

Abstract
The first highly diastereo- and enantioselective additions of aryl nitromethane pronucleophiles to aryl aldimines are described. Identification of an electron rich chiral Bis(Amidine) catalyst for this aza-Henry variant was key to this development, leading ultimately to differentially protected cis-stilbene diamines in two steps. This method then became the lynchpin for an enantioselective synthesis of (–)-Nutlin-3 (Hoffmann-LaRoche), a potent cis-imidazoline small molecule inhibitor of p53-MDM2 used extensively as a probe of cell biology and currently in drug development.
The attention provided to the aza-Henry reaction, also commonly referred to as the nitro-Mannich reaction, over the past decade has resulted in a range of catalysts that can provide the β-amino nitroalkane addition products with high enantioselection.1 These products are readily transformed to unsymmetrical vicinal diamines by straightforward reduction of the nitro functional group, thereby providing straightforward access to a key synthetic building block.2 Nitromethane additions have received the lion's share of attention in these reports, and several classes of nitroalkanes remain problematic. For example, the use of aryl nitromethanes in catalytic, enantioselective aza-Henry reactions has been reported twice, but both examples provided low enantioselection, and little diastereoselection (≤2:1) compared to otherwise high stereoselection with alternative nitroalkanes.3 We encountered similar challenges when approaching these additions with Bis(AMidine) catalysis but report here the successful realization of highly diastereo- and enantioselective aryl nitromethane additions to azomethine using catalyst control. The value of this work lies in its ability to furnish unsymmetrical cis-stilbene diamine derivatives, and this is further grounded by our development of the first fully stereocontrolled synthesis of the potent p53/MDM2 inhibitor (–)-Nutlin-3 (2) discovered by Hoffmann-La Roche (HLR).

We initially targeted β-amino nitroalkane 1a (Table 1). Nitroalkane 4 was prepared in one step from para-chlorobenzyl bromide using the method of Kornblum,4 and imine 3 was formed from the corresponding α-amido sulfone using potassium carbonate to effect the elimination to azomethine.5 Our first attempts to promote the addition of nitroalkane 4 to imine 3 utilized a symmetrical chiral proton catalyst (5•HOTf), leading to the adduct in 13:1 dr and 64% ee (major) (Table 1, entry 1).6,7 Since the Brønsted acid salt of the BisAMidine ligand has always been the most selective catalyst in our past work, we did not anticipate the finding that the free base was equally effective;8 use of bis(amidine) 5 under otherwise identical conditions led to product with nearly equal diastereoselection and enantioselection (Table 1, entry 2). It should be noted that protonation of the Brønsted basic bis(amidine) by aryl nitromethane 4 is possible, and the aryl nitromethane salt that would form (e.g [BAM•H]+•[ArCHNO2]–) could itself be a catalyst in these reactions. Regardless, this observation broadened our investigation of catalysts to include both free base and 1:1 BAM:TfOH. Using unsymmetrical ligand 6, we found that the salt led to the desired addition product with good diastereoselection (7:1 dr), and slightly improved enantioselection (80% ee) (Table 1, entry 3), whereas the free base was similarly diastereoselective (6:1 dr), but less enantioselective (72% ee) (Table 1, entry 4). The binding pocket in catalyst 7 is presumably more open than those of the two previous ligands (5-6), leading to low enantioselection while diastereoselection remained high (Table 1, entries 5-6). However, the free base now provided enantioselection at a slightly improved level relative to the use of its triflic acid salt, emphasizing the need to optimize this transformation using a rather empirical approach. It was not until the Pyrrolidine BisAMidine (8a)6a was used that the enantioselection increased significantly to 85% ee (Table 1, entry 7). The behavior of this bis(amidine) was consistent with its parent HQuin-BAM (5) in that the free base performed equally well relative to the triflic acid salt (Table 1, c.f. entries 7-8). Due to the increase in enantioselection upon increasing the Brønsted basicity/electron rich nature of the aromatic rings, we prepared three additional derivatives in which the quinoline periphery is substituted by methoxy groups (8b-d). These catalysts provided improvements over 8a, with 8d leading to the addition product in 13:1 dr, 91% ee, and nearly quantitative yield (Table 1, entries 10-12). A single fractional recrystallization of this material from toluene provided the adduct as a single diastereomer (>200:1) with 97% ee.
Table 1. Development of a Diastereo- and Enantioselective BisAMidine-Catalyzed Aryl Nitromethane Addition to Azomethinea.
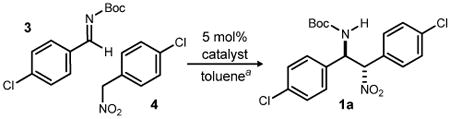 |
|||||||
|---|---|---|---|---|---|---|---|
| entry | BisAMidine | temp (°C) | TfOH (equiv) | dr | ee (%) | yieldb (%) | |
| 1 | 5c | −20 | 1 | 13:1 | 64 | 81 | 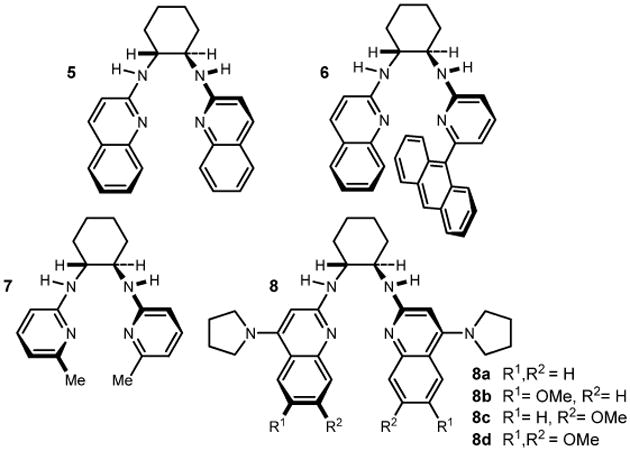 |
| 2 | 5c | −20 | 0 | 10:1 | 65 | 99 | |
| 3 | 6c | −20 | 1 | 7:1 | 80 | 87 | |
| 4 | 6c | −20 | 0 | 6:1 | 72 | 98 | |
| 5 | 7c | −20 | 1 | 5:1 | 58 | 95 | |
| 6 | 7c | −20 | 0 | 6:1 | 63 | 96 | |
| 7 | 8a | −78 | 1 | 5:1 | 85 | 99 | |
| 8 | 8a | −78 | 0 | 9:1 | 86 | 97 | |
| 9 | 8a | −20d | 0 | 6:1 | 83 | 99 | |
| 10 | 8b | −78 | 0 | 13:1 | 87 | 99 | |
| 11 | 8c | −78 | 0 | 16:1 | 89 | 99 | |
| 12 | 8d | −78 | 0 | 13:1 | 91 | 97 | |
Reactions employed 1 equivalent of α-bromo nitroalkane (0.2 M in THF), with NIS added as the final reagent at 25 °C.
Isolated yields.
2 Equivalents of imine used.
1.2 Equivalents of imine used.
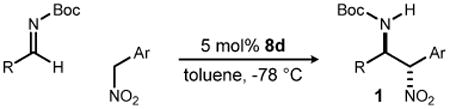
Catalyst 8d was applied to a range of variants to determine preliminary scope (Table 2). Electron rich aryl aldimines led to addition products with higher diastereoselection and similar enantioselection (Table 2, entries 2-4), whereas electron deficient aldimines provided lower, but still good diastereoselection at 14:1 or better (Table 2, entries 5-6). Several aryl nitromethane pronucleophiles were also prepared and subjected to the standard reaction conditions. These provided the adducts with high diastereo- and enantioselection as well (Table 2, entries 9-12), with the exception of the most electron deficient case (Table 2, entry 13). The acidity of the product in this case renders it more susceptible to epimerization at the nitro-substituted carbon. In most every case examined, the addition products were crystalline and were isolated in very good yield.
Table 2. Catalyzed Additions of Arylnitromethanes to Aldiminesa.
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | Ar | 1 | drb | eeb (%) | Yieldb (%) |
| 1c | pClC6H4 (3) | Ph (16) | b | 19:1 | 90 | 93 |
| 2d | pAllylO (9) | 16 | c | 131:1 | 87 | 70 |
| 3e | pMeOC6H4 (10) | 16 | d | 81:1 | 85 | 83 |
| 4d | pMeC6H4 (11) | 16 | e | 38:1 | 91 | 93 |
| 5d | pFC6H4 (12) | 16 | f | 15:1 | 87 | 87 |
| 6d | pCF3C6H4 (13) | 16 | g | 14:1 | 84 | 99 |
| 7d | pPhC6H4 (14) | 16 | h | 44:1 | 93 | 99 |
| 8d | 2Naphth (15) | 16 | i | 25:1 | 91 | 99 |
| 9 | 3 | 4 | a | 13:1 | 91 | 97 |
| 10 | 3 | mBrC6H4 (17) | j | 13:1 | 89 | 91 |
| 11 | 3 | 2Np (18) | k | 10:1 | 80 | 99 |
| 12e | 3 | pMeOC6H4 (19) | l | 17:1 | 86 | 90 |
| 13 | 3 | pNO2C6H4 (20) | m | 2:1 | 76 | 99 |
All reactions were employed 1.1 equiv of nitroalkane in toluene (0.1 M) and 18-26 h reaction time unless otherwise noted. Configuration assigned by analogy to an adduct whose absolute and relative configuration were assigned by X-ray crystallography.
Ee and dr were determined by HPLC. Isolated yield after column chromatography.
For comparison, using 10 mol% of the Takemoto or Deng thiourea at –20 °C provided the adduct (1b) in 4:1 dr/57% ee and 5:1 dr/-31% ee, respectively.
1.2 Equiv of imine used.
Warmed to −20 °C for one hour before workup.
The successful development of an anti-selective addition to generate key intermediate 1a left our remaining aim to convert this masked diamine into Nutlin-3 (2). Nutlin-3 is a cis-imidazoline that can restore the pathway to apoptosis in cancerous cells where the p53 gene is wild type.9 The Nutlins have been shown to inhibit p53/MDM2,10 and the more potent enantiomer, (–)-Nutlin-3, has been used extensively as a small molecule probe of cell biology and remains in clinical development as a chemotherapeutic.11 Its synthesis has not been reported outside of a collection of patents for the preparation of rac-Nutlin-3.12,13 In order to avoid a meso intermediate without unduly telescoping the synthesis using deprotection techniques, conversion of β-amino nitroalkane 1a into the unsymmetrical imidazoline would need to use the inherent chemoselectivity offered by this intermediate. This was accomplished by reduction of the nitro to amine using cobalt boride formed in situ,14 and subsequent acylation with acid 21 to produce Boc-protected amide 22 (Scheme 1). Straightforward deprotection of the Boc group using trifluoroacetic acid revealed the secondary amine in 88% yield. Acylation of the amine with carbonyl diimidazole led to an intermediate isocyanate that was treated with piperazinone 23.
Scheme 1. Preparation of (–)-Nutlin-3.
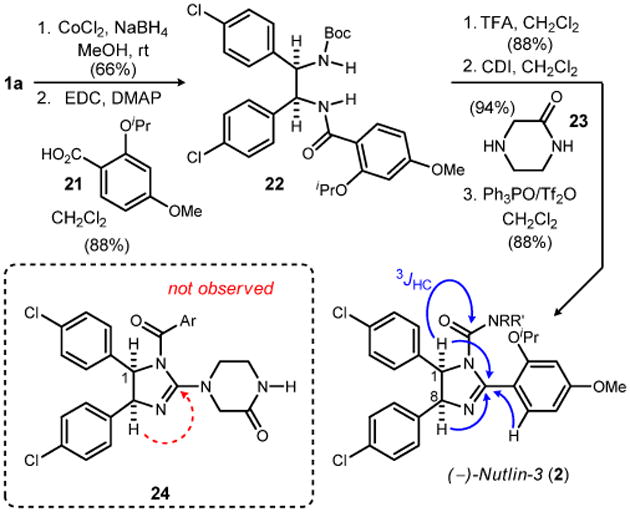
This set the stage to attempt a chemoselective cyclizative dehydration to the desired imidazoline. For this purpose we turned to the powerful dehydrating property of a phosphonium anhydride formed by the combination of triphenylphosphine oxide and triflic anhydride (Hendrickson's reagent).15 Faced with the question of chemoselectivity in the context of a mixed amide/carbamate substrate,16 we applied the Hendrickson protocol and observed a single imidazoline product that was retrieved in 88% isolated yield.
At present, the exact mass for Nutlin-3 determined by high resolution mass spectrometry is the only available analytical data for comparison. As a result, we sought evidence to support whether the desired imidazoline (Nutlin-3) or an alternative isomer (24) formed during this dehydrative cyclization. Support for the imidazoline depicted for Nutlin-3 was obtained by an HMBC (600 MHz) experiment. Among the 3 JHC couplings observed was the diagnostic crosspeak to the urea carbon from H1 of the imidazoline; a crosspeak between H8 and an amide carbonyl carbon was not observed. Nutlin-3 produced using this synthesis and catalyst 8d is levorotatory.
Conclusion
In conclusion, we have developed the first highly diastereo- and enantioselective aza-Henry additions using aryl nitromethanes. This transformation was used to prepare a key cis-stilbene diamine intermediate that served as a precursor to the potent chemotherapeutic (–)-Nutlin-3. As a consequence of this synthesis, complete spectroscopic and analytical data for synthetic intermediates and (–)-Nutlin-3 are available.17 This synthesis removes the current reliance on preparatory chromatography using a chiral stationary phase and substitutes it with a readily prepared chiral catalyst that furnishes intermediate 1a with high stereocontrol. This improved accessibility of cis-imidazolines may stimulate their broader use as probes, if not provide a practical synthesis to fuel drug development.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM 084333). We are grateful to Dr. Maren Pink (Indiana University Molecular Structure Center) for determination of absolute and relative configuration by X–ray diffraction for a related addition product. We are grateful to Clayton Marshall and Dr. Jennifer Pietenpol (VICC) for confirming the activity of both (–)- and (+)-Nutlin-3 (synthetic).
Footnotes
Electronic Supplementary Information (ESI) available: Complete experimental details and analytical data for all new compounds, as well as a summary of the HLR synthesis of rac-Nutlin-3.
Notes and References
- 1.Reviews: Marques-Lopez E, Merino P, Tejero T, Herrera RP. Eur J Org Chem. 2009:2401.Akiyama T, Itoh J, Fuchibe K. Adv Synth Catal. 2006;348:999.Ting A, Schaus SE. Eur J Org Chem. 2007:5797.Westermann B. Angew Chem Int Ed. 2003;42:151. doi: 10.1002/anie.200390071.Selected examples: Adams H, Anderson JC, Peace S, Pennell AMK. J Org Chem. 1998;63:9932.Yamada K, Harwood SJ, Groger H, Shibasaki M. Angew Chem Int Ed. 1999;38:3504. doi: 10.1002/(sici)1521-3773(19991203)38:23<3504::aid-anie3504>3.0.co;2-e.Knudsen KR, Risgaard T, Nishiwaki N, Gothelf KV, Jorgensen KA. J Am Chem Soc. 2001;123:5843. doi: 10.1021/ja010588p.Okino T, Nakamura S, Furukawa T, Takemoto Y. Org Lett. 2004;6:625. doi: 10.1021/ol0364531.Bernardi L, Fini F, Herrera RP, Ricci A, Sgarzani V. Tetrahedron. 2005;62:375.Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A. Angew Chem Int Ed. 2005;44:7975. doi: 10.1002/anie.200502646.Palomo C, Oiarbide M, Laso A, Lopez R. J Am Chem Soc. 2005;127:17622. doi: 10.1021/ja056594t.Xu X, Furukawa T, Okino T, Miyabe H, Takemoto Y. Chem--Eur J. 2006;12:466. doi: 10.1002/chem.200500735.Rampalakos C, Wulff WD. Adv Synth Catal. 2008;350:1785. doi: 10.1002/adsc.200800214.
- 2.For reviews describing vic-diamines and their preparation, see: Lucet D, Le Gall T, Mioskowski C. Angew Chem Int Ed. 1998;37:2580. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2580::AID-ANIE2580>3.0.CO;2-L.Faugeroux V, Genisson Y. Curr Org Chem. 2008;12:751.Kotti S, Timmons C, Li GG. Chem Biol Drug Des. 2006;67:101. doi: 10.1111/j.1747-0285.2006.00347.x.Mortensen MS, O'Doherty GA. Abstracts of Papers of the American Chemical Society. 2005;230:U3433.For metal-catalyzed diamination reactions of alkenes, see: Cardona F, Goti A. Nature Chemistry. 2009;1:269. doi: 10.1038/nchem.256.
- 3.Enantioselective additions of aryl nitromethane additions to azomethine (both with α-imino esters): 2:1 dr, 84% ee, 64% yield – Rueping M, Antonchick AP. Org Lett. 2008;10:1731. doi: 10.1021/ol8003589.1.2:1 dr, 74/77% ee, 59% yield – Nishiwaki N, Knudsen KR, Gothelf KV, Jorgensen KA. Angew Chem Int Ed. 2001;40:2992. doi: 10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3.
- 4.Kornblum N, Larson HO, Blackwood RK, Mooberry DD, Oliveto EP, Graham GE. J Am Chem Soc. 1956;78:1497. [Google Scholar]
- 5.Marianacci O, Micheletti G, Bernardi L, Fini F, Fochi M, Pettersen D, Sgarzani V, Ricci A. Chemistry-a European Journal. 2007;13:8338. doi: 10.1002/chem.200700908. [DOI] [PubMed] [Google Scholar]
- 6.Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; Davis TA, Wilt JC, Johnston JN. J Am Chem Soc. 2010;132:2880. doi: 10.1021/ja908814h. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wilt JC, Pink M, Johnston JN. Chem Commun. 2008:4177. doi: 10.1039/b808393b. [DOI] [PubMed] [Google Scholar]; Singh A, Johnston JN. J Am Chem Soc. 2008;130:5866. doi: 10.1021/ja8011808. [DOI] [PubMed] [Google Scholar]; Shen B, Johnston JN. Org Lett. 2008;10:4397. doi: 10.1021/ol801797h. [DOI] [PubMed] [Google Scholar]; Singh A, Yoder RA, Shen B, Johnston JN. J Am Chem Soc. 2007;129:3466. doi: 10.1021/ja068073r. [DOI] [PubMed] [Google Scholar]; Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418. doi: 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]
- 7.See the Supporting Information for methods used to assign absolute and relative configuration for addition products 1. The absolute configuration is also consistent with all other cases using nitroalkanes and bis(amidine) catalysts (ref. 6) and with suggestions of the absolute configuration made by HLR (ref. 17).
- 8.Our first exception to this was recently published: Dobish MC, Johnston JN. Org Lett. 2010;12:5744. doi: 10.1021/ol1025712.
- 9.Overview: Harris CC. Proc Natl Acad Sci U S A. 2006;103:1659. doi: 10.1073/pnas.0510948103.Reviews: Fischer PM, Lane DP. Trends Pharmacol Sci. 2004;25:343. doi: 10.1016/j.tips.2004.04.011.Bond GL, Hu WW, Levine AJ. Curr Cancer Drug Targets. 2005;5:3. doi: 10.2174/1568009053332627.
- 10.Lowe SW, Cepero E, Evan G. Nature. 2004;432:307. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 11.Over 1000 publications have cited the HLR paper in the intervening six years: Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472.Additional selected references: Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472.Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Cancer Res. 2005;65:1918. doi: 10.1158/0008-5472.CAN-04-3576.Fry DC, Graves B, Vassilev LT. Methods Enzymol. 2005;399:622. doi: 10.1016/S0076-6879(05)99041-1.Fry DC, Graves BJ, Vassilev LT. Protein-Protein Interact. (2nd) 2005:893.Vassilev LT. Cell Cycle. 2004;3:419.
- 12.See Supporting Information for details of the HLR synthesis.
- 13.Kong N, Liu EA, Vu BT. WO 03/051360 A1. 2003 [Google Scholar]
- 14.Satoh T, Suzuki S, Suzuki Y, Miyaji Y, Imai Z. Tetrahedron Lett. 1969:4555. [Google Scholar]
- 15.Hendrickson JB, Schwartzman SM. Tetrahedron Letters. 1975;16:277. [Google Scholar]; Hendrickson JB, Hussoin MS. J Org Chem. 1989;54:1144. [Google Scholar]; Hendrickson JB, Hussoin MS. J Org Chem. 1987;52:4137. [Google Scholar]
- 16.Leading references: You SL, Kelly JW. Org Lett. 2004;6:1681. doi: 10.1021/ol049439c.You SL, Razavi H, Kelly JW. Angew Chem Int Ed. 2003;42:83. doi: 10.1002/anie.200390059.Petersson MJ, Jenkins ID, Loughlin WA. Org Biomol Chem. 2009;7:739. doi: 10.1039/b818310d.Petersson MJ, Jenkins ID, Loughlin WA. J Org Chem. 2008;73:4691. doi: 10.1021/jo800447v.
- 17.Absolute configuration has been suggested in the patent literature: Haley GJ, Kong N, Liu EA, Vu BT. WO 2005/123691 Al. 2005
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.