

Summary
We show that T7 RNA polymerase can efficiently transcribe DNA containing gaps from one to five bases in the template strand. Surprisingly, broken template strands missing up to 24 bases can still be transcribed, although at reduced efficiency. The resulting transcripts contain the full template sequence with the RNA deleted for the gapped region missing on the template strand. These findings indicate that the end of a downstream template strand can be brought into the polymerase and transcribed as if it were a part of an intact polynucleotide chain by utilizing the unpaired nontemplate strand. This, as well as transcription of an intact template strand, relies heavily upon the nontemplate strand, suggesting that a duplex DNA-binding site on the leading edge of RNA polymerase is required for RNA chain elongation on DNA templates. This work contributes substantially to the emerging picture that the nontemplate strand is an important element of the transcription elongation complex.
Introduction
Bacteriophage T7 RNA polymerase is a single polypeptide of 99 kDa. This relatively simple enzyme is capable of carrying out DNA-directed RNA synthesis analogous to its multisubunit counterparts in bacteria using the same general mechanism, although it transcribes DNA templates more efficiently than Escherichia coli RNA polymerase in vitro and it binds DNA less tightly (Sousa et al., 1992; Chamberlin and Ryan, 1982). T7 RNA polymerase recognizes a specific 17 bp promoter sequence (Oakley and Coleman, 1977; Dunn and Studier, 1983; Rosa, 1979) and goes through an initial abortive phase of transcription before entering the elongation phase (Morris et al., 1986; Martin et al., 1988). During elongation, it unwinds double-stranded DNA, terminates transcription at specific sequences (Jeng et al., 1990, 1992; Macdonald et al., 1993, 1994), and is similar to other RNA polymerases in many respects. Its relatively small size and the availability of its crystal structure (Sousa et al., 1993) make this enzyme an ideal model for studying the transcription process.
To carry out DNA template–dependent transcription, RNA polymerase has to melt the duplex DNA template so that RNA can be synthesized via complementary base pairing with the DNA template strand (Saucier and Wang, 1972). The melting of duplex DNA is accomplished by the formation of the transcription bubble, which is maintained throughout the entire transcription process (Yager and von Hippel, 1987). One component of the transcription bubble is the nontemplate strand, but its role in transcription elongation is not well understood (Yager and von Hippel, 1987). Several studies suggest that the nontemplate strand is dispensable for transcription. For example, RNA polymerase can synthesize short pieces of RNA from single-stranded DNA in a DNA template–dependent fashion (Milligan et al., 1987; Chamberlin and Berg, 1964). Transcription elongation can be blocked by covalent base modifications, such as a psoralen monoadduct or cyclobutane pyrimidine dimer when present on the template strand but not the nontemplate strand (Selby and Sancar, 1990; Donahue et al., 1994; Shi et al., 1988). The termination efficiency of E. coli RNA polymerase at the trp attenuator is determined by the DNA sequence on the template strand, not the nontemplate strand (Ryan and Chamberlin, 1987). Recently, however, a role for the nontemplate strand in the regulation of elongation was suggested in λ phage (Ring and Roberts, 1994) in which a specific pause occurs at position +16 and +17 in the template strand. This pause is controlled by nontemplate sequences in the region of +6 and is crucial for Q protein–mediated antitermination.
Our group has previously shown that T7 RNA polymerase can bypass a 1 nt gap on the template strand with high efficiency (Zhou and Doetsch, 1994), generating a full-length runoff transcript with a 1 nt deletion opposite the gap. This observation indicates that physical breakage in the template strand does not disrupt the transcription bubble during transcription elongation. A large template gap, however, should theoretically have a different effect on transcription elongation by T7 RNA polymerase. Here, we have increased the size of the template gaps up to 24 nt, which is considerably larger than the 15 nt footprint of elongating T7 RNA polymerase on the template strand (Shi et al., 1988). When the catalytic site of T7 RNA polymerase encounters such large template gaps, the downstream component of the broken template strand should be located outside the leading edge of the RNA polymerase contact boundary, and the “transcription bubble” model with its current components would predict that transcription will be halted at such gaps because of the lack of downstream template strand continuity. Our results demonstrate that T7 RNA polymerase is capable of bypassing these large template gaps, generating internally deleted runoff transcripts, and that the downstream component of the broken template strand can be threaded into the RNA polymerase during the transcription bypass process. Our data also indicate that the nontemplate strand plays an important role in transcription bypass of template gaps, and disruption of the interaction between T7 RNA polymerase and the nontemplate strand reduces RNA chain elongation activity, suggesting that T7 RNA polymerase interacts with the nontemplate strand to maintain efficient transcription elongation of an intact DNA template.
Results
T7 RNA Polymerase Bypasses 1–24 nt Gaps in the Template Strand
By annealing three different types of oligonucleotides, we constructed DNA templates 1–24, all of which contained a 17 bp T7 RNA polymerase promoter (Figure 1A). This method also allowed us to increase sequentially the size of the template gaps from 1 to 24 nt. The identities of these DNA templates were verified by a DNA template analysis procedure described previously (Zhou and Doetsch, 1994; data not shown). DNA templates 5–9 contained 5–9 nt gaps on the template strand, respectively, and in each case, the template gaps start 20 nt downstream from the transcription initiation site (Figure 1A). Transcription of these templates was initially carried out in the absence of UTP, and T7 RNA polymerase was stalled at nucleotide position 14, forming a stable ternary complex. A single round of chain extension by this 14 nt RNA-bearing ternary complex was examined for these templates (Figure 1B). Surprisingly, T7 RNA polymerase was capable of bypassing gaps in templates 5–9, generating runoff transcripts 35–39 nt in length. The length of the runoff transcripts is consistent with the theoretical length of the runoff transcripts produced from the unbroken template minus the gap size (Figure 1B). The nature of these runoff products was established by direct RNA sequence analysis, which indicated that RNA polymerase accurately transcribed the broken template strand flanking the gap site. The runoff transcripts contained the full template sequence with the RNA deleted for the gapped region present on the template strand (data not shown). However, for some but not all internally deleted runoff transcripts, the RNA sequence corresponding to the two nucleotides flanking the gap site was difficult to determine because of a low level of background cleavage at these sites (data not shown).
Figure 1. Transcription Bypass of Template Gaps.

(A)Gapped templates 0–24. These templates contain a 17 bp T7 RNA polymerase promoter region (stippled box) and a gap on the template strand 20 nt downstream from the transcription start site (indicated by horizontal arrows) except for templates 0, 24, and 0′. Template 24 contains a 24 nt gap on the template strand starting at nucleotide position 16. Templates 0 and 0′ contain an intact template strand.
(B)Transcription bypass of 5–9 nt template gaps. T7 RNA polymerase ternary complexes were formed on templates 5–9 at nucleotide position 14, and aliquots were removed for transcript analysis before the addition of UTP (lanes 1, 6,11,16, and 21), and at 5 s (lanes 2, 7,12,17, and 22), 10 s (lanes 3, 8, 13, 18, and 23), 30 s (lanes 4, 9, 14, 19, and 24) and 120 s (lanes 5, 10, 15, 20, and 25) following the addition of UTP. L lanes contain RNA size markers (see Experimental Procedures). Migration positions of runoff (R035-39) and shortened transcripts (ST 19–21) are indicated. Bands at the top of the gel are labeled duplex DNA template. Bands in the size range of 14 nt represent stalling transcripts (component of the ternary complex at nucleotide position 14), and bands 7 nt in length (located at gel bottom) are aborted transcription products (Zhou and Doetsch, 1994).
(C)Transcription bypass of a 24 nt template gap. Multiple-round transcription experiments were carried out on templates 24 (24 nt gap) and 0′ (unbroken template control), and aliquots were removed for transcript analysis at 0 min (lanes 1 and 4), 5 min (lanes 2 and 5), and 30 min (lanes 3 and 6) after the start of transcription. Lane L contains RNA size markers, and the migration position and the size of runoff (R057 and R058 and R033 and R034) and shortened (ST13–21) transcripts are indicated.
(D)Effect of template gap size on transcription bypass efficiency. Single-round transcription experiments were carried out on templates 1–19, and the transcription bypass efficiency was determined on aliquots that were removed at 2 min following the addition of UTP (see Experimental Procedures).
Transcription pausing or termination at the gap site on templates 5–9 should generate shortened transcripts 19 nt in length. Such 19 nt transcripts, as well as transcripts 20 and 21 nt in length, were generated with all five templates, indicating that a certain percentage of RNA polymerase is stopped at the gap site (Figure 1B). A significant portion of the shortened 19 nt transcripts disappeared at later times, 30 and 120 s (Figure 1B, lanes 2–5, 7–10,12–15, 17–20, and 22–25), indicating that the 19 nt species is capable of further RNA chain extension. The 20 and 21 nt species, however, remained throughout the entire time course, suggesting that they are not capable of further RNA chain extension, and represent termination products. The sequences of the 20 and 21 nt species were DNA template dependent from nucleotide positions 1–19 (data not shown) and appeared to have been extended by the previously described nontemplated additions of nucleoside monophosphates by T7 RNA polymerase (Jacques and Kolakofsky, 1991; Milligan et al., 1987).
The largest template gap size investigated was 24 nt in length (Figure 1A, template 24). The position of the gap started at nucleotide position 16 on the template strand, and transcription bypass of such a gap should generate a 33 nt runoff transcript. Transcription (multiple rounds) of the control, unbroken template (template 0′) generated primarily runoff transcripts 57 or 58 nt in length as expected (Figure 1C). Transcription (multiple rounds) of template 24 generated a series of transcripts 13–21 nt in length, indicating some degree of transcription pausing or termination in the vicinity of the gap site. Remarkably, T7 RNA polymerase bypassed this gap with reasonable efficiency and produced runoff transcripts 33 and 34 nt in length (Figure 1C). Hence, T7 RNA polymerase can bypass a 24 nt gap on the template strand and generate runoff transcripts that are 24 nt shorter than that of the control transcripts.
The size of the template gaps affected the extent of bypass efficiency, ranging from greater than 78% when the template gap size is 5 nt or less, to 31% with an additional 4 nt increase in the gap size (Figure 1D). Further increases in the template gap size by up to 10 nt did not result in a further decrease in the bypass efficiency, which remained constant at approximately 30% (Figure 1D). Therefore, the bypass efficiency was high for small gaps (5 nt or less) and relatively lower for larger gaps (9 nt or more). This decrease in the transcription bypass efficiency probably reflects the location of the downstream component of the broken template strand with respect to an RNA polymerase contact boundary or groove when the catalytic site encounters the template gap.
The Downstream Component of the Broken Template Strand Can Be Threaded into RNA Polymerase during Bypass of Gaps
When T7 RNA polymerase encounters a 24 nt gap, the downstream portion of broken template strand should lie distal to the leading edge of RNA polymerase. Presumably, the template strand is threaded into RNA polymerase for gap bypass to occur. To test this possibility, a template thread-in experiment was designed. Templates A and B both contained a shortened template strand, terminating at nucleotide position 19 (Figure 2). The addition of oligo D to templates A and B should generate templates 1 and 13, respectively, which contain template gaps 1 nt or 13 nt in length, starting at nucleotide position 20, and which should support the generation of 37 nt runoff transcripts. In the absence of UTP in the transcription mixture, T7 RNA polymerase will stall on templates A and B at nucleotide position 14, forming a stable ternary complex (Figure 2, arrow 1). The addition of UTP to the ternary complex allows elongation to proceed, but T7 RNA polymerase will be halted at the 3′ terminus of the shortened template strand (Figure 2, arrow 2). The thread-in experiment allows us to establish whether oligo D can be utilized by T7 RNA polymerase stalling at nucleotide positions 14 or 19 on either template. Transcription of template A alone generated shortened species 20 and 21 nt in length as expected (Zhou and Doetsch, 1994; data not shown). Addition of oligo D to the stalled RNA polymerase at nucleotide position 14 (Figure 2, reaction group 1) followed by addition of UTP generated runoff transcripts in addition to the shortened transcripts. Hence, the stalled ternary complex at nucleotide position 14 on template A can utilize oligo D for complete RNA chain extension. Oligo D was also added to the stalled ternary complex 5 s after the addition of UTP (Figure 2, reaction group 2). During this period of time, T7 RNA polymerase should be chased to nucleotide position 19 on the template strand (Zhou and Doetsch, 1994). The generation of the runoff transcripts in reaction group 2 allows us to deduce that a template strand located outside the T7 RNA polymerase contact boundary can still be threaded into the RNA polymerase and be utilized to generate runoff transcripts.
Figure 2. The Downstream Component of the Broken Template Strand Can Be Threaded into RNA Polymerase in a Template Gap Size–Independent Fashion.
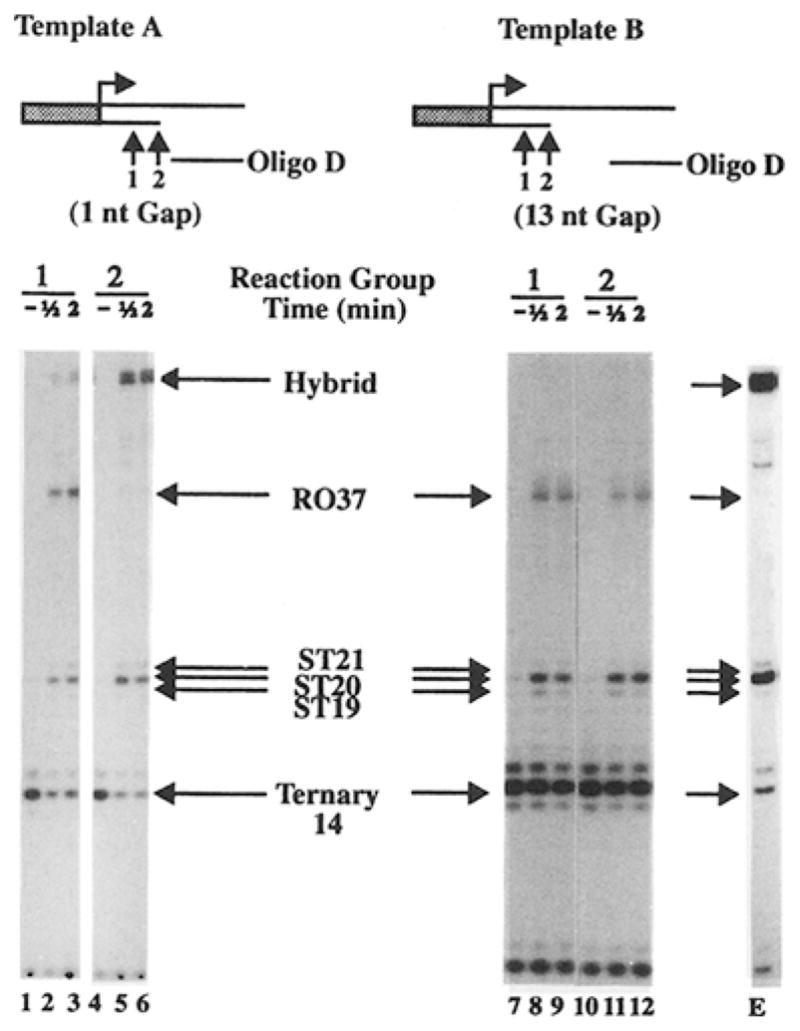
Templates A and B and oligo D. Nucleotide positions 14 and 19 on DNA templates A and B are indicated by arrows 1 and 2, respectively. Single-round transcription experiments were carried out on templates A or B in the absence of UTP, and the reaction mixture was divided into reaction groups 1 and 2. Aliquots were removed from each reaction group before the addition of UTP and/or oligo D as a control (lanes 1, 4, 7, and 10). Oligo D was added to the ternary complex at nucleotide position 14 (reaction group 1, lanes 2–3 and 8–9) or to the halted complex at nucleotide position 19, 5 s (reaction group 2, lanes 5–6 and 11–12) after the addition of UTP. Oligo E was added to the ternary complex on template A at nucleotide position 14 (lane E) before the addition of UTP. Aliquots were removed for transcript analysis at 30 s (lanes 2, 5, 8, and 11) or at 2 min (lanes 3, 6, 9, 12, and E) after the addition of all reaction components. The migration positions for runoff (R037), shortened (ST19–21), and ternary (14) transcripts are indicated. R.037 is absent in lane E. The bands at the top of the gel on the left panel are DNA–RNA hybrids (Zhou and Doetsch, 1994).
Owing to the nontemplated addition activity of T7 RNA polymerase to the transcripts in the halted complex at nucleotide position 19, we could only achieve 15% gap bypass efficiency when oligo D was added in trans, even though it was added immediately after UTP (Figure 2, reaction group 2). Presumably, this is due to the longer length of time required for annealing of oligo D to template A compared with the length of time required for nontemplated additions to the existing shortened transcripts. Gap bypass was not observed if oligo D was added 30 s after the addition of UTP (data not shown) because of the nontemplated additions to the initial 19 nt transcript during the 30 s time interval. This results in the exclusive generation of shortened 20 and 21 nt terminal transcripts, neither of which are capable of further RNA chain extension. Therefore, the thread-in/chain extension process competes with the nontemplated addition process for the 19 nt species during RNA polymerase transcription bypass of template gaps.
The template thread-in experiment was also carried out on template B, and oligo D can be utilized by 17 RNA polymerase at either position 14 or 19 for the generation of runoff transcripts (Figure 2, lanes 8–11). Interestingly, 15% bypass efficiencies were observed for both templates A (template 1 precursor) and B (template 13 precursor) under the conditions of the template thread-in experiments (Figure 2, reaction group 2). This result is in contrast with the one in Figure 1D that indicated that 17 RNA polymerase can bypass the gap in template 1 with a 99% efficiency, but can bypass the gap in template 13 with only a 27% efficiency (Figure 1D). Therefore, when oligo D was threaded into RNA polymerase, the actual size of the template gap did not affect the overall RNA polymerase bypass efficiency, and we conclude that, under these conditions, the bypass efficiency is primarily determined by the thread-in process.
As a control, similar template thread-in experiments were carried out on templates A and B, but with two oligonucleotides that do not contain any sequence complementary to the nontemplate strand in templates A and B (Experimental Procedures, oligos E and F). Addition of either oligo E (Figure 2, lane E) or oligo F (data not shown) to the stalled or halted RNA polymerase on template A or template B did not produce runoff transcripts. Hence, only an oligonucleotide that contains a complementary sequence to the nontemplate strand can be utilized in a successful thread-in experiment with T7 RNA polymerase. These results indicate that complementary base pairing between a template and a nontemplate strand is required for transcription bypass of template gaps.
“Gap Closing” Is Required for Transcription Bypass
The success of the template thread-in experiments suggested that the template gap was closed for the synthesis of internally deleted runoff transcripts. To investigate this possibility, we constructed 1 nt and 19 nt gap-containing DNA templates with modified termini flanking the gap site (Figure 3A). As reported previously (Zhou and Doetsch, 1994), when templates contain either two hydroxyl groups or a single hydroxyl and a phosphoryl group flanking the 1 nt gap site, T7 RNA polymerase bypasses these template gaps with high efficiency (Figures 3B and 3C). The electrostatic charge repulsion between the two negatively charged phosphoryl termini flanking the gap on template 1d (Figure 3A), however, impaired the “gap closing” so that T7 RNA polymerase progression is inhibited at the gap site (Figure 3B, lanes 18–20), and the bypass efficiency was only 31 % (Figure 3C). Such 5′ and 3′ terminal phosphoryl groups contain two negatively charged oxygens, each at pH 8.0, and the pKa value for secondary phosphate ionization in a nucleoside monophosphate is 6.6 (Saenger, 1984). Thus, at pH 6.0, one of the two oxygens will be approximately 80% protonated, leaving the terminal phosphoryl group with essentially one negatively charged oxygen, and the electrostatic charge repulsion between the two terminal phosphoryl groups should be diminished during the gap closing event.
Figure 3. The Effect of 3′ and 5′ Terminal Phosphoryl Groups Flanking the Gap Site on Transcription Bypass Efficiency.

(A)Templates 1a, 1b, 1c, 1d, 19a, 19b, 19c, and 19d contain the same DNA sequence as templates 1 and 19, respectively; however, the 3′ and 5′ termini flanking the gap site contain either phosphoryl groups (P) or hydroxyl groups (OH) (see Experimental Procedures).
(B)Single-round transcription experiments were carried out on templates 1a (lanes 1–10) and 1d (lanes 11–20) at either pH 6.0 (lanes 1–5 and 11–15) or 8.0 (lanes 6–10 and 16–20). Aliquots were removed for transcript analysis at stage 1 (lanes 1, 5, 11, and 15), stage 2 (lanes 2, 6, 12, and 16), and at 5 s (lanes 3, 8,13, and 18), 2 min (lanes 4, 9,14, and 19), and 4 min (lanes 5,10,15, and 20) after the addition of UTP (stage 3). L lanes contain RNA size markers. Migration positions of the runoff (R037) and shortened (ST19, ST20, ST21) transcripts are indicated.
(C)The gap bypass efficiency of T7 RNA polymerase was determined as described in Experimental Procedures for templates 1 a, 1 b, 1c, and 1 d at pH 8.0 and pH 6.0.
(D)Effect of differentially phosphorylated 5′ and 3′ gap termini contained in a 19 nt–gapped template on transcription bypass efficiency. The gap bypass efficiency was determined for templates 19a, 19b, 19c, and 19d at pH 8.0. The results shown here are averages of two experiments, and the percentage variation obtained between a given template is less than 3%.
Lowering the pH to 6.0 reduces the overall transcription efficiency, and this did not affect gap bypass on templates containing none or one phosphoryl group flanking the gap (Figure 3C). The transcription bypass efficiency for a template containing two phosphoryl groups flanking a one base gap (template 1 d), however, was increased from 31 % to 58% at pH 6.0 (Figure 3B, lane 15; Figure 3C). Hence, T7 RNA polymerase bypasses the template gap on template 1 d with substantially higher efficiency at pH 6.0 compared with pH 8.0. This result suggests that the electrostatic charge repulsion between the 3′ and 5′ terminal phosphoryl groups on template 1d is responsible for preventing the template gap from closing at pH 8.0 and results in inhibition of T7 RNA polymerase progression at the gap site. We conclude that the proper juxtaposition of the 5′ end of the upstream template strand and the 3′ end of the downstream template strand (gap closing) can occur in a specific template-binding groove in RNA polymerase.
We reasoned that bringing a distal part of the template strand that was expected to reside outside the T7 RNA polymerase footprint (19 nt–gapped template) into this template groove might be difficult to achieve if it contained a phosphoryl group. Indeed, the presence of a single phosphoryl group at the 3′ terminus of a 19 nt–gapped template decreased the transcription bypass efficiency from 30% to 9% (Figure 3D). Two phosphoryl termini flanking the gap site further decreased bypass efficiency to 4%. Hence, the 3′ terminal phosphoryl group effectively blocks transcription with a 19 nt–gapped template (templates 19c and 19d). A 3′ terminal phosphoryl group is bulkier than a 3′ hydroxyl group and is negatively charged. During the transcription bypass of a 19 nt gap, this terminus is probably located outside the RNA polymerase contact boundary and is likely to interfere with the thread-in process of the downstream component of the template strand.
The Role of the Nontemplate Strand in Transcription Elongation
The requirement for a complementary nontemplate strand for the thread-in process to succeed suggests that the nontemplate strand plays an important role in the RNA polymerase template gap bypass process. This is probably because of the need to form duplex DNA with the downstream component of the broken template strand for T7 RNA polymerase to bind (duplex DNA–binding site). A direct interaction of the nontemplate strand alone with T7 RNA polymerase may also be important. To explore this, we constructed DNA templates containing various discontinuities on the nontemplate strand. Template Z was similar to template 1, except it contained an intact template strand and a 1 nt gap on the nontemplate strand at position 20 (Figure 4). Template 0 contained intact duplex DNA and was used as a positive control. Template Y contained a shortened upstream nontemplate strand, terminating at position 20. Template X lacked duplex DNA in the promoter region, but contained duplex DNA starting at position 21. Single-round transcription experiments were carried out on these templates, and transcripts were analyzed before and after the addition of UTP. Template X did not contain a duplex promoter region and did not support promoter-dependent transcription (Figure 4, lanes 1–7). Template 0 supported T7 RNA polymerase transcription and generated runoff transcripts as expected (Figure 4, lanes 24 to 28). Template Y supported the generation of the ternary complex at nucleotide position 14 in the presence of ATP, CTP, and GTP (Figure 4, lane 9), but runoff transcripts were not generated following the addition of UTP (lanes 10–14) even though template Y contained an intact template strand. The majority of ternary complex generated from template Y at nucleotide position 14 was not converted into longer transcripts after the addition of UTP. Therefore, the stalled ternary complex at nucleotide position 14 on template Y is not capable of efficient RNA chain extension. This result is in contrast with the transcription of template A, which contains an intact nontemplate strand and a shortened template strand, and the stalled T7 RNA polymerase at nucleotide position 14 on template A can easily resume transcription elongation with the addition of UTP to generate shortened transcripts, 20 and 21 nt in length (Zhou and Doetsch, 1994). Therefore, the stalled T7 RNA polymerase ternary complex at nucleotide position 14 requires the presence of the downstream component of the nontemplate strand, but not the distal portion of the noncontiguous template strand, to resume elongation. This nontemplate strand requirement was further supported by the transcription experiment with template Z because the stalled T7 RNA polymerase at nucleotide position 14 could resume transcription elongation efficiently to generate runoff transcripts when the downstream component of the broken nontemplate strand was present (Figure 4, lanes 17–21). Efficient transcription of template Z also indicated that a 1 nt gap on the nontemplate strand did not affect transcription elongation by T7 RNA polymerase.
Figure 4. The Nontemplate Strand Is Required for the Ternary Complex to Resume Transcription Elongation from a Stalling State.
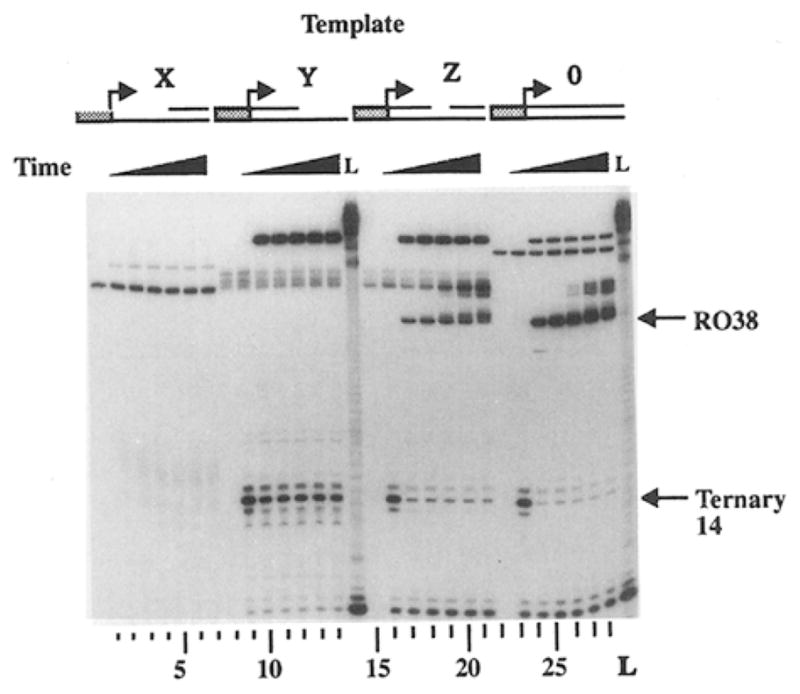
DNA template Z was similar to template 1 (Figure 1A), except that it contains a 1 nt gap on the nontemplate strand 20 nt downstream from the transcription start site. Template Y contains a shortened nontemplate strand upstream from nucleotide position 19. Template X does not contain a duplex promoter, but it contains a shortened nontemplate strand downstream starting from position 21. Single-round transcription experiments were carried out on templates X, Y, Z, and 0, and aliquots were removed for transcript analysis at stage 1 (lanes 1, 8, 15, and 22), stage 2 (2, 9, 16, and 23), and at 5 s (lanes 3, 10, 17, and 24), 10 s (lanes 4,11,18, and 25), 30 s (lanes 5, 12, 19, and 26), 1 min (lanes 6, 13, 20, and 27), and 2 min (lanes 7, 14, 21, and 28) following the start of stage 3. Lane L contains RNA size markers, and the migration position for runoff (R038) and shortened (ST14) transcripts are indicated. The bands at the top of the gel are DNA–RNA hybrids (Zhou and Doetsch, 1994).
Discussion
Using templates with missing portions of the template or nontemplate strand, we provide direct evidence that efficient elongation requires the entry of duplex DNA into RNA polymerase. It appears that continued chain extension requires the occupancy of duplex DNA in a specific binding site at the leading edge of polymerase (Figure 5, site A). The existence of this site in a ternary complex has been postulated in several transcription elongation models, and it emphasizes the importance of protein–nucleic acid interactions in the transcription bubble (von Hippel et al., 1984; Chamberlin, 1995).
Figure 5. Transcription Bypass of a Large Gapped Template.

(1) Transcription of a gapped (>9 nt in length) template by T7 RNA polymerase within the regions that contain intact, duplex DNA. The duplex DNA–binding site (A) on T7 RNA polymerase contacts DNA to maintain transcription elongation efficiency. (2) The catalytic site (B) of T7 RNA polymerase encounters the beginning of a template gap. (3)The nontemplate strand is looped out and brings the downstream portion of the noncontiguous template strand into T7 RNA polymerase. (4) Template gap closes, and RNA synthesis continues over the discontinuous template, ultimately producing runoff transcription products containing an internal deletion.
It is remarkable that T7 RNA polymerase is capable of bypassing a 24 nt template gap, even though an elongating T7 RNA polymerase only protects 15 nt on the template strand (Shi et al., 1988). The structure of such a 24 nt gap-containing DNA template is not known, and the single-stranded DNA portion in the gap region might contain an unusual secondary structure. Thus, the actual distance between the 3′ and 5′ termini flanking the gap site is likely to be less than the actual distance corresponding to a 24 bp segment of duplex B-DNA. However, it should be pointed out that the DNA sequence utilized in this study does not favor the formation of hairpin structures in the nontemplate strand, suggesting the enzyme mediates apposition of the broken ends flanking the gap (Figure 1A). The decrease in the bypass efficiency as the gap size exceeds 5 nt in length might reflect the position of the distal portion of the broken template strand relative to the RNA polymerase contact boundary when the catalytic site of T7 RNA polymerase encounters the beginning of the template gap. It is likely that when the gap size is less than 5 nt, both the proximal (upstream) and distal (downstream) portions of the template strand flanking the gap are located within RNA polymerase, and that the gap can be easily closed with a resulting high transcription bypass efficiency. When the gap size is larger than 9 nt, the distal portion of the template strand is probably located outside the RNA polymerase contact boundary, and it has to be threaded into the proper groove of RNA polymerase for transcription bypass to occur. This thread-in process appeared slower than the nontemplated addition of residues to stalled transcripts that competed with templated chain elongation. Thus, the efficiency of gap closure is limited by the competing process of nontemplated base additions. The major point demonstrated by the template thread-in experiment was that a template strand located outside the RNA polymerase boundary can be threaded into RNA polymerase to allow the continuation of transcription elongation, and this process determines the transcription bypass efficiency.
Transcription bypass of large template gaps requires the presence of a complementary nontemplate strand, and one role for the nontemplate strand appears to be as a guide to establish the coding register for a noncontiguous template strand. For the generation of internally deleted runoff transcripts, the template gap must be closed and the nontemplate strand must be looped out, and it is not clear how such a looping out process is achieved mechanistically. One possibility is that the nontemplate strand is only utilized as a guiding element for downstream template contacts and to restrict the RNA polymerase search for a downstream duplex DNA template.
Our results also indicate that the interaction between the downstream component of the nontemplate strand and RNA polymerase is crucial for T7 RNA polymerase to resume transcription elongation from a stalling state (Figure 4). This finding differs from a study in which runoff transcripts were synthesized from a synthetic DNA template containing a duplex 17 RNA polymerase promoter and single-strand DNA template strand (Milligan et al., 1987), and could be due to the reaction conditions (high concentration of heparin and no spermidine) used in this study. Our results are consistent with those of Daube and von Hippel (1992) who show that the interaction between T7 RNA polymerase and the nontemplate strand is important to support efficient RNA synthesis. Hence, T7 RNA polymerase might establish its transcription elongation efficiency on normal duplex DNA via a specific site that accommodates downstream duplex DNA, effectively pulling in on the nontemplate strand (Figure 5). When RNA polymerase encounters a small or large template gap, such an activity will close the gap and loop out the nontemplate strand. When the template gap exceeds 9 nt in length, the downstream component of the broken template strand can associate with the RNA polymerase contact boundary through its base-pairing interactions with the nontemplate strand, resulting in a thread in process for the distal part of the template strand to enter the proper groove of RNA polymerase. Therefore, the template thread-in experiment only works when the downstream portion of the noncontiguous template strand is complementary to the nontemplate strand. The presence of a 3′ bulky or charged group impairs such a thread-in process with large template gaps. Furthermore, the need for the enzyme to recognize downstream duplex DNA might be responsible for the transition from the transcription initiation to the transcription elongation state and might generally be used to maintain the transcription bubble during the elongation stage. In this regard, we have also observed template gap bypass by SP6 RNA polymerase (Liu and Doetsch, unpublished data).
In conclusion, the surprising result that T7 RNA polymerase bypasses large gaps in the template strand can be best understood in terms of the interaction between the polymerase and the nontemplate strand. We propose that such an interaction may be a key component for efficient elongation for RNA polymerases in general.
Experimental Procedures
Generation of Gapped Templates
To construct a 1 nt gap-containing DNA template (template 1), the nontemplate strand of template 1, oligo 1 (5′-TAATACGACTCACTA-TAGGGAGACCGGAAGCTTGGGATGGAGTTGGAGACGGGTG-3′), was first 5′ end labeled with T4 polynucleotide kinase and [γ-32P]ATP (Maniatis et al., 1982). 5′ end labeled oligo 1 (100 pmol) was added to 300 pmol of oligo 3 (5′-CCCAAGCTTCCGGTCTCCCTATAGTGAG-TCGTATTA-3′) and 500 pmol of oligo D (5′-CACCCGTCTCCAACT-CCA-3′) in 10 μl of 10 mM MgCl2. The mixture was heated to 70°C for 10 min and cooled to room temperature over 4 hr (standard annealing conditions). DNA template 1 was purified from a 20% polyacrylamide nondenaturing gel as described previously (Zhou and Doetsch, 1994). A similar approach was used to construct DNA templates 0–24 as depicted in Figure 1A. For the generation of templates 1a, 1b, 1c, 1d, 19a, 19b, 19c, and 19d, oligo D was modified to contain either a 3′ hydroxyl or a 3′ phosphoryl group, and oligo 3 was modified to contain either a 5′ hydroxyl group or a 5′ phosphoryl group (Zhou and Doetsch, 1994). Templates were constructed by standard annealing conditions described above (Figure 1). Template 1a is equivalent to template 1, except that the terminal hydroxyl groups at the gap site are generated by enzymatic methods (Zhou and Doetsch, 1994).
In Vitro Transcription by T7 RNA Polymerase
Single-round transcription experiments with T7 RNA polymerase were carried out in three stages. In stage 1, 0.05 μM T7 RNA polymerase was added to each 0.05 μM DNA template in 40 mM HEPES (pH 8.0), 10 mM NaCl, 6 mM MgCl2 (transcription buffer), 500 μM ATP and CTP, and 3 μCi [α-32P]CTP (3000 Ci/mmol) at 20°C for 8 min to form the transcription initiation complex in a volume of 20 μl. Because there is no promoter-dependent transcription at this stage, aliquots removed from the reaction mixture contain only 32P end-labeled DNA templates. In stage 2, 500 μM GTP and 1.25 mg/ml heparin were added to the stage 1 reaction mixture for 2 min to allow transcription initiation and elongation to proceed until T7 RNA polymerase was stalled immediately before the incorporation of the first uracil at nucleotide position 14 (Figure 1), and aliquots were removed to identify the transcripts in the ternary complex. In stage 3, 500 μM UTP was added to the stage 2 reaction mixture, and aliquots were removed at different time intervals (5, 10, 30, 120 s) following UTP addition to identify the runoff transcription products. Transcription was stopped by the addition of the RNA-loading dye (9.8 M urea, 0.2 mM EDTA, 0.1 % xylene cyanol), and the 32P-labeled transcripts were analyzed on 15% polyacrylamide gels (7 M urea, 45 mM Tris–borate, and 1 mM EDTA) as described previously (Zhou and Doetsch, 1994). For transcription with T7 RNA polymerase at pH 6.0 or pH 8.0, 40 mM NaH2PO4 (pH 6.0 or pH 8.0) was used in the transcription buffer instead of 40 mM HEPES (pH 8.0). Multiple-round transcription experiments with T7 RNA polymerase were carried out on DNA template 24 and 0′ using the same transcription buffer component minus heparin at 37°C for 5 or 30 min, and transcription was terminated by the addition by RNA-loading dye. To generate the RNA size marker ladder, transcription experiments were carried out with templates 0 or 0′ under multiple round transcription conditions. The resulting reaction mixture was then subjected to alkaline hydrolysis with 50 mM Na3PO4 (pH 12) for 20 min at 70°C.
For transcript sequencing, multiple round transcription conditions were used in the absence of [α-32P]CTP. Run-off and shortened transcripts were dephosphorylated by calf intestinal phosphatase, gel purified, and 5′ 32P end labeled (Maniatis et al., 1982). RNA sequencing was conducted with base-specific ribonucleases as described in the RNA Sequence Kit (Nuclease Method, United States Biochemicals).
Template Thread in Experiments
Single-round transcription experiments were carried out on template A or template B in the absence of UTP to form a ternary complex at nucleotide position 14 (stage 2 reaction conditions; Figure 2, arrow 1). The reaction mixture was divided into reaction groups 1 and 2. In reaction group 1, 0.15 μM of oligo D was added to the ternary complex first, and 500 μM UTP was added 10 s later. In reaction group 2,0.15 μM oligo D was added 5 s after the addition of 500 μM UTP. Aliquots were removed at 30 or 120 s following the addition of all reaction components, and transcription was terminated by the addition of the RNA-loading dye. Oligos E (5′-GAATACACGGAATTCGAGC-3′) and F (5′-AATAGCACTCACTATAG-3′) were also used in the template thread in experiments in place of oligo D.
Quantitation of RNA Polymerase Bypass Efficiency
To calculate gap bypass efficiency, single-round transcription experiments were carried out with DNA templates 0–19 in three stages with [α-32P]CTP as described above. Aliquots were removed 2 min after the addition of UTP (stage three reaction conditions) and were analyzed on a 15% polyacrylamide denaturing gel. The radioactivity in the resulting 32P-labeled RNA transcripts (runoff or shortened transcripts) were determined by phosphorimager analysis (Molecular Dynamics). Run-off transcripts, R038 (38 nt) and R037 (37 nt) contain four cytosine residues, and the amount of radioactivity in these transcripts is defined as RO. The shortened transcripts, 19–21 nt contain three cytosine residues, and the amount of radioactivity in these transcripts is defined as ST. T7 RNA polymerase bypass efficiency was calculated as (RO)/[RO + ST × 4/3] × 100%.
Acknowledgments
We would like to thank Drs. Michael J. Chamberlin, Robert Landick, and Charles Turn-bough for critical reading of the manuscript and helpful discussions. This work was supported by research grants CA55896 (P. W. D.) and GM46311 (D. R.) from the National Institutes of Health.
References
- Chamberlin MJ. New models for the mechanism of transcription elongation and its regulation. Harvey Lect. 1995;88:1–21. [PubMed] [Google Scholar]
- Chamberlin MJ, Berg P. Mechanism of RNA polymerase action: formation of DNA-RNA hybrids with single-stranded templates. J Mol Biol. 1964;8:297–313. doi: 10.1016/s0022-2836(64)80139-x. [DOI] [PubMed] [Google Scholar]
- Chamberlin MJ, Ryan T. Bacteriophage DNA-dependent RNA polymerase. In: Boyer PD, editor. The Enzymes. 15B. New York: Academic Press; 1982. pp. 87–109. [Google Scholar]
- Daube SS, von Hippel PH. Functional transcription elongation complexes from synthetic RNA-DNA bubble duplexes. Science. 1992;258:1320–1324. doi: 10.1126/science.1280856. [DOI] [PubMed] [Google Scholar]
- Donahue BA, Yin S, Taylor JS, Reines D, Hanawalt PC. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci USA. 1994;91:8502–8506. doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JJ, Studier FW. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- Jacques JP, Kolakofsky D. Pseudo-templated transcription in prokaryotic and eukaryotic organisms. Genes Dev. 1991;5:707–713. doi: 10.1101/gad.5.5.707. [DOI] [PubMed] [Google Scholar]
- Jeng ST, Gardner JF, Gumport RI. Transcription termination by bacteriophage T7 RNA polymerase at rho-independent terminators. J Biol Chem. 1990;265:3823–3830. [PubMed] [Google Scholar]
- Jeng ST, Gardner JF, Gumport RI. Transcription termination in vitro by bacteriophage T7 RNA polymerase: the role of sequence elements within and surrounding a rho-independent transcription terminator. J Biol Chem. 1992;267:19306–19312. [PubMed] [Google Scholar]
- Macdonald LE, Zhou Y, McAllister WT. Termination and slippage by bacteriophage T7 RNA polymerase. J Mol Biol. 1993;232:1030–1047. doi: 10.1006/jmbi.1993.1458. [DOI] [PubMed] [Google Scholar]
- Macdonald LE, Durbin RK, Dunn JJ, McAllister WT. Characterization of two types of termination signal for bacteriophage T7 RNA polymerase. J Mol Biol. 1994;238:145–158. doi: 10.1006/jmbi.1994.1277. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1982. [Google Scholar]
- Martin CT, Muller DK, Coleman JE. Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry. 1988;27:3966–3974. doi: 10.1021/bi00411a012. [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Withevell GW, Uhlenbeck OC. Oligonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucl Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CE, McGraw NJ, Joho K, Brown JE, Klement JF, Ling ML, McAllister WT. Mechanism of promoter recognition by the bacteriophage T3 and T7 RNA polymerase. In: Reznikoff WS, Burgess RR, Dahlberg JE, Gross CA, Record T, Wichens MD, editors. RNA Polymerases and the Regulation of Transcription. Amsterdam: Elsevier; 1986. pp. 47–58. [Google Scholar]
- Oakley JL, Coleman JE. Structure of a promoter for T7 RNA polymerase. Proc Natl Acad Sci USA. 1977;74:4266–4270. doi: 10.1073/pnas.74.10.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring BZ, Roberts JW. Function of a nontranscribed DNA strand site in transcription elongation. Cell. 1994;78:317–324. doi: 10.1016/0092-8674(94)90300-x. [DOI] [PubMed] [Google Scholar]
- Rosa MD. Four T7 RNA polymerase promoters contain an identical 23 bp sequence. Cell. 1979;76:815–825. doi: 10.1016/0092-8674(79)90097-7. [DOI] [PubMed] [Google Scholar]
- Ryan T, Chamberlin MJ. Transcription analyses with heteroduplex trp attenuator templates indicate that the transcripts stem and loop structure serves as the termination signal. J Biol Chem. 1987;258:4690–4693. [PubMed] [Google Scholar]
- Saenger W. Principles of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- Saucier JM, Wang JC. Angular alteration of the DNA helix by E coli RNA polymerase. Nature. 1972;239:167–170. doi: 10.1038/newbio239167a0. [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J Biol Chem. 1990;265:21330–21336. [PubMed] [Google Scholar]
- Shi YB, Gamper H, Hearst JE. Interaction of T7 RNA polymerase with DNA in an elongation complex arrested at a specific psoralen adduct site. J Biol Chem. 1988;263:527–534. [PubMed] [Google Scholar]
- Sousa R, Patra D, Lafer EM. Model for the mechanism of bacteriophage T7 RNA polymerase transcription initiation and termination. J Mol Biol. 1992;224:319–334. doi: 10.1016/0022-2836(92)90997-x. [DOI] [PubMed] [Google Scholar]
- Sousa R, Chung YJ, Rose JP, Wang BC. Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature. 1993;364:593–599. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- von Hippel PH, Bear DG, Morgan WD, McSwiggen JA. Protein-nucleic acid interactions in transcription: a molecular analysis. Annu Rev Biochem. 1984;53:389–446. doi: 10.1146/annurev.bi.53.070184.002133. [DOI] [PubMed] [Google Scholar]
- Yager TD, von Hippel PH. Transcript elongation and termination in Escherichia coli. In: Neidhardt FC, editor. E. coli and S. typhimurium: Cellular and Molecular Biology. Washington, D.C: American Society for Microbiology Publications; 1987. pp. 1241–1275. [Google Scholar]
- Zhou W, Doetsch PW. Transcription bypass or blockage at single-strand breaks on the DNA template strand: effect of different 3′ and 5′ flanking groups on the T7 RNA polymerase elongation complex. Biochemistry. 1994;33:14926–14934. doi: 10.1021/bi00253a032. [DOI] [PubMed] [Google Scholar]