

Abstract
Background
Nitric oxide (NO) and its oxidative reaction products have been repeatedly shown to block steroid receptor function via nitrosation of zinc finger structures in the DNA-binding domain (DBD). In consequence NO-donors could be of special interest for the treatment of deregulated androgen receptor(AR)-signaling in castration resistant prostate cancer (CRPC).
Methods
Prostate cancer (PCa) cells were treated with JS-K, a diazeniumdiolate derivate capable of generating large amounts of intracellular NO following activation by glutathione S-transferase. Generation of NO was determined indirectly by the detection of nitrate in tissue culture medium or by immunodetection of nitrotyrosine in the cytoplasm. Effects of JS-K on intracellular AR-levels were determined by western blotting. AR-dimerization was analyzed by mammalian two hybrid assay, nuclear translocation of the AR was visualized in PCa cells transfected with a green fluorescent AR-Eos fusion protein using fluorescence microscopy. Modulation of AR- and WNT-signalling by JS-K was investigated using reporter gene assays. Tumor cell proliferation following JS-K treatment was measured by MTT-Assay.
Results
The NO-releasing compound JS-K was shown to inhibit AR-mediated reporter gene activity in 22Rv1 CRPC cells. Inhibition of AR signaling was neither due to an inhibition of nuclear import nor to a reduction in AR-dimerization. In contrast to previously tested NO-donors, JS-K was able to reduce the intracellular concentration of functional AR. This could be attributed to the generation of extremely high intracellular levels of the free radical NO as demonstrated indirectly by high levels of nitrotyrosine in JS-K treated cells. Moreover, JS-K diminished WNT-signaling in AR-positive 22Rv1 cells. In line with these observations, castration resistant 22Rv1 cells were found to be more susceptible to the growth inhibitory effects of JS-K than the androgen dependent LNCaP which do not exhibit an active WNT-signaling pathway.
Conclusions
Our results suggest that small molecules able to inhibit WNT- and AR-signaling via NO-release represent a promising platform for the development of new compounds for the treatment of CRPC.
Background
Nitric oxide (NO), a free radical gas, is a pleiotropic molecule critical to a number of physiological and pathological processes. NO-releasing drugs are a growing class of promising new therapeutics with applications in a large variety of diseases like cardiovascular and respiratory disorders, osteoporosis, Alzheimer's disease, inflammatory lesions and urinary incontinence [1-4]. Moreover there is increasing evidence that NO donors could have potential in the prevention and therapy of various malignant tumors like myeloma, breast cancer, ovarian cancer, pancreatic cancer or prostate cancer (PCa) [5-11].
PCa is the most commonly diagnosed neoplasm in elderly men and the second cause of cancer related deaths in the Western world [12]. Current treatment for advanced PCa is mainly based on androgen ablation therapies like orchiectomy, systemic administration of LHRH analog/blocker or anti-androgens. Unfortunately, the benefit of androgen ablation is only transitory. Within a few years many PCa progress to a state of the disease termed castration resistant prostate cancer (CPRC) where tumor cells grow and survive under subphysiological levels of androgens [13]. Although in vitro the development of a castration resistant phenotype is mostly based on the loss of the AR in PCa cells, several clinical studies demonstrated that the AR is rarely lost in CRPC cells in vivo [14-16]. Indeed, CRPC cells continue to depend on AR-signalling but bypass the requirements for physiological levels of circulating androgens. Various molecular mechanisms that promote AR-dependent growth of CRPC cells growing under androgen deprived conditions have been identified: over-expression/amplification of the AR (hypersensitive pathway), AR mutations that broaden ligand specificity (promiscious pathway), AR-activation by non steroid ligands like growth factors or cytokines (outlaw pathway) [17] as well as the expression of C-terminally truncated AR variants lacking vast parts of the ligand binding domain (LBD). These AR-variants, termed ARΔLBD, are either products of alternative splicing (AR-V), point mutations leading to premature stop codons or proteolytic cleavage of the AR protein [18-21]. In contrast to a full length AR which is activated upon androgenic stimuli, previous in vitro studies were able to show that most ARΔLBDs, devoid of a ligand binding domain, are constitutively active [18-21]. As ARΔLBDs lack most parts of the LBD situated in the C-terminus of the AR, they are insensitive to any form of androgen ablation.
Nitric oxide (NO) and its oxidative reaction products have been repeatedly shown to block nuclear receptors via nitrosation of their zinc finger structures in the DNA-binding domain (DBD). The DBD is an essential part of functional full length AR as well as the constitutively active ARΔLBDs. In consequence NO-donors could be of special interest for the treatment of deregulated AR-signalling in CRPC cells. Inhibition of AR-functions following treatment with the long living spontaneous NO-donor (Z)-1-[N-(2-Aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolat (DETA/NO) has recently been demonstrated [22]. However, due to the short half-life of the free radical NO in cell culture medium, relatively high concentrations of DETA/NO were necessary to induce effects on intracellular nuclear receptors. In order to generate large amounts of intracellular NO, new compounds, able to deliver NO in a spatially controllable manner, are needed. In our study we used the non-ionic, nitroaromatic diazeniumdiolate JS-K, a glutathione S-transferase(GST)-activated NO-prodrug able to generate high intracellular levels of NO in the micromolar range [23,24].
Studying the effects of JS-K on AR- and WNT-signalling in the AR-positive PCa cell lines 22Rv1 and LNCaP we were able to demonstrate that only a 100-fold lower concentration of the GST-activated JS-K are necessary to reach comparable biological effects in PCa cells than the commonly used DETA/NO that spontaneously generates NO in physiological fluids. Our data show that NO-prodrugs, capable of generating large intracellular amounts of NO, may be suitable for the treatment of advanced prostate cancer.
Methods
Plasmids, antibodies, chemicals
Probasin promoter driven luciferase-reporter plasmid (pGL3E-Probasin) was kindly provided by Prof. Dr. Z. Culig (Innsbruck, Austria). The pARt1EosFP expression plasmid coding for a green fluorescent AR-Eos fusion protein (AR-EosFP) was a gift from Prof. F. Oswald (Ulm, Germany). The CMV-promoter driven β-catenin expression plasmid pbCAT, containing human β-catenin cDNA with an activating S33Y mutation was provided by Dr. H. Clevers (Utrecht, The Netherlands). TCF-reporter plasmids pTopFlash (TOP) and pFopFlash (FOP), containing three copies of wild-type or mutant TCF-binding sites upstream of a thymidine kinase minimal promoter driving a luciferase gene, were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Renilla reniformis luciferase reporter plasmid pRL-tk-LUC was a product of Promega (Mannheim, Germany). Mouse monoclonal antibodies directed against beta actin (ab8224) and nitrotyrosine (ab7048) were products of Abcam (Cambridge, UK). Mouse monoclonal antibody AR441 directed against the N-terminus of the AR was purchased from Dako (Hamburg, Germany). The NO-donors O2-(2,4-Dinitrophenyl)-1-[(4-ethoxycarbo-nyl)piperazin-1-yl]diazen-1-ium-1,2-diolate (JS-K), (Z)-1-[N-(2-Aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA/NO) and the androgen dihydrotestosterone (DHT) were a products of Sigma Aldrich (Taufkirchen, Germany).
Cell culture
22Rv1, LNCaP, DU-145 and PC-3 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). LNCaP-SSR, a castration resistant LNCaP subline [25,26], was a generous gift from Prof. Martin Burchardt, Greifswald, Germany. 22Rv1, LNCaP, DU-145 and PC-3 were grown in RPMI-1640 (PAA Laboratories GmbH, Pasching, Austria) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin (BioWest, Nuaille, France). LNCaP-SSR were routinely grown in RPMI-1640, 1% Penicillin/Streptomycin supplemented with 10% steroid free, dextran charcoal treated FBS (FBSdcc, BioWest, Nuaille, France) [26].
Transfection and reporter gene assays
22Rv1 cells were grown on 24-well plates (Sarstedt, Nümbrecht, Germany). Transfections were performed using FuGene HD transfection reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. AR- and WNT-specific reporter gene assays were performed as recently described [22,27]. In brief: (1) AR-signalling: Cells were transfected with 200 ng/well of the AR-dependent reporter-plasmid pGL3E-Probasin. The pRL-tk-LUC vector coding for a Renilla luciferase under control of a constitutively active thymidine kinase promoter was co-transfected (80 ng/well) to correct for transfection efficiency. After transfection cells were grown in RPMI 1640 with 5% dextran charcoal treated steroid free, dextran charcoal treated FBS (FBSdcc; Biowest, Nuaillé, France) and treated with 5 nM DHT and different concentrations of JS-K. (2) WNT-signalling: Luciferase reporter plasmids pTopFlash (250 ng/well) and pFopFlash (250 ng/well) were mixed either with pbCAT expression vector (250 ng/well) or insert-free vector (250 ng/well). pRL-tk-luc (80 ng/well) was co-transfected to correct for transfection efficiency. After an incubation period of 12 h in RPMI-1640 with 5% FBSdcc, medium was changed to RPMI-1640 containing 2.5% FBSdcc with increasing concentrations of JS-K.
AR- and WNT-reporter activities were analyzed after 24 hours using the Dual Luciferase Reporter Assay System (Promega, Mannheim, Germany). Experiments were repeated at least three times and performed in triplicates, unless stated otherwise.
Mammalian two hybrid assay
To investigate the dimerization of the AR we used the CheckMate/FlexiVector Mammalian Two-Hybrid System from Promega (Mannheim, Germany). VP16 activation domain (pFN10A-AR) and Gal4 DNA binding domain tagged androgen receptor fusion constructs (pFN11A-AR) were cloned according to the supplier's suggestions by using pSG5-AR as template. DU-145 and PC-3 cells were grown in 24-well plates and co-transfected with 150 ng/well of pFN10A-AR, pFN11A-AR and the Gal4 reporter construct pGL4.31. After transfection, cells were cultured and luciferase activities were determined as recently described [28].
Nuclear Translocation assay
Nuclear translocation of the AR was analyzed in AR-negative DU-145 and PC-3 cells transfected with the green fluorescent AR-EosFP [22,29]. Therefore, prostate cancer cells were transfected in a 24-well plate with pAR-t1EosFP (0.25 μg/well) for 4 hours in RPMI and allowed to grow for another 24 hours in RPMI 1640 supplemented with 5% FBSdcc and antibiotics. Thereafter, cells were grown in RPMI supplemented with 2.5% FBSdcc in the presence/absence of 5 nM DHT and JS-K for 6 hours. Localization of the AR-fusion protein was subsequently analyzed by fluorescence microscopy.
Western Blot analysis
Proteins were extracted from cells using RIPA buffer as recently described [30]. Proteins (15 μg lysate) were separated by Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-Page). Subsequently proteins were transferred onto a nitrocellulose membrane (BioTraceNT, Pall Life Sciences, Dreieich, Germany) by tank blotting (transfer buffer: 20 mM Tris/HCl, pH 8.7, 150 mM glycine and 20% (v/v) methanol). Membranes were blocked in phosphate buffered saline with 0.1% Tween20 (v/v) (PBS-T) and 5% BSA (w/v) for 1 hour at room temperature. The membrane was incubated with the primary antibody in PBS-T with 1% BSA over night at 4°C. All primary antibodies were used in a dilution of 1:1000, with the exception of the beta-actin antibody, which was diluted 1:20000. The membrane was washed with PBS-T three times before incubating with the peroxidase-coupled secondary antibody in a dilution of 1:2000 in PBS-T with 1% BSA. Signals were visualized by the SuperSignal West Pico Chemiluminescent Substrate from Pierce (Rockford, USA).
Determination of NO
NO was determined indirectly by the photometric measurement of nitrite according to Green et al. [31]
Immunocytochemistry
For immunocytochemistry 22Rv1 cells were cultured on glass coverslips (BD Falcon Culture Slides, BD Biosciences) washed (2 × 5 min) with PBS and fixed with 3.7% paraformaldehyd for 15 min at room temperature, washed (3 × 10 min) in PBS and permeabilized in 0.2% Triton X-100 with 1% BSA in PBS for 5 min at room temperature. The cells were washed (3 × 10 min) with PBS containing 1% BSA. Cells were then incubated with the monoclonal anti-tyrosine antibody (1:25 in PBS, 1% BSA for 30 min at 37°C, washed (3 × 10 min and incubated with the mouse monoclonal anti-goat antibody labeled with Alexa Fluor 488 (Invitrogen, Karlsruhe, Germany, 1:100) for 30 min at 37°C. Cells were then washed (3 × 10 min), the nuclei stained with DAPI (2.5 μg/ml in PBS) for 15 min, washed (3 × 10 min) and embedded with glycine and polyvinyle alcohol 4-88 (Fluka Analytical, Deisenhofen, Germany).
Cell viability
Cell viability was determined by means of a colorimetric MTT-assay measuring the reduction of the water soluble tetrazolium bromide into its insoluble formazan derivative by functional mitochondria [32,33].
Statistical Analysis
Data are reported as means ± standard deviation. Analysis was performed with Student's T-test (two tailed for independent samples) with p < 0.05 considered as significant.
Results
JS-K induces nitrosation of tyrosine residues in cellular proteins
In a previous paper NO generated by DETA/NO was shown to nitrosate tyrosine residues in proteins to nitrotyrosine [22]. Therefore, we tested JS-K's ability to nitrosate proteins via intracellular NO-release. In order to generate a control substance without the NO-releasing moiety, we took advantage of the observation that sera of patients were repeatedly shown to possess low GST-activity [34,35]. In our experiment 5 μM JS-K was incubated at 37°C in RPMI-1640 supplemented with 10% FBS. Generation of NO in the medium was measured indirectly by the determination of nitrite. Under these conditions maximal medium nitrite levels were detectable after nine to twelve hours (Figure 1A). The remaining inactive JS-K metabolites S-(2, 4-Dinitrophenyl)-glutathione and 4-carboxy-piperazine [23] were subsequently used as a negative control and termed JS-Kneg.
Figure 1.

Release of NO from JS-K. (A) Generation of NO from JS-K in RPMI-1640 medium containing 10% fetal bovine serum was determined indirectly by photorimetric detection of nitrite according to Green et al. [31]. (B) Detection of nitrotyrosine by fluorescence microscopy in 22Rv1 cells: Generation of intracellular NO was detected indirectly by detection of nitrotyrosine (green). Cell nuclei were stained with DAPI. [A] untreated cells, [B] 2 μM JS-Kneg, [C] 2 μM JS-K, [D] 200 μM DETA/NO.
Incubation with 2 μM JS-K as well as 200 μM DETA/NO (serving as positive control) led to a nitrosation of intracellular proteins as shown by fluorescescence microscopy of nitrotyrosine residues (Figure 1B). In contrast, neither untreated cells nor cells treated with the control solution JS-Kneg harbouring the inactive JS-K metabolites, showed any nitrotyrosine staining (Figure 1B).
JS-K inhibits AR-mediated genomic function in 22Rv1 cells
To test whether JS-K influences the transcriptional activity of the AR, we transiently co-transfected AR-positive 22Rv1 human prostate cancer cells with pGL3E-Pro, an androgen-responsive probasin-promoter driven reporter plasmid, and pRL-tk-Luc, a Renilla luciferase plasmid under control of a constitutively active thymidine kinase promoter (control of transfection efficiency). Subsequently cells were incubated for 44 hours with 5 nM DHT and increasing concentrations of JS-K or JS-Kneg. JS-K significantly inhibited DHT-induced AR-transactivation in a dose-dependent manner. Inhibition was already significant at a concentration of 2 μM JS-K. In contrast, JS-Kneg was unable to diminish AR transcriptional activity even at higher concentrations (Figure 2).
Figure 2.

JS-K inhibits AR-dependent reportergene activity in 22Rv1 cells. Cells were cotransfected with a probasin reporter gene plasmid and a Renilla luciferase plasmid serving as transfection control. Subsequently cells were grown in presence/absence of 5 nM DHT. Reportergene activity was determined using the Dual Luciferase Assay System from Promega. Data are presented as fold of untreated controls ± standard deviation (SD). Results are mean values of three independent experiments performed in quadruplicates: *p < 0.05; **p < 0.01.
JS-K does not modulate dimerization or nuclear localization of the AR
In vitro NO has been shown to repress vitamin D3 signalling through inhibition of vitamin D receptor (VDR) - retinoic X receptor (RXR)-heterodimerization [36]. To test whether JS-K can inhibit AR-homodimer formation in vivo, we performed an AR-specific mammalian two hybrid (M2H)-assay in AR-negative PC-3 and DU-145 cells (Figure 3, Additional File 1). As seen in Figure 3, JS-K was unable to repress DHT-induced AR-homodimerization at concentrations that were already sufficient to inhibit AR-transactivation (Figures 3 and 2).
Figure 3.
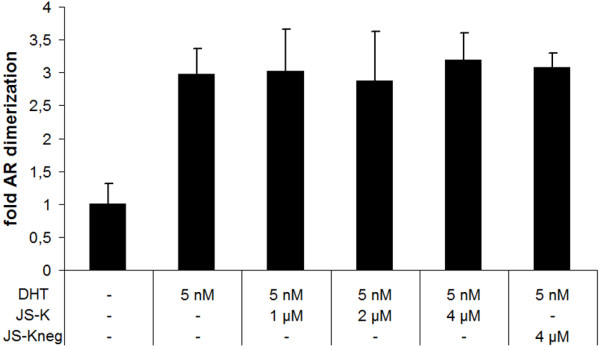
JS-K does not inhibit AR-dimerization in presence of androgens. AR-dimerization was determined in PC-3 cells using the CheckMate Mammalian Two-Hybrid System from Promega as described in Material and Methods. Results are mean values of four independent experiments performed in quadruplicates.
In order to test whether high NO concentrations are able to influence the nuclear translocation of the AR, we transfected the AR-negative PC-3 with a construct coding for a green fluorescent AR-Eos fusion protein (AR-EosFP) [22,37]. Subsequently cells were grown for six hours in the presence/absence of 5 nM DHT with or without JS-K or JS-Kneg. Under hormone-free conditions the AR was predominantly located in the cytoplasm (> 80%) whereas in presence of DHT the AR was transported to the nucleus (Figure 4). As seen in Figure 4 addition of JS-K (final concentration 4 μM) was unable to reverse the nuclear localization of the AR in DHT treated PC-3 cells (82% ± 2% to 78 ± 2%). In a complementary experiment transfection of AR negative DU-145 cells with AR-EosFP yielded similar results (Additional File 2).
Figure 4.

NO does not influence hormone-induced nuclear translocation of the AR. Prostate cancer cells (PC-3) expressing a green fluorescent AR-EosFP [37] were incubated for 6 hours in the absence/presence of DHT and JS-K/JS-Kneg. Intracellular localization of AR-EosFP was determined by fluorescence microscopy: (A) untreated controls, (B) 5 nM DHT, (C) 5 nM DHT and 4 μM JS-K, (D) 5 nM DHT and 4 μM JS-Kneg. Bars: Data presented in % cellular localization ± SD. Results are mean values of three independent experiments performed in quadruplicates.
JS-K leads to a down-regulation of the AR-protein
In contrast to the commonly used spontaneous NO-donor DETA/NO which generates NO in aquaeous solutions, JS-K is able to generate high amounts of NO within target cells after enzymatic activation by GST. The profound differences in local NO-production prompted us to analyze the effects of JS-K on the AR-concentration in the prostate cancer line 22Rv1 and LNCaP. Both cell lines express functional AR-proteins. Whereas 22Rv1 cells express a 120 kDa AR-isoform as well as a C-terminally truncated, constitutively active 79 kDa AR-V splicing variant [38] the LNCaP cells express a full length AR (119 kDa) with a point mutation at position 877 (T877A) [39]. Treatment with JS-K for 30 hours diminished the amount of all AR-forms in 22Rv1 as well as in LNCaP cells (Figure 5). The downregulation of the AR in LNCaP was less pronounced than the dramatic decrease of AR and AR-V in 22Rv1 cells (Figure 5). In both cell lines incubation with JS-Kneg had no influence on intracellular AR-levels (Figure 5).
Figure 5.
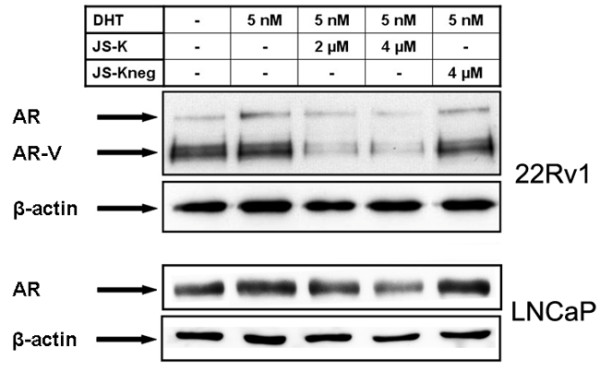
Influence of JS-K and JS-Kneg on the androgen receptor concentration in human prostate cancer cells. 22Rv1 and LNCaP cells were incubated for 30 hours with or without 5 nM dihydrotestosterone in the presence of JS-K or JS-Kneg. Subsequently cells were lysed and separated by SDS-electrophoresis (15 μg protein per lane). Proteins were transferred onto a nitrocellulose membrane. AR and β-actin (loading control) were visualized by immunodetection.
JS-K inhibits WNT/β-catenin signaling in 22Rv1 cells
Interaction of the canonical WNT/β-Catenin pathway with the AR is thought to promote progression of PCa to the terminal castrate-resistant stage [40]. Crosstalk between the canonical WNT and AR-pathways occurs at several levels: (a) β-Catenin interacts with the AR to increase its transcriptional activity [27], (b) WNT-signalling influences AR-signalling through its ability to regulate AR-mRNA as well as AR-stability [41].
Several lines of evidence indicate that NO is involved in the regulation of the WNT/β-catenin pathway in various tumors [42-44]. A common approach to analyze the canonical WNT-pathway in vitro is the over-expression of mutated stabilized β-catenin (S33Y) that activate TCF/LEF-dependent reporter gene constructs [27]. As LNCaP cells are unable to activate β-catenin dependent reporter gene expression [27] we tested the effects of JS-K on the canonical WNT-pathway in 22Rv1 cells. As seen in Figure 6, JS-K was able to diminish β-catenin signalling in the CRPC cell line 22Rv1 in a dose dependent manner. Down-regulation of the canonical WNT-pathway was already significant at JS-K concentrations of 1 μM (reduction of transcriptional activity: 42 ± 8%, p < 0.01).
Figure 6.
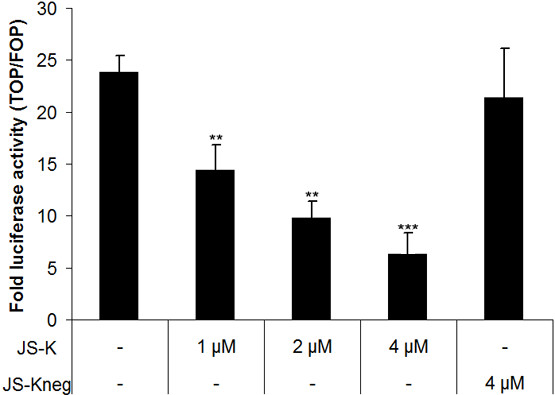
JS-K diminishes WNT-signalling in 22Rv1 cells. 22Rv1 cells were co-transfected with an expression vector for mutated, stabilized β-catenin together with either the TCF reporter construct TOP or FOP as recently described [25]. Data are presented as fold of untreated controls (TOP/FOP) ± SD, **p < 0.01, ***p < 0.001.
Growth inhibitory effects of JS-K are most pronounced in castration resistant prostate cancer cells
Based on our observations we tested the antiproliferative effects of JS-K on prostate cancer cell lines. JS-Kneg served as negative control. Cellular proliferation was assessed by means of a colorimetric MTT assay, measuring the reduction of tetrazolium salts to formazan derivatives by functional mitochondria [32,33]. Although redox sensitive viability assays have some limitations depending on the chemical nature of the compounds to be evaluated [45] they are widely used for measuring cell viability following nitrosative or oxidative stress [22,46,47]. The suitability of this method was furthermore documented by a previous study analyzing the effects of the spontaneous NO-donor DETA/NO on the viability of PCa-cells. Comparison of MTT with three commonly used methods to measure cell proliferation, i.e quantification of intracellular ATP, trypan blue dye exclusion and neutral red assay were found to generate similar results [22].
A first series of experiments indicated that castration resistant 22Rv1 cells were more susceptible to the growth inhibitory effects of JS-K than the androgen sensitive LNCaP cells (Figure 7A). Maximal growth inhibitory effects were achieved at a concentration of 4 μM JS-K for 22Rv1 (growth inhibition 50 ± 6%, p < 0.05) and 4 μM JS-K for LNCaP (growth inhibition 23 ± 9%).
Figure 7.

Effects of JS-K on the proliferation of prostate cancer cells. (A) 22Rv1 cells are more susceptible to the growth inhibitory effects of JS-K than the androgen sensitive LNCaP cells Cells were cultured in the absence/presence of increasing concentrations of JS-K (1,2,4 μM) or JS-Kneg (4 μM). Cell viability was determined after 96 hours by a colorimetric MTT assay. Results are mean values of four independent experiments performed in quadruplicates. Data are presented as % of untreated controls ± SD, *p < 0.05. (B) Effects of JS-K on the AR negative DU-145 and the AR-positive LNCaP (hormone sensitive) and LNCaP-SSR (castration resistant). Cells were cultured in the absence/presence of increasing concentrations of JS-K (1, 2, 4 μM) or JS-Kneg (4 μM). Cell viability was determined after 96 hours by a colorimetric MTT assay. Results are mean values of four independent experiments performed in quadruplicates.
Given the highly divergent nature of the AR-positive cell lines 22Rv1 and LNCaP we repeated the proliferation experiments using the androgen sensitive LNCaP and LNCaP-SSR, an AR-positive castration resistant LNCaP-subline. The AR-negative DU-145 served as control (AR off-target effects). Interestingly, the castration resistant LNCaP-SSR cells were more susceptible to the growth inhibitory effects (growth inhibition at 1 μM JS-K 31 ± 2%, p < 0.01) than the parental LNCaP cells (growth inhibition at 4 μM JS-K 33 ± 1%, p < 0.01) (Figure 7B). In contrast to the AR-positive LNCaP and LNCaP-SSR the AR-negative DU-145 were less susceptible to the growth inhibitory effects of JS-K (growth inhibition at 4 μM JS-K 17 ± 9%).
Discussion
Current treatment for advanced PCa is mainly based on androgen ablation therapies like chemical or surgical castration or application of anti-androgens. Although hormonal therapy is initially very effective, the benefit of androgen withdrawal is only transitory. While still expressing high levels of functional AR, some PCa cells acquire the ability to grow and survive under castrate levels of circulating androgens. The mechanisms involved in an altered AR-signalling of CRPC cells include an over-expression/amplification of the AR (hypersensitive pathway), point mutations broadening ligand-specificity of the receptor (promiscuous pathway) as well as activation of the AR by peptide growth factors or cytokines (outlaw pathway) [17]. A recently identified mechanism enabling CRPC cells to bypass the requirements for physiological levels of androgens is the expression of C-terminal truncated, constitutively active AR splicing variants termed AR-V. Due to the loss of the C-terminus, AR-Vs are lacking essential parts of the ligand binding domain [21]. In the absence of androgens, AR-Vs were shown to induce AR-signalling either as AR-V/AR-V-homodimers or AR-V/AR-heterodimers [18,48]. Whereas AR-V/AR-V homodimers are unaffected by hormone ablation in general, some AR-V/AR-heterodimers were insensitive to the commonly used anti-androgens [48]. Given the fact that both, AR wild type as well as AR-V, play a crucial role in a high percentage of CRPC there is an urgent need for an AR-blockade that does not involve the LBD.
Nitric oxide, a free radical gas, has been repeatedly shown to impair the function of nuclear receptors via targeting their cys4-type zinc fingers, located in the DBD of these receptors [49]. This is well exemplified by studies showing a NO-induced nitrosation of the cystein residues within the zinc finger structures of steroid receptors like the estrogen receptor (ER) [50]. As the DBD is a crucial part of functional full length AR as well as constitutively active AR-Vs, it is tempting to speculate that NO-donors able to release NO in a spatial and timely controllable manner could be of special interest for the treatment of deregulated AR-signalling in CRPC cells.
Inhibitory effects of NO on AR-function in PCa cells was first described in vitro using the spontaneous NO-donor Deta/NO [22]. In a recent clinical study Siemens et al. were able to show an increase of the PSA doubling time in patients treated with the NO-donor glyceryl trinitrate (GTN) [51]. No cardiovascular toxicities or serious side effects were encountered during treatment with GTN [51]. Although the number of patients enrolled in the study was relatively low (29 patients), our data and the clinical data with GTN [22,51] support the assumption that NO might be suitable for the treatment of advanced PCa.
Commonly used NO-Donors like Deta/NO, spontaneously dissociate in a pH dependent process thereby releasing the free radical NO. Unfortunately NO exhibits only a relatively short half-life in the tissue culture medium or body fluids. Therefore, we tested a potent NO-prodrug, the non-ionic nitroaromatic diazeniumdiolate JS-K. The compound was shown to be activated by glutathione S-transferase (GST), a key phase II detoxification enzyme that is frequently over-expressed in cancer tissue. Among the various isoforms of GSTs which are located in the cytoplasm and the mitochondria, mainly the π isoform is able to form the so called < Meisenheimer complex >[52] which finally delivers NO directly within the cell. First experimental in vivo studies using tumor xenografts in a mouse model, showed that JS-K exhibits a strong anti-tumor activity and is generally well tolerated by the host organism [5,23,24].
In order to test the effects of JS-K on AR signalling, we treated the AR-positive CRPC cell line 22Rv1 grown in presence of DHT with increasing concentrations of JS-K. Generation of intracellular NO was monitored indirectly by detection of nitrotyrosine using fluorescence microscopy. JS-K was able to inhibit the genomic functions of the AR in 22Rv1 cells. The inhibition of AR-signalling was neither due to an inhibition of AR-dimerization nor to an inhibition of nuclear translocation. Interestingly, JS-K was able to diminish intracellular levels of all AR-isoforms in PCa cells. However, in contrast to the dramatic downregulation of AR-V and AR in 22Rv1 cells the intracellular AR levels of LNCaP were only slightly affected.
So far, studies on intracellular steroid receptor levels following NO-treatment are sparse. In a previous study using Deta/NO as an NO-donor the concentration of the endogenous AR in LNCaP remained unaffected [22]. In contrast, DETA/NO was able to downregulate the intracellular levels of the Drosophila melanogaster ecdysteroid receptor (EcR) when overexpressed in chinese hamster ovary cells, CHO-K1 [53]. Although the heterologeous expression of an insect steroid receptor in CHO-K1 cells differs largely from the situation found in LNCaP cells that express high levels of intracellular AR under physiological conditions the present data suggest that there are cell specific differences in the sensitivity of cells towards NO-treatment. Moreover, the generation of NO by JS-K largely depends on the intracellular GST-activity that differs between different cell lines or cell types.
The fact that NO is able to inhibit the canonical WNT-pathway in different tumor cells [42-44] including prostate cancer cells (as seen in this study) offers a more intriguing explanation for the massive down regulation of AR and AR-V in 22Rv1 cells. There is experimental evidence that the WNT/β-catenin-pathway is able to up-regulate AR-mRNA expression in prostate cancer cell lines through interaction with TCF/LEF binding sites situated in the promoter region of the AR [41]. In contrast to the highly aggressive castration resistant 22Rv1 cells which are able to drive a WNT-typical T-cell factor (TCF)-dependent reporter gene activity, the androgen sensitive LNCaP are unable to do so [27]. In consequence, the inhibition of the canonical WNT-pathway would lead to a decrease of AR and AR-V mRNA in 22Rv1 cells but not or to a lesser extent in LNCaP.
In proliferation assays castration resistant 22Rv1 and LNCaP-SSR cells were more susceptible to the growth inhibitory effects of JS-K than LNCaP. Whereas LNCaP cells largely depend on androgenic stimuli, the 22Rv1 and LNCaP-SSR cells are able to grow under androgen deprived conditions. LNCaP cells express a full length AR with a point mutation at position 877 (T877A), enabling the AR to be stimulated by different steroids (promiscuous AR) [39]. In contrast to the hormone dependent LNCaP, the castration resistant LNCaP-SSR cells were shown to exhibit high levels of nuclear AR in the absence of hormonal stimuli [26]. Increased nuclear AR-levels were paralleled by elevated PSA-levels suggesting that the AR is active in these cells [26]. In 22Rv1 cells two AR-forms can be found: A larger AR-form expressing 3 zinc finger motifs due to the duplication of exon 3 (AREx3dup) and a C-terminally truncated, constitutively active AR-V [20]. In the absence of androgenic stimuli, some AR-Vs have been shown to form constitutively active AR-V-homodimers or AR/AR-V-heterodimers, thereby uncoupling the need of CRPC cells for physiological levels of androgens [18,48]. Although it is unknown whether AREx3dup is able to form androgen independent heterodimers with AR-V in 22Rv1 cells, its expression of 3 zinc finger structures makes it probably also more susceptible to the effects of NO. The fact that NO targets the zinc finger structures of wild type AR, mutated AR-forms as well as AR-Vs suggests that NO-donors are promising compounds for the treatment of deregulated AR-activity. Moreover, the observation that CRPC cells like 22Rv1 or LNCaP-SSR, expressing either constitutively active AR-Vs or functionally deregulated full length AR, are more susceptible to the effects of JS-K than LNCaP cells further supports this assumption.
Conclusions
From our in vitro studies we conclude that GST-activated NO-prodrugs like JS-K show features which might be suited for the treatment of advanced PCa. Therapies targeting the AR and the canonical WNT pathway may lead to a more efficient treatment of CRPC. Therefore, compounds like JS-K may serve as leader compounds for the development of NO-based therapeuticals, targeting key structures in castration resistant prostate cancer cells.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ML, K-D S and MVC conceived the study; ML, WS, AH performed the experiments, MS AJS contributed to data analysis; MVC, K-D S, AJS, MS wrote the paper. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Figure 1S: JS-K does not inhibit AR-dimerization. Complementary data to experiment presented in Figure 3: AR-dimerization was determined in DU-145 cells using the CheckMate Mammalian Two-Hybrid System described in Material and Methods. Results are mean values of three independent experiments performed in quadruplicates. (DOC 20 kb).
Figure 2S: NO does not influence hormone-induced nuclear translocation of AR-EosFP in DU-145. Complementary data to experiment presented in Figure 4. (TIFF 1541 kb).
Contributor Information
Martin Laschak, Email: martin.laschak@uni-ulm.de.
Klaus-Dieter Spindler, Email: kmspindler@t-online.de.
Andres J Schrader, Email: ajschrader@gmx.de.
Andrea Hessenauer, Email: andrea.hessenauer@web.de.
Wolfgang Streicher, Email: wolfgang.streicher@uni-ulm.de.
Mark Schrader, Email: mark.schrader@uniklinik-ulm.de.
Marcus V Cronauer, Email: marcus.cronauer@uni-ulm.de.
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (HE 6078), Action Lions Vaincre le Cancer, Luxembourg (MVC) and the International Graduate School in Molecular Medicine Ulm (ML). The authors thank K. Dengler-Wupperfeld and S. Schmidt for skillful technical assistance.
References
- Burgaud JL, Riffaud JP, Del Soldato P. Nitric-oxide releasing molecules: A new class of drugs with several major indications. Curr Pharm Design. 2002;8:201–213. doi: 10.2174/1381612023396357. [DOI] [PubMed] [Google Scholar]
- Pavlos CM, Xu H, Toscano JP. Photosensitive precursors to nitric oxide. Curr Top Med Chem. 2005;5:637–647. doi: 10.2174/1568026054679326. [DOI] [PubMed] [Google Scholar]
- Kanwar JR, Kanwar RK, Burrow H, Baratchi S. Recent advances on the roles of NO in cancer and chronic inflammatory disorders. Curr Med Chem. 2009;16:2373–2394. doi: 10.2174/092986709788682155. [DOI] [PubMed] [Google Scholar]
- Ahmad R, Rasheed Z, Ahsan H. Biochemical and cellular toxicology of peroxynitrite: implications in cell death and autoimmune phenomenon. Immunopharmacol Immunotoxicol. 2009;31:388–396. doi: 10.1080/08923970802709197. [DOI] [PubMed] [Google Scholar]
- Kiziltepe T, Anderson KC, Kutok JL, Jia L, Boucher KM, Saavedra JE, Keefer LK, Shami PJ. JS-K has potent anti-angiogenic activity in vitro and inhibits tumour angiogenesis in a multiple myeloma model in vivo. J Pharm Pharmacol. 2010;62:145–151. doi: 10.1211/jpp.62.01.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurtry V, Saavedra JE, Nieves-Alicea R, Simeone AM, Keefer LK, Tari AM. JS-K, a nitric oxide-releasing prodrug, induces breast cancer cell death while sparing normal mammary epithelial cells. Int J Oncol. 2011;38:963–971. doi: 10.3892/ijo.2011.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuaria G, Magalhaes A, Angioli R, Mendez L, Mirhashemi R, Wang J, Wang P, Penalver M, Averette H, Braunschweiger P. Antitumor activity of a novel glyco-nitric oxide conjugate in ovarian carcinoma. Cancer. 2000;88:381–388. doi: 10.1002/(SICI)1097-0142(20000115)88:2<381::AID-CNCR20>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Stevens EV, Carpenter AW, Shin JH, Liu J, Der CJ, Schoenfisch MH. Nitric oxide-releasing silica nanoparticle inhibition of ovarian cancer cell growth. Mol Pharm. 2010;7:775–785. doi: 10.1021/mp9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita H, Kaneki M, Furuhashi S, Hirota M, Takamori H, Baba H. Nitric oxide inhibits the proliferation and invasion of pancreatic cancer cells through degradation of insulin receptor substrate-1 protein. Mol Cancer Res. 2010;8:1152–1163. doi: 10.1158/1541-7786.MCR-09-0472. [DOI] [PubMed] [Google Scholar]
- Royle JS, Ross JA, Ansell I, Bollina P, Tulloch DN, Habib FK. Nitric oxide donating nonsteroidal anti-inflammatory drugs induce apoptosis in human prostate cancer cell systems and human prostatic stroma via caspase-3. J Urol. 2004;172:338–344. doi: 10.1097/01.ju.0000132367.02834.41. [DOI] [PubMed] [Google Scholar]
- Huguenin S, Fleury-Feith J, Kheuang L, Jaurand MC, Bolla M, Riffaud JP, Chopin DK, Vacherot F. Nitrosulindac (NCX 1102): a new nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibits proliferation and induces apoptosis in human prostatic epithelial cell lines. Prostate. 2004;61:132–141. doi: 10.1002/pros.20081. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180–1192. doi: 10.1111/j.1742-1241.2011.02799.x. [DOI] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Hobisch Z, Culig C, Radmayr G, Bartsch H, Klocker A, Hittmair A. Distant metastases from prostatic carcinoma express androgen receptor protein. Cancer Res. 1995;55:3068–3072. [PubMed] [Google Scholar]
- Hobisch A, Culig Z, Radmayr C, Bartsch G, Klocker H, Hittmair A. Androgen receptor status of lymph node metastases from prostate cancer. Prostate. 1996;28:129–135. doi: 10.1002/(SICI)1097-0045(199602)28:2<129::AID-PROS9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Cronauer MV, Schulz WA, Burchardt T, Anastasiadis AG, de la Taille A, Ackermann R, Burchardt M. The androgen receptor in hormone-refractory prostate cancer: relevance of different mechanisms of androgen receptor signaling (Review) Int J Oncol. 2003;23:1095–1102. doi: 10.3892/ijo.23.4.1095. [DOI] [PubMed] [Google Scholar]
- Céraline J, Cruchant MD, Erdmann E, Erbs P, Kurtz JE, Duclos B, Jacqmin D, Chopin D, Bergerat JP. Constitutive activation of the androgen receptor by a point mutation in the hinge region: A new mechanism for androgen-independent growth in prostate cancer. Int J Cancer. 2004;108:152–157. doi: 10.1002/ijc.11404. [DOI] [PubMed] [Google Scholar]
- Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007;67:9001–9005. doi: 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–R196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronauer MV, Ince Y, Engers R, Rinnab L, Weidemann W, Suschek CV, Burchardt M, Kleinert H, Wiedenmann J, Sies H, Ackermann R, Kröncke KD. Nitric oxide-mediated inhibition of androgen receptor activity: possible implications for prostate cancer progression. Oncogene. 2007;26:1875–1884. doi: 10.1038/sj.onc.1209984. [DOI] [PubMed] [Google Scholar]
- Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, Keefer LK. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther. 2003;2:409–417. [PubMed] [Google Scholar]
- Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110:709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MW, Vacherot F, De La Taille A, Gil-Diez-De-Medina S, Shen R, Friedman RA, Burchardt M, Chopin DK, Buttyan R. The emergence of protocadherin-PC expression during the acquisition of apoptosis-resistance by prostate cancer cells. Oncogene. 2002;21:7861–7871. doi: 10.1038/sj.onc.1205991. [DOI] [PubMed] [Google Scholar]
- Schütz SV, Schrader AJ, Zengerling F, Genze F, Cronauer MV, Schrader M. Inhibition of glycogen synthase kinase-3β counteracts ligand-independent activity of the androgen receptor in castration resistant prostate cancer. PLoS One. 2011;6:e25341. doi: 10.1371/journal.pone.0025341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronauer MV, Schulz WA, Ackermann R, Burchardt M. Effects of WNT/β-catenin pathway activation on signaling through T-cell factor and androgen receptor in prostate cancer cell lines. Int J Oncology. 2005;26:1033–1040. doi: 10.3892/ijo.26.4.1033. [DOI] [PubMed] [Google Scholar]
- Laschak M, Bechtel M, Spindler KD, Hessenauer A. Inability of NCoR/SMRT to repress androgen receptor transcriptional activity in prostate cancer cell lines. Int J Mol Med. 2011;28:645–651. doi: 10.3892/ijmm.2011.735. [DOI] [PubMed] [Google Scholar]
- Rinnab L, Schütz SV, Jeannine Diesch J, Schmid E, Küfer R, Hautmann RE, Spindler K-D, Cronauer MV. Inhibiton of Glycogen-Synthase Kinase-3 (GSK) in Androgen Responsive Prostate Cancer Cell Lines - Are GSK-Inhibitors Therapeutically Useful? Neoplasia. 2008;10:624–634. doi: 10.1593/neo.08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CC, Hessenauer A, Montenarh M, Götz C. p53 is dispensable for the induction of apoptosis after inhibition of protein konase CK2. Prostate. 2010;70:126–134. doi: 10.1002/pros.21044. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15 N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-X. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Park JG, Kramer BS, Steinberg SM, Carmichael J, Collins JM, Minna JD, Gazdar AF. Chemosensitivity testing of human colorectal carcinoma cell lines using a tetrazolium-based colorimetric assay. Cancer Res. 1987;47:5875–5879. [PubMed] [Google Scholar]
- Mazur W, Gonciarz M, Kajdy M, Mazurek U, Jurzak M, Wilczok T, Gonciarz Z. Blood serum glutathione alpha s-transferase (alpha GST) activity during antiviral therapy in patients with chronic hepatitis C. Med Sci Monit. 2003;9(Suppl 3):44–48. [PubMed] [Google Scholar]
- Habdous M, Vincent-Viry M, Visvikis S, Siest G. Rapid spectrophotometric method for serum glutathione S-transferases activity. Clin Chim Acta. 2002;326:131–142. doi: 10.1016/S0009-8981(02)00329-7. [DOI] [PubMed] [Google Scholar]
- Kröncke KD, Carlberg C. Inactivation of zinc finger transcription factors provides a mechanism for a gene regulatory role of nitric oxide. FASEB J. 2000;14:166–173. doi: 10.1096/fasebj.14.1.166. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Röcker C, Salih A, Spindler K-D, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, Pretlow TG, Kung HJ. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–6614. [PubMed] [Google Scholar]
- Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, DeVere White RW, Pretlow TG, Harris SE, Wilson EM, Mohler JL, French FS. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–459. doi: 10.1210/me.11.4.450. [DOI] [PubMed] [Google Scholar]
- Wang G, Wang J, Sadar MD. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008;68:9918–9927. doi: 10.1158/0008-5472.CAN-08-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chen MW, Terry S, Vacherot F, Bemis DL, Capodice J, Kitajewski J, de la Taille A, Benson MC, Guo Y, Buttyan R. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene. 2006;25:3436–3444. doi: 10.1038/sj.onc.1209366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévotat L, Filomenko R, Solary E, Jeannin JF, Bettaieb A. Nitric oxide-induced down-regulation of beta-catenin in colon cancer cells by a proteasome-independent specific pathway. Gastroenterology. 2006;131:1142–1152. doi: 10.1053/j.gastro.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Nath N, Vassell R, Chattopadhyay M, Kogan M, Kashfi K. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: effects on COX-2 expression and Wnt/beta-catenin/TCF-4 signaling. Biochem Pharmacol. 2009;78:1298–1304. doi: 10.1016/j.bcp.2009.06.104. [DOI] [PubMed] [Google Scholar]
- Nath N, Chattopadhyay M, Pospishil L, Cieciura LZ, Goswami S, Kodela R, Saavedra JE, Keefer LK, Kashfi K. JS-K, a nitric oxide-releasing prodrug, modulates β-catenin/TCF signaling in leukemic Jurkat cells: evidence of an S-nitrosylated mechanism. Biochem Pharmacol. 2010;80:1641–1649. doi: 10.1016/j.bcp.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Henning SM, Heber D. Limitations of MTT and MTS-based assays for measurement of antiproliferative activity of green tea polyphenols. PLoS One. 2010;5:e10202. doi: 10.1371/journal.pone.0010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedge M, Lortz S, Munday R, Lenzen S. Protection against the co-operative toxicity of nitric oxide and oxygen free radicals by overexpression of antioxidant enzymes in bioengineered insulin-producing RINm5F cells. Diabetologia. 1999;42:849–855. doi: 10.1007/s001250051237. [DOI] [PubMed] [Google Scholar]
- Royle JS, Ross JA, Ansell I, Bollina P, Tulloch DN, Habib FK. Nitric oxide donating nonsteroidal anti-inflammatory drugs induce apoptosis in human prostate cancer cell systems and human prostatic stroma via caspase-3. J Urol. 2004;172:338–344. doi: 10.1097/01.ju.0000132367.02834.41. [DOI] [PubMed] [Google Scholar]
- Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler K-D, Laschak M, Cronauer MV. In: Nuclear Receptors. Bates MK, Kerr RM, editor. New York: Nova Science Publishers Inc; 2011. Nitric oxide - a tool to block nuclear receptors; pp. 87–102. [Google Scholar]
- Garbán HJ, Márquez-Garbán DC, Pietras RJ, Ignarro LJ. Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc Natl Acad Sci USA. 2005;102:2632–2636. doi: 10.1073/pnas.0409854102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens DR, Heaton JP, Adams MA, Kawakami J, Graham CH. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology. 2009;74:878–883. doi: 10.1016/j.urology.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Bico P, Chen CY, Jones M, Erhardt J, Dirr H. Class pi glutathione S-transferase: Meisenheimer complex formation. Biochem Mol Biol Int. 1994;33:887–892. [PubMed] [Google Scholar]
- Cronauer MV, Braun S, Tremmel Ch, Kröncke KD, Spindler-Barth M. Nuclear localization and DNA binding of ecdysone receptor and ultraspiracle. Arch Insect Biochem Physiol. 2007;65:125–133. doi: 10.1002/arch.20184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S: JS-K does not inhibit AR-dimerization. Complementary data to experiment presented in Figure 3: AR-dimerization was determined in DU-145 cells using the CheckMate Mammalian Two-Hybrid System described in Material and Methods. Results are mean values of three independent experiments performed in quadruplicates. (DOC 20 kb).
Figure 2S: NO does not influence hormone-induced nuclear translocation of AR-EosFP in DU-145. Complementary data to experiment presented in Figure 4. (TIFF 1541 kb).