

Abstract
Purpose.
Although endothelin-1 (ET-1) is a potent vasoconstrictor peptide implicated in several retinal pathologies, the underlying mechanism of vasoconstriction is understood incompletely. We addressed this issue by assessing the contributions of extracellular calcium (Ca2+), L-type voltage-operated calcium channels (L-VOCCs), Rho kinase (ROCK), and protein kinase C (PKC) to ET-1-induced constriction of porcine retinal arterioles, all of which have been implicated commonly in vascular smooth muscle contraction.
Methods.
Porcine retinal arterioles (∼50–100 μm) were isolated for vasomotor study and molecular assessment of ROCK isoforms.
Results.
Isolated arterioles developed stable basal tone at 55 cmH2O luminal pressure and constricted to ET-1 (0.1 nM) with a 40 ± 6% reduction in resting diameter in 20 minutes. In the absence of extraluminal Ca2+, arterioles lost basal tone and failed to constrict to ET-1. Although L-VOCC inhibitor nifedipine reduced basal tone and blocked vasoconstriction to PKC activator PDBu, vasoconstriction to ET-1 was unaffected. The broad-spectrum PKC inhibitor Gö-6983 abolished vasoconstriction to PDBu, but did not alter ET-1-induced vasoconstriction or basal tone. Incubation of arterioles with ROCK inhibitor H-1152 abolished basal tone and vasoconstrictions to ET-1 and PDBu. Both ROCK1 and ROCK2 isoforms were expressed in the retinal arteriolar wall.
Conclusions.
Extracellular Ca2+ entry via L-VOCCs and basal ROCK activity play important roles in the maintenance of basal tones of porcine retinal arterioles. ET-1-induced constriction is mediated by extracellular Ca2+ entry independent of L-VOCCs and by ROCK activation without the involvement of PKC. However, direct PKC activation can cause vasoconstriction via L-VOCC and ROCK signaling.
In this report it is demonstrated that porcine retinal arteriolar constriction to endothelin-1 is dependent upon extracellular Ca2+ and Rho kinase activation, but is independent of L-type voltage-operated calcium channels and protein kinase C activation.
Introduction
Endothelin-1 (ET-1) is a 21 amino acid peptide synthesized by vascular endothelial cells.1 It is a potent vasoconstrictor with roles in both physiologic and pathophysiologic contexts in the cardiovascular system.2 In the eye, increased ET-1 has been implicated in the pathogenesis of retinal vein occlusion,3 open angle glaucoma,4 and diabetic retinopathy.5,6 Since retinal arterioles are capable of synthesizing and releasing ET-1,7 elevated levels of ET-1 may contribute to retinal hypoxia or ischemia and subsequently manifest as each of these diseases. However, the mechanistic action of ET-1 in retinal arterioles remains understood incompletely.
It has been well characterized that vascular smooth muscle contraction is calcium (Ca2+)-dependent, with Ca2+ entry occurring through several types of channels, including L-type voltage-operated calcium channels (L-VOCCs).8,9 It also is known that the process of contraction is coupled to the level of myosin light chain (MLC) phosphorylation,10–12 which is regulated by the Ca2+-dependent activity of MLC kinase (MLCK)13–15 and by the MLC phosphatase (MLCP).16 Activation of Rho kinase (ROCK) has been implicated in vascular smooth muscle contraction, with its predominant role being enhancement of MLC phosphorylation via inhibition of MLCP.17,18 However, the role of ROCK activation in mediating vasoconstriction to ET-1 and the expression/distribution of ROCK isoforms in retinal arterioles remain unknown.
We demonstrated previously the presence of the requisite components for ET-1 synthesis as well as this peptide's vasoconstrictor action in porcine retinal arterioles mediated by activation of the ETA receptor subtype on vascular smooth muscle cells.7 The ETA receptor is a G-protein-coupled receptor, which upon stimulation leads to activation of several different downstream pathways, including activation of phospholipase C to produce inositol 1,4,5-triphosphate (IP3) and diacylglycerol.2 These molecules generally lead to increased intracellular Ca2+,19 and activation of protein kinase C (PKC),20 respectively, to elicit vasoconstriction.21,22 Although this classic explanation conceptually is legitimate, there is a paucity of experimental studies regarding the signaling molecules and pathways involved in vasoconstriction to ET-1 in the retinal circulation at the level of small resistance arterioles.
In the present study, we investigated the contribution of extracellular Ca2+ entry through L-VOCCs, the role of ROCK, and the possible involvement of PKC in the maintenance of basal tone and mediation of ET-1-induced constriction of retinal arterioles. To address these issues directly, we used an isolated vessel approach, thereby eliminating confounding influences from surrounding neuroglial tissue and hemodynamic changes that are inherent commonly in in vivo preparations.
Methods
Animal Preparation
All animal procedures were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Scott and White Institutional Animal Care and Use Committee. Pigs of either sex (age range 8–12 weeks, weight 8–21 kg) purchased from Real Farms (San Antonio, TX) were sedated with Telazol (4–8 mg/kg, intramuscularly) and intubated. The procedure used for harvesting eyes has been described previously.7
Isolation and Cannulation of Microvessels
The techniques used for identification, isolation, cannulation, pressurization and visualization of the retinal vasculature have been described previously.7,23 In brief, the isolated retinal arterioles (∼80 μm in situ) were cannulated with a pair of glass micropipettes and pressurized to 55 cmH2O intraluminal pressure without flow by two independent pressure reservoir systems.24 Vasomotor activity of isolated vessels was recorded continuously using videomicroscopic techniques25 throughout the experiments.
Study of Vasomotor Function
Cannulated, pressurized arterioles were bathed in physiological saline solution with albumin (PSS-albumin, 0.1%) at 36°C to 37°C to allow the development of basal tone (stable within 90 minutes). In the first series of studies, the involvement of extracellular Ca2+, the role of Ca2+ entry via L-VOCCs, and the activation of PKC and ROCK as signaling molecules in the maintenance of resting tone and in the initiation of vasoconstriction to ET-1 were assessed. The arterioles with tone were incubated in Ca2+-free PSS-albumin containing 1 mM EDTA, or in normal PSS-albumin containing the dihydropyridine L-VOCC blocker nifedipine (1 μM), the broad-spectrum PKC inhibitor Gö-698326 (3 μM), or the non-selective ROCK inhibitor H-1152 (10 μM). The resulting diameter changes over a period of 20 minutes were recorded. After establishing a new stable level of vascular tone, these vessels subsequently were exposed to ET-1 (0.1 nM) and the changes in vascular diameter were monitored for an additional 20 minutes. Because some of the aforementioned inhibitors caused a reduction in vascular tone, in another set of experiments the vessels were incubated with the endothelium-independent vasodilator sodium nitroprusside (SNP, 30 μM) for 20 minutes before administration of ET-1 (0.1 nM, 20 minutes) for comparison.
In the second series of studies, the contributions of extracellular Ca2+, L-VOCCs, PKC and ROCK to the maintenance of vasoconstriction to ET-1 were examined. Arterioles with tone were exposed first to ET-1 (0.1 nM) for 20 minutes to establish stable vasoconstriction and then incubated with Ca2+-free PSS-albumin or with nifedipine (1 μM), Gö-6983 (3 μM), or H-1152 (3 and 10 μM) for an additional 20 minutes.
In the final series of studies, the signaling pathway for vasoconstriction in response to direct PKC activation was investigated. Following development of basal tone, arterioles were incubated with the PKC activator phorbol-12,13-dibutyrate (PDBu, 0.1 μM) for 20 minutes before or after a 20-minute exposure to Gö-6983 (3 μM), nifedipine (1 μM) or H-1152 (3 μM). At the end of each experiment above, the maximum diameter of the vessels was obtained by incubating with a Ca2+-free PSS-albumin solution containing 0.3 mM SNP.
Chemicals
ET-1 was obtained from BaChem (Bubendorf, Switzerland), PDBu from Tocris Bioscience (Ellisville, MO), albumin from USB (Cleveland, OH), H-1152 and Gö-6983 from EMD Chemicals (Gibbstown, NJ), and nifedipine and SNP from Sigma (St. Louis, MO). ET-1, SNP, and H-1152 were dissolved initially in water. PDBu and Gö-6983 were dissolved in dimethyl sulfoxide, and nifedipine in ethanol. All subsequent dilutions of drugs for use in experiments were performed using PSS.7 The final concentrations of dimethyl sulfoxide in the vessel bath with PDBu and Gö-6983 were 0.001% and 0.03% by volume, respectively. The final concentration of ethanol in the vessel bath with nifedipine was 0.01% by volume. These solvent concentrations had no significant effect on vessel viability or maintenance of tone (data not shown).
Western Blot Analysis
Retinal arterioles of similar size to those used for functional studies were isolated and homogenized in lysis buffer. The protein content of each sample was quantified and separated by electrophoresis as described previously.7 For electrophoresis, 20 μg of protein were loaded in each lane. Blotting and detection of proteins was carried out as described previously using the following primary antibodies: rabbit anti-ROCK1 or anti-ROCK2 (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) or mouse anti-smooth muscle actin (anti-SMA, 1:20,000; Sigma).7 After incubation with an appropriate secondary antibody (anti-mouse or anti-rabbit, 1:2000; Sigma), the proteins were visualized via enhanced chemiluminescence (Pierce, Rockford, IL).
Immunohistochemical Analysis
The immunohistochemical detection of ROCK1 and ROCK2 isoforms in the vascular wall was performed after the preparation of cryomicrotome sections of retinal arterioles. Techniques for immunohistochemical staining of the isolated retinal vasculature were described in our previous work.7 Herein, we used four primary antibodies: goat anti-ROCK1 (1:100; Santa Cruz Biotechnology), rabbit anti-ROCK2 (1:100; Santa Cruz Biotechnology), mouse anti-endothelial nitric oxide synthase (eNOS, 1:100; BD Biosciences, San Diego, CA), or mouse anti-SMA (1:100; Sigma). Secondary antibodies that were used included FITC-conjugated anti-goat IgG (1:150; Jackson ImmunoResearch Laboratories, West Grove, PA) and Cy3-conjugated anti-rabbit IgG (1:150; Jackson ImmunoResearch Laboratories). Slides were observed using a fluorescence microscope (Axiovert 200, Zeiss, Thornwood, NY) for red (Cy3) and green (FITC) images. Merged images were created with ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html).
Data Analysis
Vessel diameters observed during experiments were normalized to resting vessel diameter following development of basal tone and are reported as percentages. A D'Agostino-Pearson test for normality was used to confirm normal distribution of the data. As appropriate, Student's t-test or repeated measures ANOVA with Tukey's multiple comparisons test were used to determine the level of significance of diameter changes in response to pharmacological interventions. Statistical analyses were carried out using Prism software (GraphPad, San Diego, CA). Data are reported as mean ± SEM. P < 0.05 was considered significant and n represents number of vessels (1 per pig per treatment group) used in functional studies.
Results
Roles of L-VOCC, PKC, and ROCK in Maintenance of Basal Tone
All vessels (n = 112) developed a similar level of stable basal tone (42 ± 1% of maximum diameter) in PSS-albumin. The average resting and maximum diameters of the vessels were 36 ± 1 μm (range 18–68 μm) and 84 ± 1 μm (range 51–109 μm), respectively. Changing the vessel bath from PSS-albumin to a Ca2+-free solution elicited a significant vasodilation, increasing resting diameter about 2.5-fold (i.e., reaching 97 ± 1% of maximum diameter, Fig. 1A). Incubation of vessels with the dihydropyridine L-VOCC blocker nifedipine (1 μM) led to a 41 ± 3% increase in resting diameter (Fig. 1A). The broad-spectrum PKC inhibitor Gö-698326 (3 μM) did not cause a significant change in resting diameter (Fig. 1B). In contrast, the ROCK inhibitor H-1152 (10 μM) and the endothelium-independent vasodilator SNP (30 μM) both caused approximately 2-fold increases in diameter, reaching near maximum dilation of the vessels (i.e., 98 ± 1% and 92 ± 2% of maximum diameter, respectively, Fig. 1B).
Figure 1.

Prevention of ET-1-induced vasoconstriction. (A) Temporal course of vasoconstriction induced by ET-1 (0.1 nM, n = 7) in the presence of basal tone shown as the control response. Pre-treatment of vessels with basal tone with nifedipine (1 μM, n = 4) or pre-incubation in a Ca2+-free solution (n = 3) for 20 minutes was followed by ET-1 (0.1 nM) treatment for 20 minutes. (B) Vessels with basal tone were pre-treated with SNP (30 μM, n = 3), H-1152 (10 μM, n = 6) or Gö-6983 (3 μM, n = 5) for 20 minutes before incubation with ET-1 (0.1 nM) for 20 minutes. R, resting diameter of vessels. *P < 0.05 versus percent resting diameter at R. #P < 0.05 versus percent resting diameter observed at time of addition of 0.1 nM ET-1 (0 minutes).
Roles of L-VOCC, PKC, and ROCK in Vasoconstriction to ET-1
As shown in Figure 1A, ET-1 (0.1 nM) caused a gradual vasoconstriction, stabilizing within 15–20 minutes and yielding an average of 40 ± 6% reduction in resting diameter. In the absence of extraluminal Ca2+, ET-1-induced constriction was not observed. In contrast, retinal arteriolar constriction to ET-1 remained in the presence of nifedipine (1 μM) and vasoconstriction reached the same magnitude as that of control vessels (Fig. 1A). Pre-treatment of vessels with the PKC inhibitor Gö-6983 had no effect on the ability of vessels to constrict to ET-1, but the ROCK inhibitor H-1152 (10 μM) blocked ET-1-induced vasoconstriction (Fig. 1B). In contrast, while pre-treatment of vessels with SNP (30 μM) caused a reduction in tone similar to that observed with H-1152, no inhibition of ET-1-induced constriction was observed (Fig. 1B).
As shown in Figure 2, ET-1 (0.1 nM) caused a similar reduction in diameter in all groups of vessels tested. This vasoconstriction was reversed by a 20-minute exposure to Ca2+-free solution, with vessel diameters increased to 227 ± 18% of the original resting level, corresponding to 95 ± 1% of maximum diameter. Treatment of ET-1-constricted vessels with nifedipine (1 μM) yielded vasodilation within one minute of drug administration, followed by a gradual constriction, whereas Gö-6983 did not cause any significant change in diameter (Fig. 2). Dose-dependent vasodilations to H-1152 (3 μM and 10 μM) were observed in vessels constricted with ET-1 (Fig. 2).
Figure 2.

Reversal of ET-1-induced vasoconstriction. Vessels treated with ET-1 (0.1 nM) for 20 minutes constricted to a stable diameter, then were exposed to Ca2+-free solution (n = 6) or treated with 10 μM (n = 8) or 3 μM (n = 14) H-1152, nifedipine (1 μM, n = 6), or Gö-6983 (3 μM, n = 8) for 20 minutes. R, resting diameter of vessels. *P < 0.05 versus percent resting diameter at R. #P < 0.05 for percent resting diameter values at 20 minutes versus percent resting diameter observed at time of addition of 0.1 nM ET-1 (0 minutes).
Roles of L-VOCC, PKC, and ROCK in Vasoconstriction to PDBu
Administration of PDBu (0.1 μM) caused significant constriction of retinal arterioles with an average of 59 ± 5% reduction in diameter, and this vasoconstriction was reversed by Gö-6983 (3 μM), nifedipine (1 μM), or H-1152 (3 μM) (Fig. 3). In the presence of Gö-6983 (3 μM), basal vascular tone was unaltered, but the vessels failed to constrict to PDBu (Fig. 4A). Nifedipine (1 μM) pre-treatment led to a decrease in tone of retinal arterioles, and subsequent addition of PDBu caused further dilation (Fig. 4B). As shown in Figure 4C, treatment of the vessels with H-1152 (3 μM) caused vasodilation and prevented PDBu-induced constriction.
Figure 3.

Reversal of PDBu-induced vasoconstriction. Vessels with basal tone were treated with PDBu (0.1 μM) for 20 minutes to produce vasoconstriction, followed by treatment with Gö-6983 (3 μM, n = 5), nifedipine (1 μM, n = 6), or H-1152 (3 μM, n = 8) for 20 minutes. #P < 0.05 versus control. *P < 0.05 versus PDBu treatment.
Figure 4.

Prevention of PDBu-induced vasoconstriction. Vessels with basal tone were pre-treated for 20 minutes with (A) Gö-6983 (3 μM, n = 6), (B) nifedipine (1 μM, n = 5), or (C) H-1152 (3 μM, n = 7), followed by treatment with PDBu (0.1 μM) for 20 minutes. *P < 0.05 versus control. #P < 0.05 versus nifedipine pre-treatment.
ROCK Isoform Expression in Retinal Arterioles
Retinal arterioles express both ROCK1 and ROCK2 isoforms (Fig. 5A), and as shown by tissue immunofluorescence analysis, ROCK1 and ROCK2 staining in the arteriolar wall overlap with SMA (Fig. 6A) and eNOS (Fig. 6B) staining.
Figure 5.
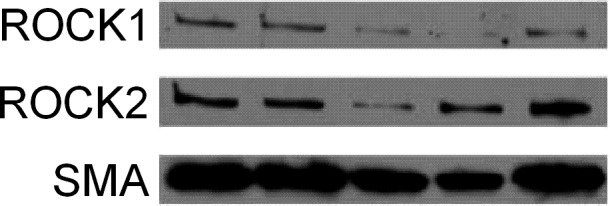
ROCK1 and ROCK2 protein expression in retinal arterioles. Immunoblot shows both ROCK isoforms expressed in arterioles with SMA as a loading control.
Figure 6.

Immunohistochemical analysis of ROCK1 and ROCK2 isoforms in retinal arterioles. (A) Staining with anti-ROCK1 (green) or anti-ROCK2 (green) and anti-SMA (red) antibodies demonstrates expression of both ROCK isoforms and SMA. Merged images show overlap staining (yellow) of ROCK isoforms with SMA. (B) Staining with anti-ROCK1 (green) or anti-ROCK2 (green) and anti-eNOS (red) antibodies demonstrates expression of both ROCK isoforms and eNOS. Merged images show overlap staining (yellow) of ROCK isoforms with eNOS. Images are representative of three separate experiments.
Discussion
Elevated vitreous and/or plasma levels of ET-1 have been implicated in several retinal pathologies, including diabetic retinopathy,6 glaucoma4 and retinal vein occlusion.3 Although increased levels of ET-1 may contribute to ischemia in various disease contexts due to its marked potency as a vasoconstrictor,3,4,27,28 the signaling pathways underlying retinal vasoconstriction remain elusive.7 Herein, we have demonstrated a central role for extracellular Ca2+ and ROCK activation, and a lack of either L-VOCC or PKC involvement in ET-1-induced constriction of the porcine retinal vasculature. Moreover, we found that L-VOCC activity is important in maintenance of basal vascular tone and can be induced by PKC activation, thereby contributing to PKC-mediated vasoconstriction. PKC activation also leads to ROCK activation as part of its signaling cascade, causing constriction of the retinal vessels. The PKC-independent vasoconstriction to ET-1 is a unique feature in retinal arterioles because it is distinct from several other vascular beds wherein PKC activation has been shown to be involved in the signaling pathway for vasoconstriction to ET-1.29,30
It has been shown that ocular tissues, including the retinal vasculature, highly express ET-1,31 which has been shown to decrease ocular blood flow in vivo.32 Moreover, ET-1 participates in retinal blood flow regulation in humans because administration of an ET-1 receptor antagonist specifically blunts retinal arteriolar constriction in response to systemic blood pressure elevation.33 Physiologic plasma concentrations of ET-1 have been reported in the low picomolar range.2,34 Normal vitreous concentrations of ET-1 are up to seven-fold higher than those in plasma,35 and a range of elevated vitreous concentrations from as low as ∼7 pM to as high as ∼46 pM has been reported in patients with glaucoma,36,37 branch retinal vein occlusion,38 or diabetic retinopathy.35,39 While the exact local concentration of ET-1 at the level of the microvasculature is unknown, the concentration of ET-1 (0.1 nM) used in the present study is around the clinical and experimental range (nM) predicted.40
It has been suggested that the mechanisms involved with maintenance of basal tone are either distinct41–43 or overlapping44,45 with those for agonist-induced constriction. Hence, we characterized some of the signaling mechanisms involved with tone maintenance in an effort to obviate potential misinterpretation of our ET-1 constriction studies. We showed previously that extracellular Ca2+ is necessary for maintenance of basal tone at physiologic intraluminal pressure.23 Administration of the L-VOCC blocker nifedipine caused a significant vasodilation, suggesting the importance of this channel for extracellular Ca2+ entry in maintenance of basal tone of retinal arterioles. It appears that initiation of basal tone also is dependent in part on activation of L-VOCC because nifedipine has been shown to diminish the development of pressure-induced tone in porcine46 and bovine47 retinal arterioles in vitro. However, the involvement of other Ca2+ entry pathways, including Na+/Ca2+ exchangers,48,49 and some members of the transient receptor potential channel family50,51 have been reported for other vasculatures, and these may have a role in the retinal circulation as well.
As is the case for maintenance of basal tone, ET-1-induced vasoconstriction also is dependent upon extracellular Ca2+ because 1) ET-1 failed to elicit vasoconstriction in the absence of extraluminal Ca2+ (Fig. 1A), and 2) ET-1-induced vasoconstriction was abolished upon vessel exposure to Ca2+-free solution (Fig. 2). However, entry of extracellular Ca2+ via L-VOCCs apparently has little role in mediating ET-1-induced constriction because nifedipine did not prevent this constriction. In fact, after 20 minutes of exposure of nifedipine-pre-treated vessels to ET-1, the amount of constriction observed was indistinguishable from that seen under control conditions (i.e., without nifedipine, Fig. 1). Notably, constriction of retinal arterioles (i.e., reduction of diameter from a resting level of 40 ± 4% to 24 ± 4% of maximum diameter) to the L-VOCC activator Bay K 8644 (1 μM) was abolished by nifedipine (1 μM) in our pilot studies (n = 8, data not shown), supporting the specificity and efficacy of the nifedipine used in the present study. In light of nifedipine's inability to prevent vasoconstriction to ET-1, L-VOCCs appear not to contribute to the initiation of ET-1-induced vasoconstriction. Furthermore, nifedipine failed to reverse ET-1-induced vasoconstriction, as shown in Figure 2. In this series of experiments, the vessels were constricted to ∼40% of their original diameter by ET-1 (Fig. 2). Based on the data shown in Figure 1, nifedipine is capable of reducing basal tone by ∼40%. Taking into account the counteraction of these two vasomotor activities, it is predictable that the final vascular diameter observed after combined treatment with both compounds should be maintained at the original resting level if vasoconstriction to ET-1 is not affected by nifedipine. As shown in Figure 2, the steady state diameter of the vessels was returned to, and subsequently maintained at, the original resting level by offsetting constriction (due to ET-1) with dilation (due to loss of basal tone by nifedipine) of the vessels. It appears that the pathway of Ca2+ entry for vasoconstriction in response to ET-1 in retinal arterioles is distinct from that used for basal tone maintenance in that the former does not involve L-VOCC activation. In contrast to the present findings, a study addressing Ca2+ entry in ET-1-induced constriction of the bovine retinal vasculature showed that blockade of L-VOCCs with nitrendipine abolished tension development in response to ET-1.53 This discrepancy may be related to the species difference, size of vessels (i.e., ∼200 μm vs. ∼80 μm in the present study) and/or the absence of basal tone in the previous bovine vessel preparations.53 Nevertheless, our results are consistent with the minimal role of L-VOCCs in the sustained intracellular Ca2+ increase induced by ET-1 (0.1 nM) reported for freshly isolated smooth muscle cells from rabbit internal carotid artery.52
In addition to L-VOCC, store-operated calcium channels have been shown to contribute to Ca2+ entry in retinal arteriolar smooth muscle cells.54 Depletion of IP3-sensitive Ca2+ stores in the sarcoplasmic reticulum following agonist stimulation can lead to activation of store-operated calcium channels. Interestingly, recent evidence using confocal microscopic imaging demonstrates that ET-1 elicits an increase in intracellular Ca2+ via phospholipase C/IP3 signaling in smooth muscle cells of rat retinal arterioles in situ.55 Because the functional impact of this signaling pathway on vasoconstriction was not assessed, future studies investigating the Ca2+ entry mechanisms contributing to ET-1-induced constriction in retinal arterioles are warranted.
A role for PKC in myogenic tone has been suggested for vascular beds from various tissues in several different species.44,56,57 However, in rat ophthalmic artery it was shown that PKC inhibition had no effect on vascular tone generated by a range of pressures between 75 mmHg and 160 mmHg, suggesting a relatively minimal role for PKC in tone maintenance.58 The data presented in Figure 1B are in agreement with this finding because treatment with Gö-6983 had no effect on resting tone. This apparent lack of PKC involvement also is evident for ET-1-induced constriction because no prevention or reversal of constriction was observed with Gö-6983 treatment (Figs. 1B, 2). Importantly, we found that PKC activation by PDBu did cause retinal vasoconstriction, which was reversed (Fig. 3) and prevented (Fig. 4A) by treating vessels with Gö-6983. These data demonstrate the specificity of Gö-6983 for PKC inhibition and further support the conclusion that PKC is not involved in the retinal arteriolar constriction to ET-1, although its activation is capable of eliciting vasoconstriction. Contrastingly, a role for PKC in vascular contraction to ET-1 has been reported in several other vascular beds,29,30,59,60 possibly due to the downstream production of diacylglycerol.2,61 It appears that ET-1 elicits a distinct signaling pathway for vasoconstriction in the retinal microcirculation, at least in the porcine model, which recently has been shown to be similar to humans in vasomotor regulation.23
Given the comparable magnitude of vasoconstriction to PDBu with that to ET-1, we also tested the effects of nifedipine on PDBu-induced constriction to ascertain whether L-VOCC flux has a role in this context. As shown in Figure 3, nifedipine not only reversed PDBu-induced vasoconstriction but also caused vasodilation, that is, increased the vessel diameter beyond its resting level. Moreover, in the presence of nifedipine, PDBu failed to constrict retinal arterioles and instead, a vasodilation to PDBu was observed (Fig. 4B). Thus retinal vasoconstriction in response to PKC activation is dependent upon L-VOCC activation. Interestingly, some studies have implicated PKC isoforms in activation of eNOS.62–64 Hence, it is possible that blocking L-VOCC-mediated contraction of smooth muscle cells might unmask an endothelium-mediated response to PDBu, thus explaining the observed vasodilation. In light of a minimal role for L-VOCCs in ET-1-induced constriction, but a prominent role in PDBu-induced constriction, it seems that PKC activation linking to L-VOCC opening, although capable of evoking vasoconstriction, is not a part of the vasomotor signaling pathway induced by ET-1.
As shown in Figure 1B, ROCK inhibition led to a significant loss of basal tone, a finding in agreement with several studies from other vascular beds implicating ROCK in maintenance of vascular tone.65–67 Pre-treatment with H-1152 prevented vasoconstriction to ET-1 (Fig. 1B), indicating that the development of ET-1-induced vasoconstriction depends on ROCK activity. The prevention of ET-1-induced vasoconstriction by ROCK inhibition was unlikely the result of vascular tone loss because vasoconstriction to ET-1 persisted in the presence of SNP, which produced a reduction of vascular tone comparable to that elicited by H-1152. As shown in Figure 2, vasoconstriction to ET-1 was reversed by H-1152 in a dose-dependent manner. Hence, these data show a prominent role of ROCK in maintenance of basal tone and ET-1-induced vasoconstriction. Interestingly, vasoconstriction to PDBu was reversed (Fig. 3) and prevented (Fig. 4C) with H-1152, suggesting a pivotal role of ROCK in mediating vasoconstriction to PKC activation. This conclusion is in agreement with studies showing that PDBu-induced contraction of rat aortic rings involves activation of the RhoA/ROCK pathway68 and that contraction of bovine coronary artery rings in response to PDBu is sensitive to ROCK inhibition.69 At the concentrations used herein, H-1152 is capable of inhibiting Ca2+/calmodulin-dependent protein kinase II (CaMKII),70 which has been suggested to have a role in vascular smooth muscle contraction.71,72 However, in our pilot studies, pre-treatment of vessels with the CaMKII inhibitor KN-93 (3 μM) had no effect on either basal tone or ET-1-induced vasoconstriction (n = 3, data not shown). Hence, H-1152 is unlikely to be exerting the effects described here through inhibition of CaMKII.
Although the present functional studies suggest that ROCK is a convergent target for vasoconstriction to ET-1 and PKC activation, the expression of ROCK isoforms in the retinal microvasculature is unknown. The immunoblot data presented in Figure 5 show that ROCK1 and ROCK2 are expressed in retinal arterioles, and the immunofluorescence data in Figure 6 demonstrate expression of both isoforms in smooth muscle (Fig. 6A) and endothelium (Fig. 6B). Wang et al. demonstrated a direct interaction between the myosin binding subunit of MLCP and ROCK2, but not ROCK1 in A7r5 and primary rat aortic smooth muscle cells.73 Moreover, silencing of either isoform led to reduced inhibitory phosphorylation of MLCP and reduced phospho-MLC levels, but there was significantly less contraction in ROCK2-silenced vascular smooth muscle cells relative to that in ROCK1-silenced or control cells,73 suggesting a major role for ROCK2 in mediating vasomotor function. However, whether one isoform is responsible predominantly for ROCK-mediated tone maintenance or constriction to ET-1 and PDBu in the retinal vasculature is unknown and will be the subject of future investigation.
In summary, we demonstrated a central role for ROCK in maintenance of basal tone as well as in constriction of the retinal vasculature to ET-1 and PKC activation. We also showed that ET-1-induced constriction does not use the PKC/L-VOCC signaling pathway, although activation of this pathway does lead to vasoconstriction. To our knowledge, this is the first report of ET-1-induced vasoconstriction that does not lead to PKC activation. It is apparent that some of the classical mechanisms invoked for smooth muscle contraction may not be applicable necessarily to the constriction of retinal microvessels in response to ET-1. This study provides important insight into the ET-1-induced constriction mechanisms used by the retinal microcirculation, and lays a foundation for future, more detailed studies of this pathway. Understanding key components of this vasoconstriction mechanism and manipulating the ROCK signaling pathway may help in future development of more targeted therapeutics that could address the pathologic effects of ET-1 and PKC activation in the retina.
Footnotes
Supported by NIH EY018420 (TWH), Retina Research Foundation (LK), and Kruse Chair Endowment Fund (LK).
Disclosure: L.B. Potts, None; Y. Ren, None; G. Lu, None; E. Kuo, None; E. Ngo, None; L. Kuo, None; T.W. Hein, None
References
- 1. Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988; 332:411–415 [DOI] [PubMed] [Google Scholar]
- 2. Khimji A-K, Rockey DC. Endothelin-biology and disease. Cell Signal. 2010; 22:1615–1625 [DOI] [PubMed] [Google Scholar]
- 3. Iannaccone A, Letizia C, Pazzaglia S, Vingolo EM, Clemente G, Pannarale MR. Plasma endothelin-1 concentrations in patients with retinal vein occlusions. Br J Ophthalmol. 1998; 82:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emre M, Orgül S, Haufschild T, Shaw SG, Flammer J. Increased plasma endothelin-1 levels in patients with progressive open angle glaucoma. Br J Ophthalmol. 2005;89:60–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng D, Evans T, Mukherjee K, Downey D, Chakrabarti S. Diabetes-induced vascular dysfunction in the retina: role of endothelins. Diabetologia. 1999; 42:1228–1234 [DOI] [PubMed] [Google Scholar]
- 6. Ergul A. Endothelin-1 and diabetic complications: Focus on the vasculature. Pharmacol Res. 2011; 63:477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hein TW, Ren Y, Yuan Z, et al. Functional and molecular characterization of the endothelin system in retinal arterioles. Invest Ophthalmol Vis Sci. 2009; 50:3329–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001; 91:973–983 [DOI] [PubMed] [Google Scholar]
- 9. Berridge MJ. Smooth muscle cell calcium activation mechanisms. J Physiol. 2008; 586:5047–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985; 25:593–620 [DOI] [PubMed] [Google Scholar]
- 11. He WQ, Peng YJ, Zhang WC, et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. 2008; 135:610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isotani E, Zhi G, Lau KS, et al. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci. 2004; 101:6279–6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology. 2009; 24:342–356 [DOI] [PubMed] [Google Scholar]
- 14. Mizuno Y, Isotani E, Huang J, Ding H, Stull JT, Kamm KE. Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am J Physiol Cell Physiol. 2008; 295:C358–C364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994; 372:231–236 [DOI] [PubMed] [Google Scholar]
- 16. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003; 83:1325–1358 [DOI] [PubMed] [Google Scholar]
- 17. Loirand G, Guérin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334 [DOI] [PubMed] [Google Scholar]
- 18. Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010; 67:3823–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berridge MJ, Irvine RF. Inositol phosphates and cell signalling. Nature. 1989; 341:197–205 [DOI] [PubMed] [Google Scholar]
- 20. Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988; 334:661–665 [DOI] [PubMed] [Google Scholar]
- 21. Bootman MD, Lipp P, Berridge MJ. The organisation and functions of local Ca2+ signals. J Cell Sci. 2001; 114:2213–2222 [DOI] [PubMed] [Google Scholar]
- 22. Görlach C, Benyó Z, Wahl M. Endothelin-1-induced contraction in cerebral vessels mediated by phospholipase C/protein kinase C cascade. Kidney Int. 1998;67:S224–S225 [DOI] [PubMed] [Google Scholar]
- 23. Hein TW, Rosa RH, Jr, , Yuan Z, Roberts E, Kuo L. Divergent roles of nitric oxide and Rho kinase in vasomotor regulation of human retinal arterioles. Invest Ophthalmol Vis Sci. 2010; 51:1583–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuo L, Davis MJ, Chilian WM. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol. 1990; 259:H1063–H1070 [DOI] [PubMed] [Google Scholar]
- 25. Hein TW, Yuan Z, Rosa RH, Jr, , Kuo L. Requisite roles of A2A receptors, nitric oxide, and KATP channels in retinal arteriolar dilation in response to adenosine. Invest Ophthalmol Vis Sci. 2005; 46:2113–2119 [DOI] [PubMed] [Google Scholar]
- 26. Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C μ by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996; 392:77–80 [DOI] [PubMed] [Google Scholar]
- 27. Kalani M. The importance of endothelin-1 for microvascular dysfunction in diabetes. Vasc Health Risk Manag. 2008; 4:1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takagi C, Bursell SE, Lin YW, et al. Regulation of retinal hemodynamics in diabetic rats by increased expression and action of endothelin-1. Invest Ophthalmol Vis Sci. 1996; 37:2504–2518 [PubMed] [Google Scholar]
- 29. Feng J, Liu Y, Khabbaz KR, et al. Endothelin-1-induced contractile responses of human coronary arterioles via endothelin-A receptors and PKC-α signaling pathways. Surgery. 2010; 147:798–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feng J, Chu LM, Robich MP, et al. Effects of cardiopulmonary bypass on endothelin-1-induced contraction and signaling in human skeletal muscle microcirculation. Circulation. 2010; 122:S150–S155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wollensak G, Schaefer HE, Ihling C. An immunohistochemical study of endothelin-1 in the human eye. Curr Eye Res. 1998; 17:541–545 [DOI] [PubMed] [Google Scholar]
- 32. Pang IH, Yorio T. Ocular actions of endothelins. Proc Soc Exp Biol Med. 1997; 215:21–34 [DOI] [PubMed] [Google Scholar]
- 33. Luksch A, Wimpissinger B, Polak K, Jandrasits K, Schmetterer L. ETa-receptor blockade, but not ACE inhibition, blunts retinal vessel response during isometric exercise. Am J Physiol Heart Circ Physiol. 2006; 290:H1693–H1698 [DOI] [PubMed] [Google Scholar]
- 34. Highsmith RF, Blackburn K, Schmidt DJ. Endothelin and calcium dynamics in vascular smooth muscle. Annu Rev Physiol. 1992; 54:257–277 [DOI] [PubMed] [Google Scholar]
- 35. Adamiec-Mroczek J, Oficjalska-Mlyńczak J, Misiuk-Hojlo M. Roles of endothelin-1 and selected proinflammatory cytokines in the pathogenesis of proliferative diabetic retinopathy: analysis of vitreous samples. Cytokine. 2010;49:269–274 [DOI] [PubMed] [Google Scholar]
- 36. Ghanem AA, Elewa AM, Arafa LF. Endothelin-1 and nitric oxide levels in patients with glaucoma. Ophthalmic Res. 2011; 46:98–102 [DOI] [PubMed] [Google Scholar]
- 37. Iwabe S, Lamas Vásquez Pélaez CG, Carrasco FG. Aqueous humor endothelin-1 (ET-1), vascular endothelial growth factor (VEGF) and cyclooxygenase-2 (COX-2) levels in Mexican glaucomatous patients. Curr Eye Res. 2010;35:287–294 [DOI] [PubMed] [Google Scholar]
- 38. Sin BH, Song BJ, Park SP. Aqueous vascular endothelial growth factor and endothelin-1 levels in branch retinal vein occlusion associated with normal tension glaucoma. J Glaucoma. 2011. September 22 [Epub ahead of print] [DOI] [PubMed]
- 39. Roldán-Pallarés M, Rollin R, Mediero A, et al. Immunoreactive ET-1 in the vitreous humor and epiretinal membranes of patients with proliferative vitreoretinopathy. Mol Vis. 2005;11:461–471 [PubMed] [Google Scholar]
- 40. Masaki T, Yanagisawa M, Goto K. Physiology and pharmacology of endothelins. Med Res Rev. 1992; 12:391–421 [DOI] [PubMed] [Google Scholar]
- 41. Liu J, Hill MA, Meininger GA. Mechanisms of myogenic enhancement by norepinephrine. Am J Physiol. 1994;266:H440–H446 [DOI] [PubMed] [Google Scholar]
- 42. Lombard JH, Eskinder H, Kauser K, Osborn JL, Harder DR. Enhanced norepinephrine sensitivity in renal arteries at elevated transmural pressure. Am J Physiol. 1990;259:H29–H33 [DOI] [PubMed] [Google Scholar]
- 43. El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol. 2010; 588:1747–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Massett MP, Ungvari Z, Csiszar A, Kaley G, Koller A. Different roles of PKC and MAP kinases in arteriolar constrictions to pressure and agonists. Am J Physiol Heart Circ Physiol. 2002; 283:H2282–H2287 [DOI] [PubMed] [Google Scholar]
- 45. Metting PJ, Stein PM, Stoos BA, Kostrzewski KA, Britton SL. Systemic vascular autoregulation amplifies pressor responses to vasoconstrictor agents. Am J Physiol. 1989; 256:R98–R105 [DOI] [PubMed] [Google Scholar]
- 46. Jeppesen P, Aalkjaer C, Bek T. Myogenic response in isolated porcine retinal arterioles. Curr Eye Res. 2003; 27:217–222 [DOI] [PubMed] [Google Scholar]
- 47. Delaey C, Van de Voorde J. Pressure-induced myogenic responses in isolated bovine retinal arteries. Invest Ophthalmol Vis Sci. 2000; 41:1871–1875 [PubMed] [Google Scholar]
- 48. Zhang J, Lee MY, Cavalli M, et al. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005; 569:243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang J, Ren C, Chen L, et al. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol. 2010; 298:H1472–H1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. 2002; 90:248–250 [DOI] [PubMed] [Google Scholar]
- 51. Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004; 95:922–929 [DOI] [PubMed] [Google Scholar]
- 52. Kawanabe Y, Hashimoto N, Masaki T. Ca2+ channels involved in endothelin-induced mitogenic response in carotid artery vascular smooth muscle cells. Am J Physiol Cell Physiol. 2002;282:C330–C337 [DOI] [PubMed] [Google Scholar]
- 53. Nyborg NC, Prieto D, Benedito S, Nielsen PJ. Endothelin-1-induced contraction of bovine retinal small arteries is reversible and abolished by nitrendipine. Invest Ophthalmol Vis Sci. 1991;32:27–31 [PubMed] [Google Scholar]
- 54. Scholfield CN, Curtis TM. Heterogeneity in cytosolic calcium regulation among different microvascular smooth muscle cells of the rat retina. Microvasc Res. 2000; 59:233–242 [DOI] [PubMed] [Google Scholar]
- 55. Tumelty J, Hinds K, Bankhead P, et al. Endothelin 1 stimulates Ca2+-sparks and oscillations in retinal arteriolar myocytes via IP3R and RyR-dependent Ca2+ release. Invest Ophthalmol Vis Sci. 2011; 52:3874–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999; 79:387–423 [DOI] [PubMed] [Google Scholar]
- 57. Wesselman JP, Spaan JA, van der Meulen ET, VanBavel E. Role of protein kinase C in myogenic calcium-contraction coupling of rat cannulated mesenteric small arteries. Clin Exp Pharmacol Physiol. 2001; 28:848–855 [DOI] [PubMed] [Google Scholar]
- 58. Ito I, Jarajapu YP, Grant MB, Knot HJ. Characteristics of myogenic tone in the rat ophthalmic artery. Am J Physiol Heart Circ Physiol. 2007; 292:H360–H368 [DOI] [PubMed] [Google Scholar]
- 59. Mueed I, Zhang L, MacLeod KM. Role of the PKC/CPI-17 pathway in enhanced contractile responses of mesenteric arteries from diabetic rats to α-adrenoceptor stimulation. Br J Pharmacol. 2005; 146:972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Snow JB. Gonzalez Bosc LV, Kanagy NL, Walker BR, Resta TC. Role for PKCβ in enhanced endothelin-1-induced pulmonary vasoconstrictor reactivity following intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2011; 301:L745–L754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Griendling KK, Tsuda T, Alexander RW. Endothelin stimulates diacylglycerol accumulation and activates protein kinase C in cultured vascular smooth muscle cells. J Biol Chem. 1989; 264:8237–8240 [PubMed] [Google Scholar]
- 62. Partovian C, Zhuang Z, Moodie K, et al. PKCα activates eNOS and increases arterial blood flow in vivo. Circ Res. 2005; 97:482–487 [DOI] [PubMed] [Google Scholar]
- 63. Michell BJ, Chen Z, Tiganis T, et al. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001; 276:17625–17628 [DOI] [PubMed] [Google Scholar]
- 64. da Silva CG, Specht A, Wegiel B, Ferran C, Kaczmarek E. Mechanism of purinergic activation of endothelial nitric oxide synthase in endothelial cells. Circulation. 2009; 119:871–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol. 2009; 587:2537–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gokina NI, Park KM, McElroy-Yaggy K, Osol G. Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J Appl Physiol. 2005; 98:1940–1948 [DOI] [PubMed] [Google Scholar]
- 67. Platts SH, Martinez-Lemus LA, Meininger GA. Microtubule-dependent regulation of vasomotor tone requires Rho-kinase. J Vasc Res. 2002; 39:173–182 [DOI] [PubMed] [Google Scholar]
- 68. Baek I, Jeon SB, Kim J, et al. A role for Rho-kinase in Ca2+-independent contractions induced by phorbol-12,13-dibutyrate. Clin Exp Pharmacol Physiol. 2009; 36:256–261 [DOI] [PubMed] [Google Scholar]
- 69. Gupte SA, Kaminski PM, George S, et al. Peroxide generation by p47phox-Src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am J Physiol Heart Circ Physiol. 2009; 296:H1048–H1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tamura M, Nakao H, Yoshizaki H, et al. Development of specific Rho-kinase inhibitors and their clinical application. Biochim Biophys Acta. 2005; 1754:245–252 [DOI] [PubMed] [Google Scholar]
- 71. Kim I, Je HD, Gallant C, et al. Ca2+-calmodulin-dependent protein kinase II-dependent activation of contractility in ferret aorta. J Physiol. 2000; 526:367–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rokolya A, Singer HA. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol. 2000; 278:C537–C545 [DOI] [PubMed] [Google Scholar]
- 73. Wang Y, Zheng XR, Riddick N, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009; 104:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]