

Abstract
The inflammatory response represents a first line of defense against invading pathogens and is important to human health. Chronic inflammation contributes to the etiology of multiple diseases, especially those associated with aging, such as cancer and cardiovascular disease. The chemistry of the inflammatory response is complex and involves the generation of highly reactive oxidants and electrophiles designed to kill the pathogen as well as the release of small molecule and protein mediators of intercellular signaling, chemotaxis, vasoconstriction, and wound-healing. Oxidation of unsaturated fatty acids—either nonenzymatic or enzymatic—contributes to the inflammatory response and associated cellular pathologies. The current perspective summarizes our research on unsaturated fatty acid oxidation in the context of inflammation and cancer. In addition to understanding the consequences of DNA and protein modification by lipid electrophiles, our research has focused on the development of molecularly targeted agents to image and treat cancer.
Inflammation is a systemic response to pathogen challenge and injury. It is characterized by the influx of inflammatory cells (e.g., macrophages and neutrophils), induction of vasoconstriction, edema (swelling), erythema (redness), and sensitivity to pain.1 The logic of inflammation is to defend against the invading pathogen by conducting chemical warfare while limiting damage to the region of the initial infection. Ultimately, inflammatory lesions resolve, and local wounds heal. Acute inflammation is a critical element of host defense, and individuals with genetic mutations that disable the inflammatory response are at elevated risk of infection.2 Although acute inflammation is beneficial to the organism (albeit perhaps painful and annoying), chronic inflammation contributes to the etiology of many diseases. The list is too long to tabulate but includes cancer, cardiovascular disease, and diabetes. There are many mediators generated during an inflammatory response. Some contribute to the toxicological events that kill the invading pathogen, whereas others recruit additional inflammatory cells to the site of the lesion, induce vasoconstriction, or promote resolution and wound healing. Oxidized lipids, particularly those derived from polyunsaturated fatty acids, contribute to all of these events.3 Our group studies the chemical events that contribute to inflammation, especially as they relate to cancer. This perspective will highlight some of the key chemical reactions associated with inflammation and cancer that may be mediated by oxygenated metabolites of polyunsaturated fatty acids.
Reactive Species Generated During Inflammation
During the inflammatory response, professional killer cells such as macrophages generate a panoply of highly reactive oxidants as part of the chemical warfare waged on an invading pathogen (Figure 1).4,5 All of these oxidants derive from the production of two free radical species—the superoxide anion radical (O2–) and nitric oxide (•NO). Activation of macrophages or neutrophils by particulate or soluble stimuli triggers a burst of O2 consumption catalyzed by the cell surface protein, NADPH oxidase, which transfers an electron from NADPH to oxygen to form O2–.6 There are a number of NADPH oxidases in humans, but the enzyme in neutrophils and macrophages catalyzes a particularly robust reduction of O2.7 Concomitant with O2 reduction, arginine is oxidized to •NO by nitric oxide synthase (NOS).8 There are three NOS’s in human tissue, but the inducible form in macrophages (NOS-2 or iNOS) is of particular interest with respect to inflammation.9
Figure 1.
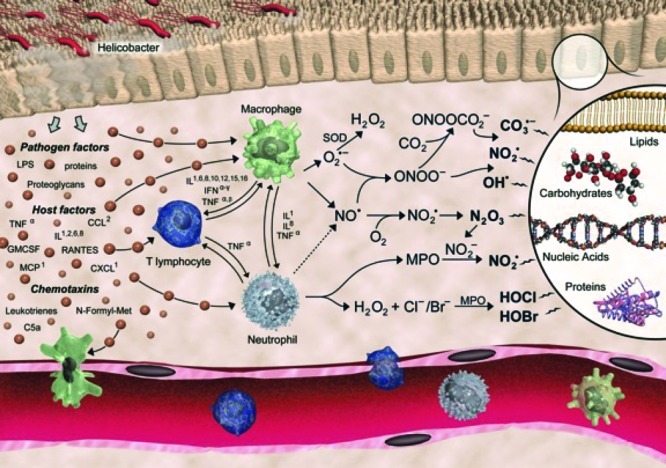
Production of reactive species by activated macrophages and neutrophils during the inflammatory response. Reproduced with permission from ref (5). Copyright 2011. Wiley. The artist is Jeff Dixon.
Coupling of •NO to O2– occurs at a near diffusion-controlled rate to form peroxynitrite (NOOO–).10 NOOO– is a strong nucleophile, and its protonated form, peroxynitrous acid (NOOOH), is an extremely strong oxidant.11 It is capable of directly reacting with organic moieties or undergoing homolysis to nitrogen dioxide (NO2•) and hydroxyl radical (HO•).12 NOOO– is also capable of coupling to carbon dioxide to form peroxynitrosocarbonate (NOOOCO2–).13 Homolysis of NOOOCO2– generates the carbonate radical (•OCO2–) and NO2•.14 One-electron oxidation of an organic donor (e.g., amino acids or nucleic acids) by the carbonate radical generates a new product radical that can couple to NO2•, resulting in nitration.15 Nitrated proteins have been detected at sites of chronic inflammation.16
O2– is not a strong oxidant and, in fact, acts as a reductant of Fe3+ to form Fe2+.17 O2– dismutates to H2O2, which is also a relatively weak oxidant (Figure 1).18 However, reduction of H2O2 by Fe2+ results in the production of HO•, which is an extremely strong oxidant.19 In inflammatory cells, H2O2 is also a substrate for peroxidases that oxidize halogens to form hypohalous acids. Macrophages and neutrophils primarily make HOCl, whereas eosinophils produce HOBr.20,21 A fraction of HOCl reacts with Cl– to form Cl2.22
Table 1 summarizes the properties of the ultimate reactive species generated during the inflammatory response. Most of them are oxidants; in fact, HO• and •OCO2– are among the most potent oxidants known and are capable of reacting with most biomolecules. N2O3 is the only “pure electrophile” in Table 1, although both HOCl and HOBr are capable of acting as oxidants or electrophiles (via halide transfer). A rough indication of the cellular targets is provided in Table 1, but the breadth of actual targets is far too extensive to summarize in a short format. This array of reactive oxidants, nitrating agents, and halogenating agents is key to killing invading pathogens, but the collateral damage of this carpet-bombing approach can be substantial.
Table 1. Properties of Reactive Species Generated During the Inflammatory Response.
| species | reactivity | reaction | cellular target(s) |
|---|---|---|---|
| OH• | oxidant | OH• + H+ + e– → H2O | most protein functional groups |
| Eo′ = +2.31 V (pH 7.0, NHE)23 | addition to alkenes | ||
| CO3•- | oxidant | CO3•– + H+ + e– → HCO3– | most protein functional groups |
| Eo′ = +1.78 V (pH 7.0, NHE)24 | nucleic acids (guanine) | ||
| HOCl | oxidant | 2HClO + 2H+ + 2e– → Cl2 + 2H2O | thiols, amines |
| Eo′ = +1.63 V (pH 7.0, NHE)25 | nucleic acids (guanine) | ||
| alkenes | |||
| HOBr | oxidant | 2HBrO + 2H+ + 2e– → Br2 + 2H2O | thiols, amines |
| Eo′ = +1.59 V (pH 7.0, NHE)25 | nucleic acids (guanine) | ||
| alkenes | |||
| ONOO– | oxidant | ONOO– + 2H+ + e– → NO2• + H2O | sulfhydryl groups |
| Eo′ = +1.4 V (pH 7.0, NHE)26 | metalloproteins | ||
| NO2• | oxidant | NO2• + e– → NO2– | sulfhydryl groups, phenols |
| Eo′ = +1.04 V (pH 7.0, NHE)27 | addition to alkenes | ||
| N2O3 | electrophile | Nuc-H + N2O3 → Nuc-NO + NO2– + H+ | thiols, amines |
Most cellular constituents can be oxidized, nitrated, or chlorinated by the species in Figure 1, but membrane constituents are particularly sensitive to reaction. This is due to the ubiquity of membranes throughout cells combined with the presence of monounsaturated or polyunsaturated fatty acids at the sn-2 position of every glycerophospholipid molecule of every membrane.28 Unsaturated fatty acid groups are prone to oxidation because of the presence of allylic H atoms. The bis-allylic H atoms in polyunsaturated fatty acids are even more sensitive to oxidation, and the reactivity increases with the number of double bonds.29 The quantitatively and biologically most significant polyunsaturated fatty acids are linoleic acid (18:2) and arachidonic acid (20:4) (Figure 2). Removal of the allylic H atom produces a carbon-centered radical that couples to O2 to form a peroxyl radical. Peroxyl radicals are reasonably strong oxidants and can oxidize neighboring polyunsaturated fatty acid residues in phospholipid membranes. These radical cascades can exhibit long chain lengths. For example, it is estimated that some 200 molecules of 20:4 can be oxidized per initial oxidation event.30 Vitamin E serves as the principal membrane-bound antioxidant that interrupts these chains, terminating autoxidation and protecting membranes from further degradation.31 The vitamin E phenoxyl radical produced by reduction of lipid peroxyl radicals decays to quinone or epoxide products that prevent propagation of lipid oxidation.32
Figure 2.
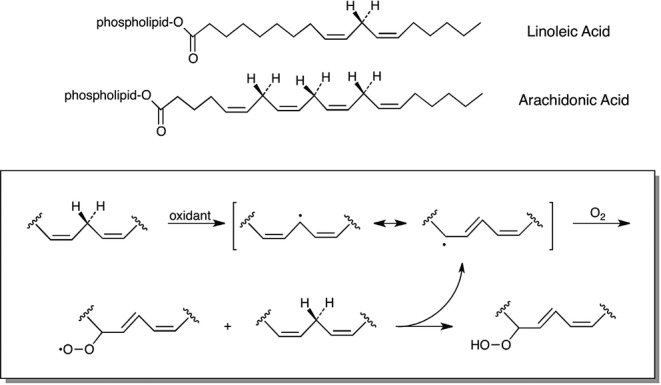
Polyunsaturated fatty acids and lipid peroxidation.
DNA Damage from Lipid Electrophiles
The fatty acid hydroperoxides generated initially during the reaction of polyunsaturated fatty acids with oxidants are subject to chemical breakdown to a variety of products (Figure 3).33 These include enals, enones, and epoxy alcohols—lipid electrophiles that react with cellular nucleophiles. The discovery that one of these aldehydes, malondialdehyde (MDA), is mutagenic and carcinogenic stimulated our interest in the possibility that inflammation-dependent or oxidative stress-dependent generation of MDA could serve as a link between chronic inflammation and the generation of DNA damage leading to genetic mutation.34,35 At the time, most of the attention in the carcinogenesis community was focused on environmental insults to DNA as the principal causative factor for cancer etiology. Thus, the possibility of “self-inflicted toxicity” was novel and presented interesting problems in chemical toxicology.
Figure 3.

Breakdown of PUFAs to electrophiles.
DNA Adducts
Our initial efforts in this area focused on the identification of chemical adducts formed by the reaction of MDA with DNA. This turned out to be much more complicated than initially anticipated because MDA, or more correctly β-hydroxyacrolein (Figure 4), is as good a nucleophile as it is an electrophile and undergoes oligomerization under conditions of its chemical generation. Thus, adducts to deoxyguanosine, deoxyadenosine, and deoxycytidine were formed by reaction of MDA as well as by reaction with dimers and trimers of MDA (Figure 4).36−40 The structures of these interesting adducts suggested that they should block DNA replication or induce mutations by virtue of the fact that they either completely disrupted or attached sizable organic functionality to exocyclic amino groups in the Watson–Crick base-pairing region of the adducted base.
Figure 4.
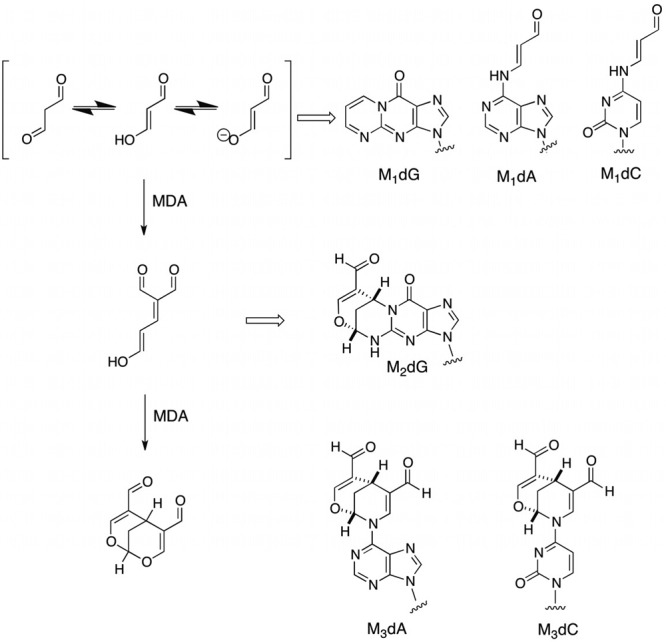
Oligomerization of MDA and the formation of DNA adducts.
Mutagenic Consequences of DNA Damage
The diversity of adducts illustrated in Figure 4 rendered it difficult to directly relate adduct structure to particular mutations induced by MDA treatment of an intact cell. Direct reaction of MDA with plasmid DNA followed by replication in E. coli demonstrated that mutations were induced at dG, dA, and dC residues (primarily dG→dT transversions, dA→dG transitions, and dC→dT transitions, respectively) (Figure 5).41
Figure 5.

Random mutagenesis experiments with MDA. A single-stranded M13 bacteriophage genome was reacted with MDA to randomly introduce adducts. The adducted vector was replicated in E. coli. and mutations were identified by a combination of screening for mutants and DNA sequencing.41
This mutation spectrum was consistent with the chemistry of DNA modification if one made the assumption that mutations at particular nucleosides resulted from the replication of an adduct formed at that nucleoside. To test this, a more direct method for relating adduct structure to mutation was employed. Site-specific mutagenesis is a technique in which a chemical adduct of known structure is built into a single position of a DNA molecule that is capable of being replicated in a bacterial or mammalian cell. Since the vectors typically used contain 5000–7000 nucleotides, this would appear to be a formidable synthetic challenge. In fact, a combination of organic synthesis and genetic engineering makes singly adducted vector construction relatively straightforward (Figure 6).
Figure 6.

Construction of vectors containing a single defined DNA adduct.
The principal challenge is the chemistry of oligonucleotide synthesis rather than engineering the vector. Many DNA adducts are not stable to the conditions of oligonucleotide synthesis or deprotection. We adopted two approaches to incorporate the MDA adduct, M1dG, into oligonucleotides for site-specific mutagenesis. The first involved a total synthesis in which enzymatically prepared M1dG was converted to a dimethoxytrityl phosphoramidite derivative for incorporation by an oligonucleotide synthesizer.42,43 The base lability of M1dG precluded ammonia deprotection, so acetoxymethybenzoyl protecting groups and sodium carbonate deprotection were employed (Figure 7).43
Figure 7.

Total synthesis of M1dG-containing oligonucleotides using AMB protecting groups.
Analysis of the product oligonucleotide revealed that the pyrimidopurinone ring-opened hydrolytically above pH 10 to an oxopropenal derivative that rapidly cyclized to the pyrimidopurinone on lowering the pH to neutrality.43 This allowed for the introduction of the pyrimidopurinone in a site-specific manner, albeit by a methodology that required the synthesis of nonstandard protected nucleotide monomers. This approach allowed us to conduct our initial biological experiments, but subsequently we adopted a postoligomerization strategy described by Rizzo and colleagues following up on the initial reports of Johnson and Harris.44 This postoligomerization methodology, illustrated in Figure 8, allows for adduct incorporation into already constructed oligonucleotides containing electrophilic nucleotides at the desired adduct position.
Figure 8.

Synthesis of M1dG-containing oligonucleotides using a postoligomerization strategy.
This approach is not only more flexible but allows incorporation of the other MDA adducts, OPdA and OPdC, which are too unstable to sodium carbonate deprotection to allow introduction by the total synthesis route. Vectors containing the M1dG adduct were replicated in Escherichia colior mammalian cells and gave comparable results.45,46 Following mutant selection and DNA sequencing, it was found that replication of M1dG resulted in transversions to dT and transitions to dA at approximately equal frequencies. The total mutation frequency was approximately 2% of all the replication events in either E. coli or mammalian cells. Although it seems puzzling that an adduct that blocks the Watson Crick base-pairing region would not have higher mutagenicity, it is important to note that a mutation frequency of 2% is some five-to-8 orders of magnitude higher than the mutation frequency observed when unmodified DNA molecules are replicated.
Part of the reason for the “low” mutagenicity of M1dG was revealed through studies of its conformation in duplex oligonucleotides.47 When single-stranded oligos containing M1dG are hybridized to complementary oligonucleotides containing dC opposite the lesion, M1dG rapidly ring-opens to an oxopropenyl derivative (Figure 9). The oxopropenyl group rotates out of the Watson–Crick base-pairing region and into the minor groove of the duplex.47 Hydrogen bonding between the adducted dG and the complementary dC is detected by NMR spectroscopy.48 When the duplex is thermally melted, the oxopropenyl group rapidly cyclizes to the pyrimidopurinone. Both the initial ring-opening and the subsequent ring-closing are catalyzed by DNA. This represents the first discovery of a DNA-catalyzed transformation of one DNA adduct into another. Interestingly, when single-stranded oligonucleotides containing M1dG are hybridized to complementary strands containing a dT opposite the lesion, no ring-opening is observed.47 Therefore, duplex vectors containing M1dG opposite dC or dT residues were constructed and replicated in parallel. The ring-closed adduct was five times more mutagenic than the ring-opened adduct.45 This probably represents an underestimate of the true differential in mutagenic potency because the position of the equilibrium between M1dG and OPdG in vivo cannot be determined. Nevertheless, in vitro experiments in which adducted template-primers containing either M1dG or OPdG were replicated by model DNA polymerases confirmed the greater ability of the ring-closed adduct to block replication and induce mutations.48
Figure 9.

Ring opening and closing of M1dG in duplex DNA. When a dC residue is placed opposite M1dG, it opens quantitatively. No ring-opening is observed with dT is placed opposite M1dG.
The chemical biology of a number of adducts derived from the reaction of lipid electrophiles other than MDA with DNA has now been evaluated in several laboratories. The structures and mutational outcomes of replication of these adducts are summarized in Figure 10.49 Since the bifunctional lipid electrophiles form exocyclic adducts in the Watson–Crick base-pairing region, it is not surprising that all of them exhibit some degree of mutagenicity. Of particular interest, however, is heptanone–ethenodeoxycytidine, a product of reaction of 4-oxononenal with dC. This adduct is highly mutagenic, exhibiting nearly a 70% total mutation frequency when replicated in E. coli.50
Figure 10.

Summary of mutations induced in bacteria or mammalian cells by different exocyclic adducts. The origin of the adduct is indicated.
Repair of Exocyclic Adducts
The high mutagenicity of the DNA adducts derived from lipid electrophiles suggests repair is an important component of the cellular response to DNA damage. Our investigations in vivo and in vitro revealed that M1dG and related exocyclic adducts are efficient substrates for nucleotide-excision repair in which oligonucleotides are excised and degraded to the level of deoxynucleosides (Figure 11).45,51
Figure 11.
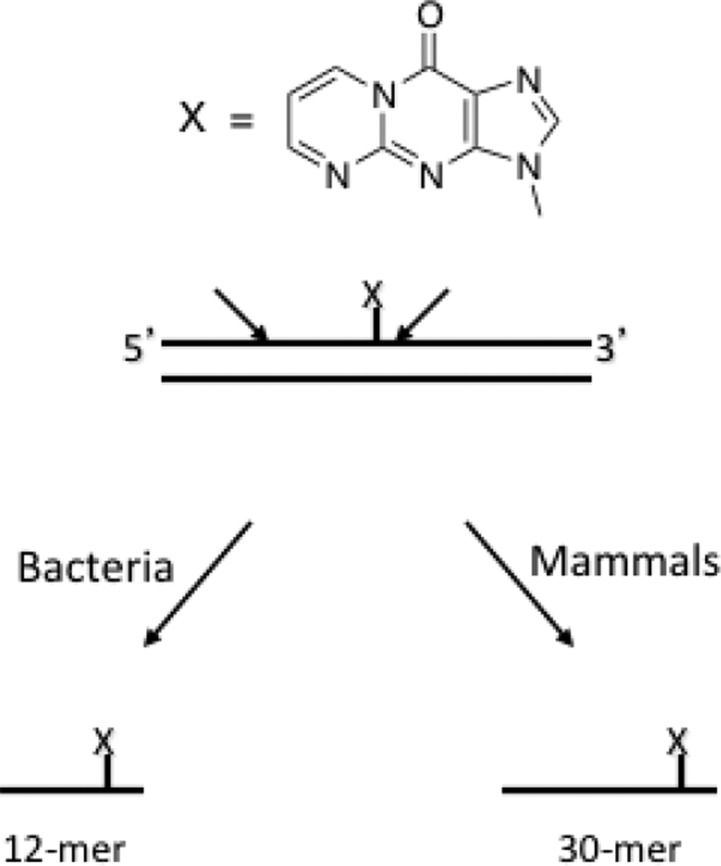
General scheme of nucleotide-excision repair of M1dG adducts. Incisions made on the 5′ and 3′ side of the adduct remove the adduct-containing single-stranded oligonucleotide.
This appears true for many of the other exocyclic adducts depicted in Figure 10. Interestingly, εdA is a relatively efficient substrate for a base-excision repair enzyme, alkyladenineglycosylase, which removes the adducted base and replaces it following excision of the deoxyribosyl unit.52 εdA is also substrate for an oxygenase, AlkB, that oxidizes the etheno ring to a vicinal diol, which decomposes to glyoxal with regeneration of dA (Figure 12).53 Thus exocyclic adducts can be removed by nucleotide-excision repair, base-excision repair, or direct repair of damaged DNA but the extent to which each pathway participates depends on the adduct.
Figure 12.

Repair of εdA by glycosylase-catalyzed base excision repair (BER) and by AlkB-catalyzed oxidation.
Detection of Exocyclic Adducts
The extent to which lipid electrophile-dependent DNA damage occurs in healthy or disease-bearing humans is a subject of great interest. Several laboratories including our own have shown that exocyclic adducts derived from endogenous electrophiles are constituents of genomic DNA of healthy human beings as well as rodents.54−60 Although early analytical methods utilized gas chromatography–mass spectrometry of volatile derivatives, contemporary methods utilize liquid chromatography–mass spectrometry of either the parental adduct or a stable conjugate of it. These studies indicate that exocyclic adducts derived from endogenous electrophiles are present at levels of approximately 1 adduct in 108–107 nucleotides or approximately 30–300 adducts per mammalian cell. Dedon and colleagues have profiled a series of adducts derived from direct oxidation of DNA, alkylation of DNA, and lipid electrophile damage to DNA and found that under conditions of oxidative stress, lipid electrophile adducts increase to a greater extent than the other adducts.61 This illustrates the potential for lipid electrophile–DNA damage to play a role in human cancer.
More recently, our laboratory has developed methods to quantify M1dG in urine and feces, providing a noninvasive way for evaluating DNA damage in population-based studies.62 These methods utilize immunoaffinity chromatography with antibodies raised against the adduct to enrich it prior to liquid chromatography–mass spectrometry. Preliminary studies illustrate that M1dG is present in human urine at levels of approximately 12 fmol/kg/day.63 The level of M1dG in human urine is much lower than that of the DNA oxidation product, 8-oxo-dG.64The low level of M1dG led us to explore the possibility that it is metabolized following excision from DNA analogous to other foreign compounds to which humans are exposed. In fact, injection of M1dG into rodents in amounts from 8 mg/kg body weight to 6 pg/kg leads to the production of a single oxidative metabolite, 6-oxo-M1dG.65−67 Experiments in rodents indicate that M1dG is preferentially excreted in urine whereas 6-oxo-M1dG is preferentially excreted in feces (Figure 13).67
Figure 13.

Repair, metabolism, and excretion of M1dG and its metabolites in urine and bile (then excreted in feces).
Very recently, we have developed an immunoaffinity and liquid chromatography–mass spectrometry-based method for quantifying 6-oxo-M1dG in urine and feces.68 Using this method, we find that 6-oxo-M1dG is present in feces of all the rats that we evaluated at levels of 350–1893 fmol/kg/day. 6-Oxo-M1dG was detected in the urine of only one rat. By contrast, M1dG was not detected in any of the animals’ urine or feces. This indicates that 6-oxo-M1dG, produced by oxidation of M1dG, is generated basally in rodents and preferentially excreted in feces.
The potential metabolic fate of other exocyclic adducts has not been profiled in a comprehensive fashion. However, we have evaluated the fate of a few of the adducts in Figure 10, and the chemistry is summarized in Figure 14.69
Figure 14.
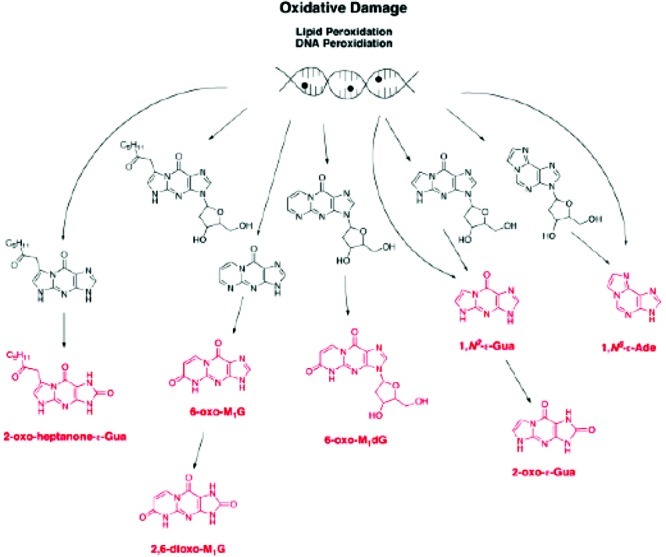
Summary of products of metabolism of exocyclic DNA adducts. The final products are indicated in red. Reproduced from ref (69). 2009. American Chemical Society.
Lipid Electrophile Modification of Protein and Its Relation to the Resulting Cellular Responses
Lipid electrophile modification of DNA is biologically important because of the genetic consequences of aberrant DNA replication. However, there are other nucleophiles in cells that are potentially as important, or more important, than DNA as targets for electrophile modification. Protein molecules appear to be quantitatively more significant targets for electrophile modification than DNA, and modification of proteins alters their function in a positive or negative way. Our interest in lipid electrophile modification of protein grew out of a comparison of the effects of bifunctional electrophiles on cell cycle progression and toxicity. We found that both MDA and 4-hydroxy-2-nonenal (HNE) induce cell cycle arrest, but only HNE induced toxicity to a colon cancer and a lung cancer cell line.70,71 In an attempt to understand the mechanism of the induction of toxicity, we examined the effects of HNE on transcription factors known to play a role in cell replication or resistance to cell death. Through these experiments, we found that HNE inhibited transcription via the NFκB pathway, which plays a role in protecting cells from toxic challenges.72 Detailed analysis of the impact of HNE on NFκB signaling revealed that it covalently modifies and inhibits the action of IκB kinase, the protein kinase responsible for release of the NFκB transcription factor from an inactive cytosolic complex.72 HNE modification of IκB kinase inhibits the transcription of NFκB-dependent genes that play a role in cell survival. Through this experience, we developed an appreciation for the chemical complexity of protein modification by lipid electrophiles and the cellular responses that they induce.
Protein Modification by Lipid Electrophiles
We decided to undertake a global analysis of protein modification and cellular response by lipid electrophiles to provide a comprehensive overview of their impact on cell signaling. It was hoped that this overview would help prioritize protein targets and cellular responses for further study. The complexity of protein modification as opposed to DNA modification prompted us to choose a single lipid electrophile, HNE, as a model and to pursue the acquisition of a complete inventory of proteins modified by this molecule. The chemistry of protein modification by HNE is summarized in Figure 15. The principal adducts are Michael addition products to cysteine, histidine, and lysine with a small percentage of lysine imino adducts.73−75 The latter adducts undergo slow transformation to pyrroles, and there is some evidence for chemical cross-linking of the initial adducts.
Figure 15.
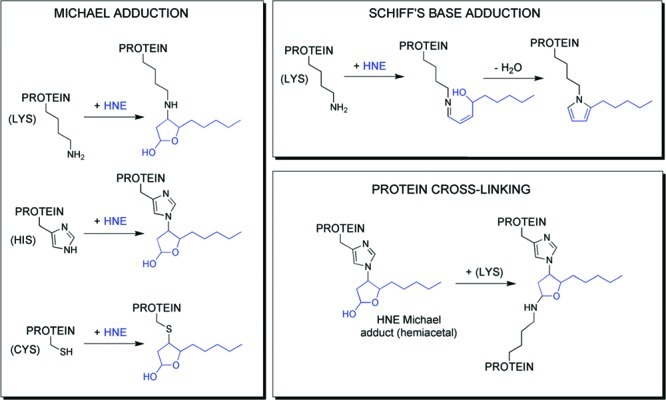
Products of amino acid adduction by HNE. Reproduced from ref (76). 2010. American Chemical Society.
The multiplicity of amino acids modified and the structures of the adducts formed suggested that the method used to inventory modified proteins needed to be independent of the structure of the chemical adduct. We chose to use click chemistry to perform this analysis. Alkynyl or azido derivatives of HNE were synthesized and shown to be comparable to HNE with regard to chemical reactivity with model peptides and toxicity as well as gene expression induction.77 This suggested that these probes were excellent models for the parent molecule. Using the protocol outlined in Figure 16, we were able to modify cellular proteins with alkynyl or azido HNE, then label them posthoc using an azido or alkynyl trap linked to biotin. Use of a copper-catalyzed click reaction provided efficient trapping of protein molecules bearing an alkynyl or azido tag. Biotinylated proteins were enriched by binding to streptavidin-coated beads followed by washing of nonspecifically adsorbed proteins and elution of the biotinylated proteins by disrupting the biotin-streptavidin complexes.77 An important adaptation of this methodology from the Porter laboratory was the incorporation of a photocleavable linker in the biotin tag.78The incorporation of this functional group allowed release of adducted proteins by irradiation of the streptavidin beads. This dramatically reduces the background of nonspecifically adsorbed proteins eluted from the beads and increases the signal-to-noise.
Figure 16.
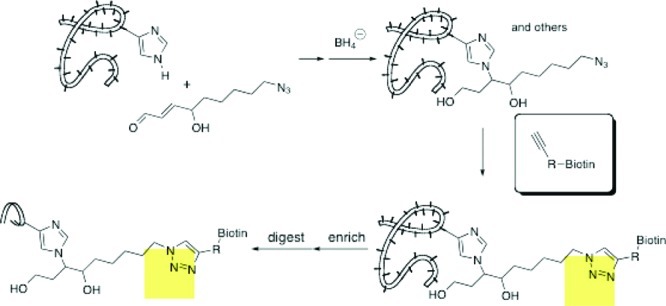
Click chemistry to enrich alkynyl-HNE-adducted proteins.
Our initial analysis of proteins in RKO cells modified by alkynyl or azido HNE indicated hundreds of molecular targets.77 These modifications were observed over a concentration range of 5–50 μM alkynyl or azido HNE, and modified proteins were detectable by SDS polyacrylamide gel electrophoresis followed by Western Blotting at submicromolar concentrations. Recent investigations suggest that the levels of lipid electrophiles generated by activated macrophages are in the high nM range.79 The identities of the proteins modified in our study revealed a broad range of targets from all major cellular components.77 It appears that the HNE derivatives freely diffuse throughout the cell. The complexity of protein modification by HNE suggests that the notion of a single molecular target explaining much of its biology is unrealistic. In fact, it seems more likely that a diversity of cellular responses is induced following protein modification, and the overall response observed represents an amalgam of parallel pathways.
Cellular Responses to Lipid Electrophile Treatment
One way to comprehensively profile cellular responses to biologically active molecules is to determine changes in gene expression by microarray analysis. When we performed such experiments with HNE, hundreds of genes were upregulated or downregulated.80 This was anticipated by the complexity of protein modification profiles.77 Detailed analysis of the gene expression changes suggested the activity of at least 14 different transcription factors were upregulated, and the activities of four transcription factors were downregulated (Table 2).81 This appears to be an underestimate of the overall cellular response, but it gives a good idea of the complexity involved. In order to prioritize transcription factor signaling pathways to study in more detail, we constructed expression vectors containing a luciferase reporter gene downstream of some of the transcription factor response elements summarized in Table 2. Parallel transfection of these vectors into recipient cells followed by HNE modification indicated that the two most dramatically affected transcription factors were Nrf2, which mediates the antioxidant response and Hsf1, which mediates the heat-shock response.80 Interestingly, Nrf2 was not identified by the bioinformatics approach summarized in Table 2 indicating the importance of utlilizing complementary biostatistical and experimental approaches to analyze complex cellular responses.
Table 2. Transcription Factors Altered in Activity by HNEa.
| up-regulated activity (14) | |||
|---|---|---|---|
| TF | enrich FDR | TF | enrich FDR |
| CREB1 | 3.83 × 10–6 | ATF2 | 4.76 × 10–5 |
| CDC5L | 8.47 × 10–5 | HSF1 | 1.65 × 10–4 |
| HSF2 | 1.93 × 10–4 | NFYB | 2.17 × 10–4 |
| CEBPA | 0.0010 | E4F1 | 0.0015 |
| SREBF1 | 0.0023 | USF1 | 0.0039 |
| CEBPD | 0.0047 | ATF4 | 0.0073 |
| ATF3 | 0.0084 | FOXO1 | 0.0090 |
| down-regulated activity (4) | |||
|---|---|---|---|
| TF | enrich FDR | TF | enrich FDR |
| MYC | 5.03 × 10–5 | E2F1 | 0.0032 |
| E2F3 | 0.0032 | NRF1 | 0.0032 |
FDR is false discovery rate, which is analogous to a p value for statistical significance.
Heat-Shock Signaling as a Response to Lipid Electrophile Stress
The antioxidant response is a well-studied cellular response to electrophile and oxidant treatment, but much less work has been done on the heat-shock response, so we focused our attention on that pathway. Validation experiments confirmed the induction of heat-shock genes by HNE. The importance of heat-shock gene expression on the cellular response to HNE was evaluated by reducing the level of the transcription factor, Hsf1, using RNA interference.82 Cells in which Hsf1 was eliminated by small interfering RNA (siRNA) treatment were much more sensitive to HNE toxicity than cells treated with a scrambled siRNA control or cells in which the levels of the transcription factor, Nrf2, were reduced. This not only suggested that heat-shock signaling in response to lipid electrophile treatment is an important adaptive response that cells use to protect themselves from toxicity, but that this pathway may be as important or more important than the pathway mediated by the antioxidant response. Microarray experiments in which gene expression changes were monitored following HNE treatment of Hsf1-knocked-down cells identified Bag3 as a critical mediator of that portion of the cellular response dependent on heat-shock.83
Bag3 is a member of a family of six different genes characterized initially by their ability to bind to members of the Bcl2 family of antiapoptopic proteins.84 Our analysis indicated that knockdown of BAG3 dramatically reduced the levels of antiapoptopic proteins, thereby increasing the sensitivity of the cells to HNE challenge.83 Dramatic reductions in the levels of Bcl2 family members were observed following lipid electrophile treatment, but knockdown of Bag3 reduced their levels even in the absence of HNE. Comparison of the sensitivity to HNE of Hsf1 knocked-down cells indicated that the increased sensitivity of the normal cells to Hsf1 knockdown was completely recapitulated by Bag3 knockdown. This suggested that despite all of the complexity of the heat-shock response induced by HNE, induction of Bag3 is a critical component. Increases in the level of Bag3 following HNE treatment leads to the formation of complexes between it, Bcl2 family members, and Hsp70 which protects the Bcl2 family members from hydrolysis in the proteasome (Figure 17). No impact of Bag3 levels is observed on the levels of mRNA of the Bcl2 family members.83
Figure 17.

Relation of HNE activation of heat shock and activation of Bag3. Reproduced with permission from ref (83). Copyright 2009. American Society for Biochemistry and Molecular Biology.
Hanahan and Weinberg have suggested that cancer cells exhibit six hallmark molecular properties—unlimited replicative potential, resistance to apoptosis, self-sufficiency in growth signals, insensitivity to antigrowth signals, sustained angiogenesis, and the ability to invade tissue and metastasize.85 Recently, Elledge proposed additional hallmarks that arise in a cancer cell because of its rapid growth and large number of genetic mutations. These hallmarks represent mechanisms to deal with metabolic stress, proteotoxic stress, mitotic stress, oxidative stress, and DNA damage stress.86 An elevated heat-shock response is an important contributor to resistance to proteotoxic stress. Genetic deletion of the transcription factor, Hsf1, produces viable mice that are highly resistant to the induction of cancer in the two-stage mouse skin initiation–promotion model.87 This illustrates that the ability of cancer cells to deal with proteotoxic stress and lipid electrophile stress is an important component of their ability to survive. Our findings linking Hsf1 to protection of cancer cells from apoptosis mediated by induction of Bag3 provides a direct linkage between two of the hallmark properties of cancer—the resistance of cancer cells to proteotoxic stress and their resistance to apoptosis. It also suggests that agents designed to lower Bag3 levels in cancer cells may be useful adjuvant therapeutic agents when administered with certain anticancer agents, especially the recently developed agents that lower the levels of Bcl2 in cancer cells.
This analysis indicates that it is possible to use the information provided by global profiling of cellular responses to lipid electrophiles to prioritize important signaling pathways for further experimentation. Furthermore, it suggests there is a treasure trove of information to be mined by analysis of individual signaling networks that may provide important new insights into cancer etiology or its treatment. We are continuing this analysis with several of the other transcription factors activated or inactivated by HNE treatment.
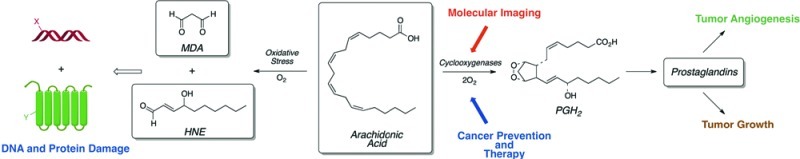
Molecular Imaging of Inflammation and Cancer
The generation of oxidants by activated macrophages and neutrophils is a critical factor in our defenses against infection. However, as indicated in the introduction, there are many other physiological components of inflammation that require the generation and release of bioactive mediators. Oxidized lipids are important constituents of the chemotactic and vasoactive events of inflammation, but these mediators are products of enzymatic transformations rather than the nonenzymatic oxidations described above.3 Mother Nature has co-opted the chemistry outlined in Figure 2 to generate multiple families of bioactive lipids. Lipoxygenases (LOX) and cyclooxygenases (COX) catalyze controlled autoxidations of polyunsaturated fatty acids to produce hydroperoxy fatty acids and prostaglandin endoperoxides, respectively.88,89 Both types of intermediates are converted to downstream metabolites—e.g., leukotrienes and prostaglandins—that bind to membrane-bound G-protein coupled receptors (Figure 18).
Figure 18.

LOX and COX catalysis as a source of bioactive lipids. Conversion of LOX products to resolvins and protectins is not shown.
Cyclooxygenases in Inflammation and Cancer
There are two COX enzymes (COX-1 and COX-2).90 COX-1 and COX-2 are approximately 60% identical in amino acid sequence and have very similar three-dimensional structures. However, they differ substantially in their regulation, tissue localization, and substrate specificity. COX-1 is constitutively expressed and oxidizes only free fatty acids, whereas COX-2 is highly inducible and oxidizes fatty acids and certain fatty acid esters and amides. COX-2 expression is activated by a diverse array of agonists including bacterial lipopolysaccharide. Thus, it is a major source of prostaglandins synthesized during the inflammatory response. This also makes COX-2 the molecular target for the anti-inflammatory action of nonsteroidal anti-inflammatory drugs (NSAIDs). Indeed, selective COX-2 inhibitors such as rofecoxib (Vioxx) and celecoxib (Celebrex) exhibit anti-inflammatory activity.91,92
COX-2 is not expressed in most untransformed epithelial cells, but early in transformation to malignancy it is expressed at a high level (Figure 19).93 In fact, the earliest premalignant lesions that lead to most solid tumors display COX-2 expression.94,95 As progression to malignancy occurs, COX-2 enzyme levels increase. The prostaglandin products of COX-2 action contribute to the cancer progression process, and COX-2 selective inhibitors prevent tumor development in animal models. These discoveries were initially made in the colon, but similar observations have been reported in most solid tumors except ovarian cancer where COX-1 appears to be induced.96
Figure 19.

COX-2 is expressed at the earliest detected premalignant phase of colon cancer. Reproduced with permission from ref (97). 1999. Nature Publishing Group.
COX-2 inhibitors have been extensively tested in human clinical trials for prevention and adjuvant therapy of cancer. Studies of rofecoxib and celecoxib in colon polyp recurrence trials demonstrated a dramatic reduction in recurrence, especially in individuals who had large polyps removed at the beginning of the trial.98,99 Celecoxib exhibits dramatic effects in the treatment of advanced lung cancer when combined with gemcitabine and carboplatin.100 Stage 3 and stage 4 lung cancer patients who express COX-2 in their tumors, demonstrated a doubling of lifespan when celexocib was added to gemcitabine and carboplatin, while individuals who did not express COX-2 in their lung cancers demonstrated a poorer outcome when celecoxib was combined with the two chemotherapeutic agents.100 This illustrates the importance of being able to determine whether a patient’s cancer expresses the molecular target for a particular therapy, in this case, COX-2. Although COX-2-selective inhibitors have demonstrated profound cancer preventive and therapeutic effects in animal models and human clinical trials, they have also demonstrated cardiovascular side effects that have limited their use for prevention and therapy. This is especially true for patients who would be on the drug for a period of years. Thus, COX-2 is a highly validated target for cancer prevention and treatment but one where careful analysis of the risk/benefit ratio is absolutely necessary before beginning therapy.101
Imaging COX-2
The established value of COX-2 as a cancer therapeutic target combined with the need to carefully select appropriate patients led us to hypothesize that it might be an attractive target for molecular imaging. Early detection remains the best way to reduce mortality from cancer, so we felt that imaging COX-2 might be an effective way to impact the disease. The structure of both COX proteins makes them nearly ideal targets for imaging (Figure 20).102−104 The active site where substrates and inhibitors bind is located deep in the interior of the protein and is connected to the exterior through a long channel. A gate separates the active site from the rest of the channel, and this gate is typically closed once an inhibitor or substrate binds.
Figure 20.

Stereodrawing of the COX-2 active site. Reproduced from ref (89). 2003. American Chemical Society.
Our laboratory described several years ago that amides and esters of certain carboxylic acid-containing NSAIDs exhibit COX-2 selectivity.105 This provided the basis for our design strategy for the construction of COX-2-targeted imaging agents. An NSAID core is tethered to a fluorophore to generate an optical imaging agent that will accumulate in COX-2 expressing cells and tissue. To test our strategy, we evaluated a number of NSAID cores, a variety of tethers, and multiple different fluorophores. Each compound was evaluated for COX-2 selectivity against purified COX-1 and COX-2, for potency in intact cells, and for the ability to image COX-2 in intact cells. Candidates that survived this gauntlet were then evaluated in mouse models of inflammation and cancer for their ability to accumulate in tissues expressing high levels of COX-2. Some 250 compounds were made and evaluated; most of them did not inhibit COX-2 or they did not penetrate the cell membrane. However, two compounds were quite effective in both in vitro and in vivo experiments.106 These compounds contained indomethacin as the NSAID core tethered through a butylenediamine linker to the fluorophore carboxy-X-rhodamine (Figure 21).
Figure 21.

Fluorocoxibs A and B.
Although these compounds were promising, purchase of the fluorophore from commercial vendors is extraordinarily expensive which limits their utility for in vivo experiments. Therefore, we developed a straightforward synthetic route to the two carboxy-X-rhodamine isomers that enables inexpensive preparation of both compounds (Figure 22).107
Figure 22.

Synthetic route to the carboxy-X-rhodamine precursors of fluorocoxibs. Reproduced from ref (107). 2008. American Chemical Society.
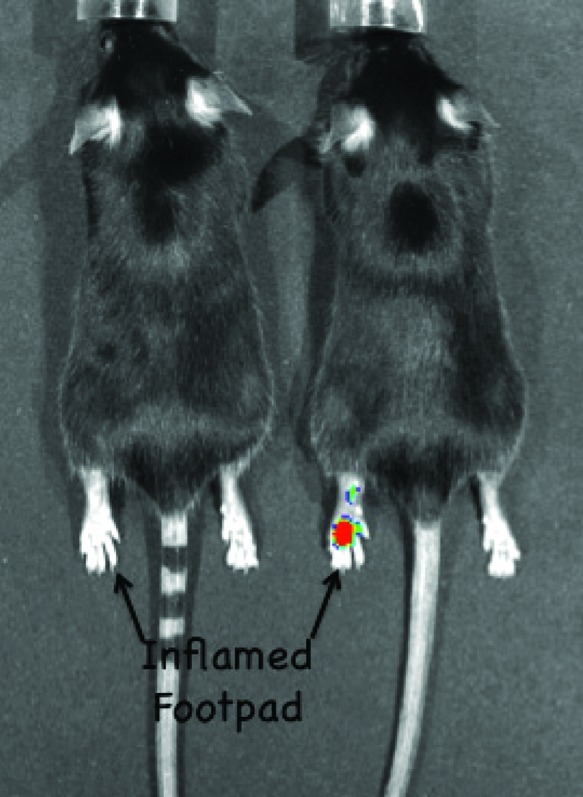
The target compounds were evaluated in multiple in vivo models.106 Since COX-2 is an important inducible component of the inflammatory response, the utility of the COX-2-targeted imaging agents (termed fluorocoxibs) was examined in the mouse footpad model of inflammation. Injection of carrageenan into one footpad of a mouse induces a profound inflammatory response, which is accompanied by the induction of high levels of COX-2 within approximately 12 h. Twenty-four hours after carrageenan injection, fluorocoxib A was injected into the peritoneal cavity of the treated mouse, and optical imaging performed 3–5 h thereafter. As Figure 23 illustrates, compounds selectively accumulated in the inflamed paw, but not in the noninflamed contralateral paw.
Figure 23.

Accumulation of fluorocoxibs in the inflamed paw. Carageenan was injected into the paw at time zero. After 24 h, the fluorocoxib was administered by intraperitoneal injection.
Indeed, this is one of the attractive features of the footpad model of inflammation; every animal serves as its own control. Parallel experiments using animals in which COX-2 had been genetically deleted (i.e., COX-2 knockouts) revealed no selective accumulation in the inflamed compared to the noninflamed paw, indicating that the uptake in the wild-type animals was dependent upon the presence of COX-2 in the tissue. This was confirmed by pretreating wild-type animals with either the nonselective NSAID, indomethacin, or the selective COX-2 inhibitor, celecoxib, prior to fluorocoxib administration. Either inhibitor prevented accumulation of fluorocoxib in the inflamed lesion. A particularly useful control compound is an analog of fluorocoxib B in which the four-carbon tether is shortened to two carbons. Because of the shortened tether, this compound is unable to inhibit COX-2. It contains the same indomethacin core and the same carboxy-X-rhodamine fluorophore but it is not a COX-2 inhibitor, so it is a very useful negative control for in vivo experiments. Comparison of the uptake of fluorocoxib B and the negative control molecule (Figure 24) illustrates that no accumulation in the inflamed lesion is observed following injection of the compound that is unable to bind to COX-2. Thus, genetic and pharmacological experiments validate the hypothesis that fluorocoxib A and B accumulate in inflamed tissue because of the presence of COX-2 in that tissue.
Figure 24.
Comparison of fluorocoxib B uptake with that of LM4752. The same protocol was followed as described in Figure 23. Reproduced with permission from ref (106). 2010. American Association for Cancer Research.
We extended these observations to human cancers grown as xenografts in nude mice.106 Figure 25 shows the comparison of uptake into a COX-2-expressing human head and neck cancer (1483) or non-COX-2-expressing human colon cancer (HCT116) grown on the flanks of a nude mouse. Compounds were injected retro-orbitally, and the residue can be seen at the site of injection. Three-and-one-half hours after administration of fluorocoxib A, fluorescence was observed in human tumors that express COX-2 but not in tumors that do not express COX-2. Furthermore, pretreatment of these animals with either indomethacin or celecoxib abolishes uptake of the fluorocoxib into the COX-2-expressing tumor. The identity of the fluorescent material in the tumor xenografts was established by extraction of the tumor and analysis by liquid chromatography and mass spectrometry. A single fluorescent peak was observed that coelutes with fluorocoxib A and displays an identical mass spectrum. Thus, the fluorescent material that accumulates in the human tumor expressing COX-2 is the parent molecule, fluorocoxib A. Pharmacokinetic analysis of the disposition of this molecule in nude mice bearing tumors illustrates rapid uptake and distribution into plasma, liver, and kidney but not into the tumor. Accumulation in the tumor requires 3–5 h postinjection to achieve maximal uptake. However, whereas elimination of fluorocoxib A from plasma, liver, and kidney is rapid, elimination from the tumor is not. Thus, fluorescence can be detected in the tumor 24 h after administration. LC/MS analysis verifies that this material is the parent compound, fluorocoxib A.
Figure 25.
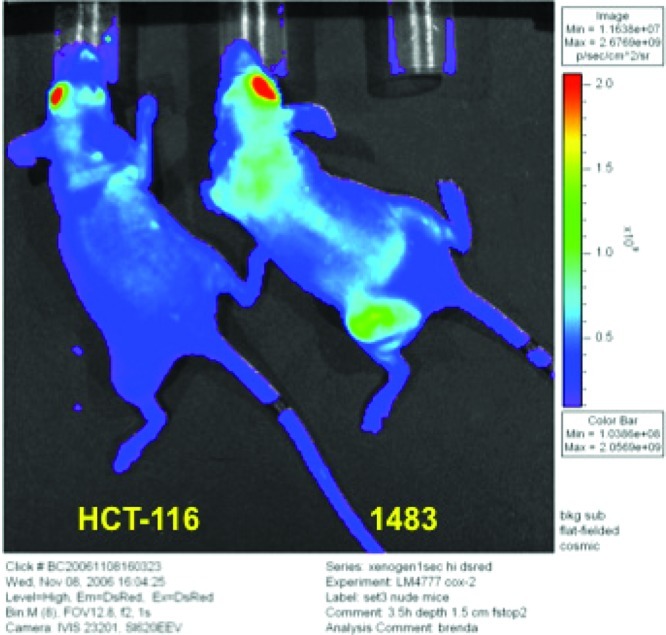
Xenograft data on uptake of fluorocoxib A. Fluorocoxib A was administered by retro-orbital injection and the animals monitored for fluorescence 3.5 h later using a Xenogen camera. The head-and-neck cancer, 1483, expresses COX-2 whereas the colon cancer, HCT116, does not express COX-2. Reproduced with permission from ref (106). 2010. American Association for Cancer Research.
The final in vivo model in which use of these compounds was validated was the APCMin+ (Min) mouse model of intestinal tumorigenesis.106 The Min mouse bears a mutation in the same APC gene that is mutated in individuals born with familial polyposis. In humans, this leads to a large number of colon tumors by midteens and ultimately the development of colon cancer. In mice, the mutation results in small intestinal tumors. Three hours following retro-orbital administration of fluorocoxib A to a Min mouse, uptake of fluorophore into these small intestinal tumors is detectable (Figure 26). Comparison of light emission from the tumors to light emission from surrounding normal tissue indicates a 50- to 100-fold uptake selectivity. This is the highest selectivity of accumulation of a targeted fluorophore into a tumor that has been reported to date.
Figure 26.
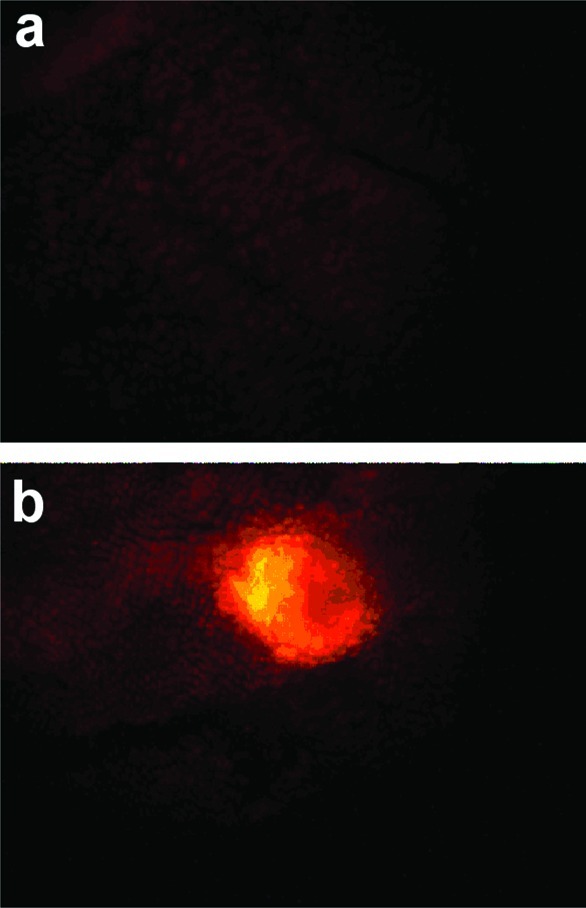
Uptake of fluorocoxib A into min mice. Compound was administered retro-orbitally, and then 3.5 h later, the animals were sacrificed, the gastrointestinal tracts were removed and cleaned, and fluorescence was detected using a fluorescence microscope. (a) Section of normal intestinal tissue. (b) Section of intestinal tissue bearing a polyp. Reproduced with permission from ref (106). 2010. American Association for Cancer Research.
These experiments validate the hypothesis that one can design and synthesize a COX-2-targeted optical imaging agent that is useful in vivo. The attrition rate of candidate compounds was extremely high (>98%), but this is not surprising when one considers that a successful imaging agent must selectively inhibit COX-2, have stability and pharmacokinetics suitable for delivery to and uptake by the tumor, be able to traverse the cell membrane to reach the enzyme in the endoplasmic reticulum and nuclear membrane, and have fluorescent properties suitable for detection with minimal autofluorescence interference.
In addition to optical imaging agents, our laboratory has reported the synthesis and in vivo validation of radiologic imaging agents for both SPECT and PET imaging.108,109 The structures of the agents are different from those of the optical imaging agents and are based on the celecoxib and rofecoxib scaffolds containing either 123I or 18F (Figure 27). The validation process for both compounds closely followed that described above for the fluorocoxibs. Thus, we have prepared an inventory of COX-2-targeted agents for in vivo imaging that includes representatives of several of the currently available modalities for in vivo detection.
Figure 27.

Structures of SPECT and PET imaging agents.
Future Opportunities
Chemical studies of DNA and protein damage by reactive species generated during the inflammatory response are being aggressively pursued in a number of laboratories. Adducts are being identified and the biological consequences of their formation explored. There is great interest in developing biomarkers of this type of damage and applying them in population-based studies to relate chemical modification to disease susceptibility. Whether direct products of chemical modification or biomolecules synthesized during the tissue response to damage will be most useful as biomarkers is uncertain.
Mechanistic investigation of the cellular responses to reactive oxidants and electrophiles is in its infancy. The complexity of the cellular response to a single electrophile revealed by the data in Table 2 suggests it will be a daunting task to establish cause-and-effect relationships between chemical modification of protein or DNA and the induction of a particular cellular response. Nevertheless, the importance of stress responses in cancer cell survival suggests there may be a payoff to studying these relationships. Based on the studies summarized above, efforts are underway to test the potential of BAG3 as a druggable target for cancer therapy.
Molecular imaging agents are anticipated to have a major effect on the detection and treatment of cancer. COX-2-targeted imaging agents represent only one class of what is already a treasure trove of targeted optical or radiological imaging agents. Fluorocoxib A and B are now commercially available for preclinical experiments and are being advanced for human clinical trials. The type of applications that one can envisage include early detection of premalignancy or malignancy, detection of tumor margins during surgery, stratification of patients for the presence of COX-2 prior to therapy, or monitoring response to therapy. A number of tissues are attractive for the use of such compounds including skin, esophagus, stomach, colon, and bladder because technology is available to deliver and collect light in these locations.
If a large bulky fluorophore, such as carboxy-X-rhodamine, can be selectively delivered to a human tumor expressing COX-2, the question to be answered in the future is whether we can also selectively deliver chemotherapy. This approach would be anticipated to increase the concentration of the chemotherapeutic agent in the tumor relative to normal tissue, thereby improving the therapeutic index. Development of targeted chemotherapeutic agents would face all of the same hurdles as optical imaging agents with the additional requirement that the chemotherapeutic moiety be active once it reaches the target tissue. This adds the complexity of incorporating the ability to release the active chemotherapeutic agent at the site of accumulation if the COX-2 targeted conjugate is not directly active. We are aggressively pursuing these challenges and opportunities.
Acknowledgments
I am deeply grateful to many talented colleagues and collaborators who have contributed to the research programs in my laboratory. Sustained funding has been provided by the National Cancer Institute through multiple research grants. I thank Carol Rouzer for a critical reading of this manuscript and editorial assistance.
Biography

Larry Marnett is Mary Geddes Stahlman Professor of Cancer Research, Professor of Biochemistry, Chemistry and Pharmacology at Vanderbilt University, Director of the A. B. Hancock Jr. Memorial Laboratory for Cancer Research and Director of the Vanderbilt Institute of Chemical Biology. His research interests include the enzymatic and nonenzymatic oxygenation of polyunsaturated fatty acids as they relate to inflammation and cancer. This article is based on his lecture for the first George and Christine Sosnovsky Award for Cancer Research from the American Chemical Society.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Ward P. A. In Fundamentals of Inflammation; Serhan C. N., Ward P. A., Gilroy D. W., Eds.; Cambridge University Press: Cambridge, 2010, p 1. [Google Scholar]
- Seger R. A. Curr.Opin. Hematol. 2011, 18, 36. [DOI] [PubMed] [Google Scholar]
- Serhan C. N.; Haeggstrom J. Z. In Fundamentals of Inflammation; Serhan C. N., Ward P. A., Gilroy D. W., Eds.; Cambridge University Press: Cambridge, 2010; p 153. [Google Scholar]
- Taghizadeh K.; McFaline J. L.; Pang B.; Sullivan M.; Dong M.; Plummer E.; Dedon P. C. Nat. Protoc. 2008, 3, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonkar P.; Dedon P. C. Int. J. Cancer 2011, 128, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F. Biochem. Biophys. Acta 1986, 853, 65. [DOI] [PubMed] [Google Scholar]
- Nauseef W. M. J. Biol. Chem. 2008, 283, 16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marletta M. A. Chem. Res. Toxicol. 1988, 1, 249. [DOI] [PubMed] [Google Scholar]
- Griffith O. W.; Stuehr D. J. Annu. Rev. Physiol. 1995, 57, 707. [DOI] [PubMed] [Google Scholar]
- Radi R.; Beckman J. S.; Bush K. M.; Freeman B. A. Arch. Biochem. Biophys. 1991, 288, 481. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G.; Radi R. ACS Chem. Biol. 2009, 4, 161. [DOI] [PubMed] [Google Scholar]
- Richeson C.; Mulder P.; Bowry V. W.; Ingold K. U. J. Am. Chem. Soc. 1998, 120, 7211. [Google Scholar]
- Lymar S. V.; Hurst J. K. J. Am. Chem. Soc. 1995, 117, 8867. [Google Scholar]
- Trujillo M.; Folkes L.; Bartesaghi S.; Kalyanaraman B.; Wardman P.; Radi R. Free Radic. Biol. Med. 2005, 39, 279. [DOI] [PubMed] [Google Scholar]
- Uppu R. M.; Squadrito G. L.; Pryor W. A. Arch. Biochem. Biophys. 1996, 327, 335. [DOI] [PubMed] [Google Scholar]
- Beckman J. S.; Koppenol W. H. Am. J. Physiol. 1996, 271, C1424. [DOI] [PubMed] [Google Scholar]
- Fee J. A.; Valentine J. S. In Superoxide and Superoxide Dismutases; Michelson A. M., McCord J. M., Fridovich I., Eds.; Academic Press: New York, 1977, p 19. [Google Scholar]
- McCord J. M.; Fridovich I. J. Biol. Chem. 1969, 244, 6049. [PubMed] [Google Scholar]
- McCord J. M.; Fridovich I. Ann. Intern. Med. 1978, 89, 122. [DOI] [PubMed] [Google Scholar]
- Klebanoff S. J. In Peroxidases in Chemistry and Biology; Everse J, Everse K. E, Grisham M. B, Eds.; CRC Press: Boca Raton, 1991; Vol. 1, p 1. [Google Scholar]
- Blanchard C.; Rothenberg M. E. Adv. Immunol. 2009, 101, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen S. L.; Hsu F. F.; Duffin K.; Heinecke J. W. J. Biol. Chem. 1996, 271, 23080. [DOI] [PubMed] [Google Scholar]
- Sawyer D. T.Oxygen Chemistry; Oxford University Press: New York, 1991. [Google Scholar]
- Augusto O.; Bonini M. G.; Amanso A. M.; Linares E.; Santos C. C.; De Menezes S. L. Free Radic. Biol. Med. 2002, 32, 841. [DOI] [PubMed] [Google Scholar]
- Handbook of Chemistry and Physics; 92nd ed.; CRC Press: Boca Raton, 2011. [Google Scholar]
- Koppenol W. H.; Moreno J. J.; Pryor W. A.; Ischiropoulos H.; Beckman J. S. Chem. Res. Toxicol. 1992, 5, 834. [DOI] [PubMed] [Google Scholar]
- Stanbury D. M. Adv. Inorg. Chem. 1989, 33, 69. [Google Scholar]
- Porter N. A.; Caldwell S. E.; Mills K. A. Lipids 1995, 30, 277. [DOI] [PubMed] [Google Scholar]
- Howard J. A. In Free Radicals; Kochi J. K., Ed.; Wiley-Interscience: New York, 1973, Vol. II, p 3. [Google Scholar]
- Porter N. A. Acc. Chem. Res. 1986, 19, 262. [Google Scholar]
- Culbertson S. M.; Antunes F.; Havrilla C. M.; Milne G. L.; Porter N. A. Chem. Res. Toxicol. 2002, 15, 870. [DOI] [PubMed] [Google Scholar]
- Liebler D. C. Crit. Rev. Toxicol. 1993, 23, 147. [DOI] [PubMed] [Google Scholar]
- Gardner H. W.; Crawford C. G. Biochim. Biophys. Acta 1981, 665, 126. [DOI] [PubMed] [Google Scholar]
- Mukai F. H.; Goldstein B. D. Science 1976, 191, 868. [DOI] [PubMed] [Google Scholar]
- Spalding J. W. NTP Tech. Rep. 1988, 331, 5. [Google Scholar]
- Nair V.; Vietti D. E.; Cooper C. S. J. Am. Chem. Soc. 1981, 103, 3030. [Google Scholar]
- Seto H.; Okuda T.; Takesue T.; Ikemura T. Bull. Chem. Soc. Jpn. 1983, 56, 1799. [Google Scholar]
- Marnett L. J.; Basu A. K.; O’Hara S. M.; Weller P. E.; Rahman A. F. M. M.; Oliver J. P. J. Am. Chem. Soc. 1986, 108, 1348. [Google Scholar]
- Stone K.; Ksebati M.; Marnett L. J. Chem. Res. Toxicol. 1990, 3, 33. [DOI] [PubMed] [Google Scholar]
- Stone K.; Uzieblo A.; Marnett L. J. Chem. Res. Toxicol. 1990, 3, 467. [DOI] [PubMed] [Google Scholar]
- Benamira M.; Johnson K.; Chaudhary A.; Bruner K.; Tibbetts C.; Marnett L. J. Carcinogenesis 1995, 16, 93. [DOI] [PubMed] [Google Scholar]
- Chapeau M. C.; Marnett L. J. Chem. Res. Toxicol. 1991, 4, 636. [DOI] [PubMed] [Google Scholar]
- Reddy G. R.; Marnett L. J. J. Am. Chem. Soc. 1995, 117, 5007. [Google Scholar]
- Wang H.; Marnett L. J.; Harris T. M.; Rizzo C. J. Chem. Res. Toxicol. 2004, 17, 144. [DOI] [PubMed] [Google Scholar]
- Fink S. P.; Reddy G. R.; Marnett L. J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen L. A.; Hashim M. F.; Shyr Y.; Marnett L. J. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H.; Schnetz-Boutaud N. C.; Weisenseel J. P.; Marnett L. J.; Stone M. P. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashim M. F.; Riggins J. N.; Schnetz-Boutaud N.; Voehler M.; Stone M. P.; Marnett L. J. Biochemistry 2004, 43, 11828. [DOI] [PubMed] [Google Scholar]
- Knutson C. G.; Marnett L. J. In The Chemical Biology of DNA Damage; Geacintov N. E., Broyde S., Eds.; Wiley-VCH: Weinheim, 2010; p 105. [Google Scholar]
- Yang I. Y.; Hashimoto K.; de Wind N.; Blair I. A.; Moriya M. J. Biol. Chem. 2009, 284, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Fink S. P.; Marnett L. J. J. Biol. Chem. 1997, 272, 11434. [DOI] [PubMed] [Google Scholar]
- Singer B.; Hang B. Chem. Res. Toxicol. 1997, 10, 713. [DOI] [PubMed] [Google Scholar]
- Delaney J. C.; Smeester L.; Wong C.; Frick L. E.; Taghizadeh K.; Wishnok J. S.; Drennan C. L.; Samson L. D.; Essigmann J. M. Nat. Struct. Mol. Biol. 2005, 12, 855. [DOI] [PubMed] [Google Scholar]
- Fedtke N.; Boucheron J. A.; Turner M. J. Jr; Swenberg J. A. Carcinogenesis 1990, 11, 1279. [DOI] [PubMed] [Google Scholar]
- Chaudhary A. K.; Nokubo M.; Reddy G. R.; Yeola S. N.; Morrow J. D.; Blair I. A.; Marnett L. J. Science 1994, 265, 1580. [DOI] [PubMed] [Google Scholar]
- Ham A. J.; Ranasinghe A.; Morinello E. J.; Nakamura J.; Upton P. B.; Johnson F.; Swenberg J. A. Chem. Res. Toxicol. 1999, 12, 1240. [DOI] [PubMed] [Google Scholar]
- Jeong Y. C.; Sangaiah R.; Nakamura J.; Pachkowski B. F.; Ranasinghe A.; Gold A.; Ball L. M.; Swenberg J. A. Chem. Res. Toxicol. 2005, 18, 51. [DOI] [PubMed] [Google Scholar]
- Jeong Y. C.; Nakamura J.; Upton P. B.; Swenberg J. A. Nucleic Acids Res. 2005, 33, 6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung F. L.; Nath R. G.; Ocando J.; Nishikawa A.; Zhang L. Cancer Res. 2000, 60, 1507. [PubMed] [Google Scholar]
- Chou P. H.; Kageyama S.; Matsuda S.; Kanemoto K.; Sasada Y.; Oka M.; Shinmura K.; Mori H.; Kawai K.; Kasai H.; Sugimura H.; Matsuda T. Chem. Res. Toxicol. 2010, 23, 1442. [DOI] [PubMed] [Google Scholar]
- Pang B.; Zhou X.; Yu H.; Dong M.; Taghizadeh K.; Wishnok J. S.; Tannenbaum S. R.; Dedon P. C. Carcinogenesis 2007, 28, 1807. [DOI] [PubMed] [Google Scholar]
- Otteneder M.; Scott Daniels J.; Voehler M.; Marnett L. J. Anal. Biochem. 2003, 315, 147. [DOI] [PubMed] [Google Scholar]
- Hoberg A. M.; Otteneder M.; Marnett L. J.; Poulsen H. E. J. Mass Spectrom. 2004, 39, 38. [DOI] [PubMed] [Google Scholar]
- Weimann A.; Belling D.; Poulsen H. E. Nucleic Acids Res. 2002, 30, E7 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otteneder M. B.; Knutson C. G.; Daniels J. S.; Hashim M.; Crews B. C.; Remmel R. P.; Wang H.; Rizzo C.; Marnett L. J. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson C. G.; Skipper P. L.; Liberman R. G.; Tannebaum S. R.; Marnett L. J. Chem. Res. Toxicol. 2008, 21, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson C. G.; Wang H.; Rizzo C. J.; Marnett L. J. J. Biol. Chem. 2007, 282, 36257. [DOI] [PubMed] [Google Scholar]
- Akingbade D.; Kingsley P. J.; Shuck S. C.; Cooper T.; Carnahan R.; Szekely J.; Marnett L. J. Chem. Res. Toxicol. 2012, 25, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson C. G.; Rubinson E. H.; Akingbade D.; Anderson C. S.; Stec D. F.; Petrova K. V.; Kozekov I. D.; Guengerich F. P.; Rizzo C. J.; Marnett L. J. Biochemistry 2009, 48, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Rouzer C. A.; Marnett L. J.; Pietenpol J. A. Carcinogenesis 1998, 19, 1275. [DOI] [PubMed] [Google Scholar]
- Ji C.; Amarnath V.; Pietenpol J. A.; Marnett L. J. Chem. Res. Toxicol. 2001, 14, 1090. [DOI] [PubMed] [Google Scholar]
- Ji C.; Kozak K. R.; Marnett L. J. J. Biol. Chem. 2001, 276, 18223. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Arora P. K.; Iyer R. S.; Salomon R. G. Chem. Res. Toxicol. 1993, 6, 19. [DOI] [PubMed] [Google Scholar]
- Nadkarni D. V.; Sayre L. M. Chem. Res. Toxicol. 1995, 8, 284. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Lin D.; Yuan Q.; Zhu X.; Tang X. Drug Metab Rev 2006, 38, 651. [DOI] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Acc. Chem. Res. 2010, 43, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila A.; Tallman K. A.; Jacobs A. T.; Liebler D. C.; Porter N. A.; Marnett L. J. Chem. Res. Toxicol. 2008, 21, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y.; Tallman K. A.; Liebler D. C.; Porter N. A. Mol. Cell. Proteomics 2009, 8, 2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeger A. L.; Cipollina C.; Cole M. P.; Woodcock S. R.; Bonacci G.; Rudolph T. K.; Rudolph V.; Freeman B. A.; Schopfer F. J. Nat. Chem. Biol. 2010, 6, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J. D.; Marnett L. J. Chem. Res. Toxicol. 2005, 18, 1642. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Shi Z.; Duncan D. T.; Prodduturi N.; Marnett L. J.; Liebler D. C.. Mol. Biosyst. 2011, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. J. Biol. Chem. 2007, 282, 33412. [DOI] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. J. Biol. Chem. 2009, 284, 9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doong H.; Vrailas A.; Kohn E. C. Cancer Lett 2002, 188, 25. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Cell 2000, 100, 57. [DOI] [PubMed] [Google Scholar]
- Luo J.; Solimini N. L.; Elledge S. J. Cell 2009, 136, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C.; Whitesell L.; Rogers A. B.; Lindquist S. Cell 2007, 130, 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldink G. A.; Vliegenthart J. F. Adv. Inorg. Biochem. 1984, 6, 139. [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Chem. Rev. 2003, 103, 2239. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Garavito R. M.; DeWitt D. L. J. Biol. Chem. 1996, 271, 33157. [DOI] [PubMed] [Google Scholar]
- Prasit P.; Riendeau D. Annu. Rep. Med. Chem. 1997, 32, 211. [Google Scholar]
- Talley J. J. Expert Opin. Ther. Pat. 1997, 7, 55. [Google Scholar]
- Marnett L. J.; DuBois R. N. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 55. [DOI] [PubMed] [Google Scholar]
- Eberhart C. E.; Coffey R. J.; Radhika A.; Giardiello F. M.; Ferrenbach S.; DuBois R. N. Gastroenterology 1994, 107, 1183. [DOI] [PubMed] [Google Scholar]
- Kargman S. L.; O’Neill G. P.; Vickers P. J.; Evans J. F.; Mancini J. A.; Jothy S. Cancer Res. 1995, 55, 2556. [PubMed] [Google Scholar]
- Dannenberg A. J.; Lippman S. M.; Mann J. R.; Subbaramaiah K.; DuBois R. N. J. Clin. Oncol. 2005, 23, 254. [DOI] [PubMed] [Google Scholar]
- Williams C. S.; Mann M.; DuBois R. N. Oncogene 1999, 18, 7908. [DOI] [PubMed] [Google Scholar]
- Bresalier R. S.; Sandler R. S.; Quan H.; Bolognese J. A.; Oxenius B.; Horgan K.; Lines C.; Riddell R.; Morton D.; Lanas A.; Konstam M. A.; Baron J. A. N. Engl. J. Med. 2005, 352, 1092. [DOI] [PubMed] [Google Scholar]
- Solomon S. D.; McMurray J. J.; Pfeffer M. A.; Wittes J.; Fowler R.; Finn P.; Anderson W. F.; Zauber A.; Hawk E.; Bertagnolli M. N. Engl. J. Med. 2005, 352, 1071. [DOI] [PubMed] [Google Scholar]
- Edelman M. J.; Watson D.; Wang X.; Morrison C.; Kratzke R. A.; Jewell S.; Hodgson L.; Mauer A. M.; Gajra A.; Masters G. A.; Bedor M.; Vokes E. E.; Green M. J. J. Clin. Oncol. 2008, 26, 848. [DOI] [PubMed] [Google Scholar]
- Marnett L. J. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265. [DOI] [PubMed] [Google Scholar]
- Picot D.; Loll P. J.; Garavito R. M. Nature 1994, 367, 243. [DOI] [PubMed] [Google Scholar]
- Luong C.; Miller A.; Barnett J.; Chow J.; Ramesha C.; Browner M. F. Nat. Struct. Biol. 1996, 3, 927. [DOI] [PubMed] [Google Scholar]
- Kurumbail R. G.; Stevens A. M.; Gierse J. K.; McDonald J. J.; Stegeman R. A.; Pak J. Y.; Gildehaus D.; Miyashiro J. M.; Penning T. D.; Seibert K.; Isakson P. C.; Stallings W. C. Nature 1996, 384, 644. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Crews B. C.; Rowlinson S. W.; Marnett A. B.; Kozak K. R.; Remmel R. P.; Marnett L. J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Blobaum A. L.; Kingsley P. J.; Gorden D. L.; McIntyre J. O.; Matrisian L. M.; Subbaramaiah K.; Dannenberg A. J.; Piston D. W.; Marnett L. J. Cancer Res. 2010, 70, 3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Marnett L. J. Org. Lett. 2008, 10, 4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Ghebreselasie K.; Tantawy M. N.; Marnett L. J. ACS Med. Chem. Lett. 2011, 2, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Ghebreselasie K.; Huda I.; Kingsley P. J.; Ansari M. S.; Tantawy M. N.; Reese J.; Marnett L. J. Cancer Prev. Res. 2011, 4, 1536. [DOI] [PMC free article] [PubMed] [Google Scholar]