40
Assessment of sulfhydryls by Ellman's reagent
The state of the sulfhydryls in purified sRI and sT-sRI complex was assessed with Ellman's reagent, also known as 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB, Sigma, St. Louis, MO), as previously described.41 Briefly, solutions of reduced glutathione, cystine, and purified sRI (40, 20, and 10 μM) for sRI analysis or sT-sRI complex (20, 10, and 5 μM) for complex analysis were prepared in a volume of 90 μL in purification buffer. Ten μL of 10 mM DTNB, dissolved in ethanol, was added to each reaction, samples mixed, and incubated in the dark at room temperature for 30 min. Samples were assayed for the presence of the anionic by-product 2-nitro-5-thiobenzoate at 412 nm on a UV–vis spectrophotometer (Hitachi, Tokyo).41
Acknowledgments
The authors are indebted to D. Pettigrew for AUC sample preparation and experiment setup. Additionally, helpful tips by B. Demeler (UTHSCSA) for AUC analysis are greatly appreciated. M. Scholtz and his laboratory very generously provided the equipment and technical knowledge needed for obtaining the CD spectra. They are thankful for technical support by L. Dangott of the Protein Chemistry Laboratory at TAMU. The clerical assistance of Daisy Wilbert is greatly appreciated.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Young R. Bacteriophage holins: deadly diversity. J Mol Microbiol Biotechnol. 2002;4:21–36. [PubMed] [Google Scholar]
- 2.Young R, Wang IN. Phage lysis. In: Calendar R, editor. The bacteriophages. Oxford: Oxford University Press; 2006. pp. 104–126. [Google Scholar]
- 3.Grundling A, Manson MD, Young R. Holins kill without warning. Proc Natl Acad Sci USA. 2001;98:9348–9352. doi: 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White R, Chiba S, Pang T, Dewey JS, Savva CG, Holzenburg A, Pogliano K, Young R. Holin triggering in real time. Proc Natl Acad Sci USA. 2011;108:798–803. doi: 10.1073/pnas.1011921108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang IN, Deaton J, Young R. Sizing the holin lesion with an endolysin-beta-galactosidase fusion. J Bacteriol. 2003;185:779–787. doi: 10.1128/JB.185.3.779-787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dewey JS, Savva CG, White RL, Vitha S, Holzenburg A, Young R. Micron-scale holes terminate the phage infection cycle. Proc Natl Acad Sci USA. 2010;107:2219–2223. doi: 10.1073/pnas.0914030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, Struck DK, Dankenbring CA, Young R. Evolutionary dominance of holin lysis systems derives from superior genetic malleability. Microbiology. 2008;154:1710–1718. doi: 10.1099/mic.0.2008/016956-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang IN, Smith DL, Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- 9.Hershey AD. Mutation of bacteriophage with respect to type of plaque. Genetics. 1946;31:620–640. doi: 10.1093/genetics/31.6.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doermann AH. Lysis and lysis inhibition with escherichia coli bacteriophage. J Bacteriol. 1948;55:257–276. doi: 10.1128/jb.55.2.257-276.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benzer S. On the Topology of the Genetic Fine Structure. Proc Natl Acad Sci USA. 1959;45:1607–1620. doi: 10.1073/pnas.45.11.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benzer S. On the Topography of the Genetic Fine Structure. Proc Natl Acad Sci USA. 1961;47:403–415. doi: 10.1073/pnas.47.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crick FH, Barnett L, Brenner S, Watts-Tobin RJ. General nature of the genetic code for proteins. Nature. 1961;192:1227–1232. doi: 10.1038/1921227a0. [DOI] [PubMed] [Google Scholar]
- 14.Hershey AD, Rotman R. Genetic Recombination between Host-Range and Plaque-Type Mutants of Bacteriophage in Single Bacterial Cells. Genetics. 1949;34:44–71. doi: 10.1093/genetics/34.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hershey AD, Rotman R. Linkage Among Genes Controlling Inhibition of Lysis in a Bacterial Virus. Proc Natl Acad Sci USA. 1948;34:89–96. doi: 10.1073/pnas.34.3.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doermann AH, Hill MB. Genetic Structure of Bacteriophage T4 as Described by Recombination Studies of Factors Influencing Plaque Morphology. Genetics. 1953;38:79–90. doi: 10.1093/genetics/38.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran TA, Struck DK, Young R. Periplasmic domains define holin-antiholin interactions in t4 lysis inhibition. J Bacteriol. 2005;187:6631–6640. doi: 10.1128/JB.187.19.6631-6640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young R. Bacteriophage holins: deadly diversity. J Mol Microbiol Biotechnol. 2002;4:21–36. [PubMed] [Google Scholar]
- 19.Tran TA, Struck DK, Young R. The T4 RI antiholin has an N-terminal signal anchor release domain that targets it for degradation by DegP. J Bacteriol. 2007;189:7618–7625. doi: 10.1128/JB.00854-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutton RJ, Boyd D, Berkmen M, Beckwith J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci USA. 2008;105:11933–11938. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner S, Baars L, Ytterberg AJ, Klussmeier A, Wagner CS, Nord O, Nygren PA, van Wijk KJ, de Gier JW. Consequences of membrane protein overexpression in Escherichia coli. Mol Cell Proteomics. 2007;6:1527–1550. doi: 10.1074/mcp.M600431-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of molecular biology. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 23.Bessette PH, Aslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy R, Weiss R, Chen G, Iverson BL, Georgiou G. Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Protein Expr Purif. 2001;23:338–347. doi: 10.1006/prep.2001.1520. [DOI] [PubMed] [Google Scholar]
- 25.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8:25. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic acids research. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collier NC, Wang K. Purification and properties of human platelet P235. A high molecular weight protein substrate of endogenous calcium-activated protease(s) The Journal of biological chemistry. 1982;257:6937–6943. [PubMed] [Google Scholar]
- 28.Ramanculov E, Young R. An ancient player unmasked: T4 rI encodes a t-specific antiholin. Mol Microbiol. 2001;41:575–583. doi: 10.1046/j.1365-2958.2001.02491.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen J, Song JL, Zhang S, Wang Y, Cui DF, Wang CC. Chaperone activity of DsbC. J Biol Chem. 1999;274:19601–19605. doi: 10.1074/jbc.274.28.19601. [DOI] [PubMed] [Google Scholar]
- 30.de Marco A. Minimal information: an urgent need to assess the functional reliability of recombinant proteins used in biological experiments. Microb Cell Fact. 2008;7:20. doi: 10.1186/1475-2859-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, Messens J, Bardwell JC, Collet JF. The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Mol Microbiol. 2008;67:336–349. doi: 10.1111/j.1365-2958.2007.06030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blasi U, Nam K, Hartz D, Gold L, Young R. Dual translational initiation sites control function of the lambda S gene. EMBO J. 1989;8:3501–3510. doi: 10.1002/j.1460-2075.1989.tb08515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park T, Struck DK, Deaton JF, Young R. Topological dynamics of holins in programmed bacterial lysis. Proc Natl Acad Sci USA. 2006;103:19713–19718. doi: 10.1073/pnas.0600943103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berry J, Savva C, Holzenburg A, Young R. The lambda spanin components Rz and Rz1 undergo tertiary and quaternary rearrangements upon complex formation. Protein Sci. 2010;19:1967–1977. doi: 10.1002/pro.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundling A, Blasi U, Young R. Biochemical and genetic evidence for three transmembrane domains in the class I holin, lambda S. J Biol Chem. 2000;275:769–776. doi: 10.1074/jbc.275.2.769. [DOI] [PubMed] [Google Scholar]
- 36.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takata T, Oxford JT, Demeler B, Lampi KJ. Deamidation destabilizes and triggers aggregation of a lens protein, betaA3-crystallin. Protein Sci. 2008;17:1565–1575. doi: 10.1110/ps.035410.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demeler B. UltraScan A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. In: Scott D, Harding S, Rowe A, editors. Modern Analytical Ultracentrifugation: Techniques and Methods. UK: Royal Society of Chemistry; 2005. pp. 210–229. [Google Scholar]
- 40.Demeler B, Brookes E. Monte Carlo analysis of sedimentation experiments. Colloid & Polymer Science. 2008;286:129–137. [Google Scholar]
- 41.Dewey JS, Struck DK, Young R. Thiol protection in membrane protein purifications: a study with phage holins. Anal Biochem. 2009;390:221–223. doi: 10.1016/j.ab.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.