

SUMMARY
Stereocilia of the inner ear play an integral role in the mechanotransduction of sound. Their structural support is derived from actin filaments and actin-binding proteins. We have identified a novel actin-binding protein, 2E4-kaptin (KPTN), which appears to be involved in this structural network. Using double label immunofluorescence, we now show that KPTN extends beyond the barbed ends of actin filaments at the tips of stereocilia, and using cloned human cDNA, we mapped KPTN to chromosome 19q13.4. A combination of FISH, radiation hybrid mapping and YAC screening localized KPTN between markers D19S412 and NIB1805, making this gene an excellent functional and positional candidate for DFNA4, a form of autosomal dominant non-syndromic hearing loss. We identified a second family with inherited deafness that also maps to the DFNA4 region. To screen KPTN for deafness-causing mutations, we first determined its genomic structure and then completed a mutational analysis by direct sequencing and SSCP in affected family members. Although no deafness-causing mutations were identified in the coding region, KPTN remains an excellent candidate gene for hearing loss; by synteny, its murine orthologue also remains a candidate gene for the Nijmegan waltzer (nv) mouse mutant, which has vestibular defects and a variable sensorineural hearing loss.
INTRODUCTION
Mechanotransduction of hearing is mediated through hair cells of the sensory epithelium of the inner ear (Hudspeth, 1997). These cells lie below the tectorial membrane, which oscillates in response to sound waves, displacing stereocilia and opening gated ion channels to give rise to an electrical signal. Each hair cell has a unique complement of stereocilia, varying in length and number, and tonotopically distributed with numerous, short stereocilia in the basal turn and fewer, longer stereocilia in the apical region of the cochlea. The length and number of stereocilia determine, in part, the optimal frequency to which each hair cell responds. Within stereocilia themselves are bundles of cross-linked actin filaments, formed by a complicated choreography during embryogenesis in which actin monomers are added to the distal tips (Tilney et al. 1988, 1992). By electronmicroscopy, the tip region appears as an electron-dense fuzz that is hypothesized to contain machinery essential for active polymerization of actin, a process that determines the eventual length of each stereocilium.
Kaptin (KPTN), an actin-binding protein, was identified originally by F-actin affinity chromatography and found in the leading edge of spreading platelets (Bearer, 1992, 1995; Bearer & Abraham, 1999). It also has been localized to sites of actin polymerization in motile fibroblasts and neuronal growth cones, implicating it in the regulation of actin polymerization. KPTN is thought to be one of several different nucleators that initiate the formation of actin filaments off membrane surfaces (Bearer, 1993). Its location in stereocilia suggests that it plays a role in producing the sensory apparatus in hair cells. It therefore seemed likely that genetic defects in KPTN might be the basis for inherited deafness syndromes.
In this report, double-label fluorescence microscopy is used to image both actin filaments and KPTN in the same stereocilia. KPTN is stained using a new polyclonal antibody raised against recombinant protein. The cDNA encoding KPTN is then used to map the KPTN gene to chromosome 19q13. We then determine by linkage analysis that the chromosomal location of an inherited deafness syndrome in two different families is also on 19q13 (Chen et al. 1995). Finally, because these data make KPTN an excellent positional and functional candidate for the DFNA4 gene, we completed a screen for coding sequence mutations in a DFNA4 family.
MATERIAL AND METHODS
Immunofluorescence of cochlear sensory epithelium
Chick sensory epithelia were prepared for immunofluorescence as described (Bearer & Abraham, 1999) except specimens were stained with a new affinity purified polyclonal antibody generated against the recombinant protein to confirm that the cloned protein was indeed that recognized by the Mab2E4 monoclonal antibody previously used to identify the KPTN protein and to screen an expression library and isolate KPTN cDNA (Bearer & Abraham, 1999). Specimens were stained in parallel with the MAb2E4 as primary antibody. After immunostaining, the cochlea were counter-stained for 30 min in 1/100 dilution of 6 μM FITC-phalloidin (Molecular Probes, Eugene, OR) in phosphate buffer. Analogue images were collected by consecutive double exposure on 35-mm film with a Nikon SA fluorescence microscope.
Fluorescence in situ hybridization
Metaphase spreads were prepared on glass microscope slides, as described (Jackson et al. 1992; Mark et al. 1994). KPTN cDNA (Bearer & Abraham, 1999) was biotin-labelled by nick translation using the Gibco-Life Technologies kit. Coverslips containing chromosome spreads were denatured for 2 min 20 s at 95°C, incubated with labelled probe (2 ng/μl in 10 μl/slide) overnight at 42°C in a humidified chamber and washed three times in 2× sodium-citrate-sodium chloride (SSC: 20× is 3 M sodium citrate, 3 M NaCl, pH 7); the probe was detected with avidin-FITC (Oncor, Gaithersburg, MD). Counter-staining with propidium iodide was performed as described (Mark et al. 1994; Oncor protocol). After detection of the labelled chromosomes, slides were denatured by drying on a 55°C hot table; the labelled chromosome was identified by the banding pattern determined by Giemsa trypsin.
Mapping KPTN by PCR
The full length KPTN cDNA (GenBank Accession number: AF105369) has previously been cloned and determined to be the same approximate length, 1.64 kb, as the mRNA by Northern blot (Bearer & Abraham, 1999). Recombinant KPTN has been expressed in bacteria and used as an immunogen to produce polyclonal antibodies. KPTN was mapped to chromosome 19 by PCR using the 3′ untranslated region of the cDNA to design primers: forward: TGCACCCACTCTTCCCAGACCT; and reverse: GTGGCCAATTCTCATCCAGAGT. Template genomic DNA was from a mapping panel of somatic cell hybrids each containing a single human chromosome (NIGMS Human/Rodent Somatic Cell Mapping Panel #2, Coriell Institute, NJ), or control DNA, either from human placenta, a positive control, or from the parent mouse and hamster cells lacking human chromosomes as negative controls. PCR conditions were: hot start with 15 min incubation of 1 μM primer and template with 1.5 mM MgCl2 at 94°C, then addition of Taq polymerase and continue for 5 min at 94°C, followed by 35 cycles of 1 min at 94°C, 1 min 60°C, 1 min 72°C and then 10 min 72°C. PCR reactions were performed with 30 ng of template DNA, 25 pM of each primer, 1.5 μl of 10× reaction buffer (160 mM (NH4)2SO4, 670 mM Tris–HCl, 0.1% Tween-20, pH 8.8 at 25°C), 22.5 pM of MgCl2, 15 pM of dATP, dCTP, dGTP, and dATP, and 0.375 U of DNA polymerase in a total reaction volume of 15 μl.
Further localization of KPTN was obtained using the Genebridge 4 radiation hybrid panel (GB4) from Research Genetics using two additional sets of primers: 1f, ATCCTGCAGCACAGCCTGAT; 1r, GAATGGGATCATCCCTGCTT; 2f, GCTGCAGTTCAACTACATTC; 2r, TACACAGAGCCGGTCTCGTA. The GB4 panel of 93 somatic cell hybrids has an effective resolution of 1 Mb (Gyapay et al. 1996). Results from GB4 were analyzed with RHMAPPER, a software program package provided by the Whitehead Institute for Biomedical Research/MIT Center for Genome Research. PCR reactions were conducted for 40 cycles beginning with 94°C for 30 s; annealing 54–64°C (specific temperatures were a function of the melting temperature of each primer used in the reaction and are provided in Table 1) for 30 s; extension at 72°C for 30 sec; and a post-PCR extension at 72°C for 10 min. To place KPTN on the physical map of the genome, the CEPH library of yeast artifical chromosomes (YACs, Research Genetics) was screened using the PCR conditions described above for the Coriell mapping panel and sub-cloned to yield a single positive colony. In addition, seven YACs (719B7, 830H9, 821G2, 761C1, 955G11, 777B3, 790A5) were selected and screened because they spanned the region of interest on the WC19.5 contig according to the Whitehead Contig Map.
Table 1.
Primers and splice sites used to obtain genomic structure of 2E4 (For genomic sequence of KPTN see GenBank Accession number AF24529)
| Forward primer | Splice-acceptor Size | Exon | Splice-donor | Reverse primer | |
|---|---|---|---|---|---|
| Intron/exon boundaries and primers used to amplify exons of 2E4 | |||||
| agggtgcttaactgaggg (58°) | exon 1 | TTCCCG/gtggg | gaatgtagttgaactgcagc | 295 bp | |
| agtctgcctgacactgaact (58°) | tcccag/TGGAT | exon 2 | ATCAAG/gtacc | taaggaggagggcgaaggga | 163 bp |
| ctcctccttacacacatggt (56°) | cgccag/GATTC | exon 3 | TGCGGA/gtgag | atttacagtggagtgggcgt | 272 bp |
| tccactaccttgggttctag (58°) | tttcag/GGTCC | exon 4 | AAGGAG/gtagg | agcccacagtgaatcccaca | 163 bp |
| agctgacggctgacttaagc (58°) | ccccag/AACGA | exon 5 | CAGTAG/gtagg | gtccccagtggcctctttc | 156 bp |
| aatggccgtccgtcctgtcc (64°) | cgccag/CGTCC | exon 6 | GTCGAG/gtgag | cctctagccctcaacccca | 196 bp |
| ttggggctctgaatcctcgt (57°) | acgcag/AGGTT | exon 7 | CCAAGG/gtgag | tcttccctcccacccgctctct | 157 bp |
| aactgatccacccgccttgg (65°) | ctccag/AGACC | exon 8 | GTATCG/gtgag | accacacccatgtgtctcag | 168 bp |
| tgcaagctgaccccagtcta (56°) | cctcag/GGACC | exon 9 | GGACAG/gtggg | tcaggtcacacaggttgatg | 251 bp |
| catcaacctgtgtgacctga (61°) | ccgcag/GAACT | exon 10 | CTGCAG/gtatc | ctgctcgaaactgctgaggt | 271 bp |
| catccccctcattctcttgt (56°) | ccacag/CACAG | exon 11 | ccaattctcatccagagtgg | 389 bp | |
| Additional primers used for SSCP: | |||||
| tcgcagaacaatgtgtacgg (58°) | gaatgtagttgaactgcagc | 154 bp | |||
| cgccacccttaaaggcaa (54°) | tctcaacaccacgttccagg | 151 bp | |||
| ccttcctgaacatttactgc (60°) | atttacagtggagtgggcgt | 158 bp | |||
| tccactaccttgggttctag (58°) | agcccacagtgaatcccaca | 161 bp | |||
| tgtaagctgaccccagtcta (56°) | ttctggccgcccatccaaat | 155 bp | |||
| tttgacagcgtcctctgcag (56°) | tcaggtcacacaggttgatg | 156 bp | |||
| catcaacctgtgtgacctga (61°) | acgtgagccatggccagcag | 152 bp | |||
| agcacgggttccatctgctgt (56°) | acgtgagccatggccagcag | 184 bp | |||
| ttgacccggcttcgaacatc (58°) | aggtgagcacgccattagct | 155 bp | |||
| atgcacccactcttcccaga (56°) | gagagaggagtgggttagct | 152 bp | |||
| cctgaaggacaggatgctc (56°) | ccaattctcatccagagtgg | 154 bp | |||
Annealing temperatures for each primer set are given in brackets after the forward primer sequence.
Pedigree
The families in this study were ascertained through the University of Iowa Department of Otolaryngology – Head and Neck Surgery. A family history was obtained by questionnaire and personal interviews. Audiometric data were collected on affected persons, and blood samples obtained from cooperative living family members. Genotyping using microsatellite polymorphism and linkage analyses were performed as described (Chen et al. 1995). About 160 different markers in total were used in the initial pass (markers were selected to be about 20 cM apart) and only chromosome 19 was identified to contain a region of polymorphism that cosegregated with the disease. The LOD score for family 1070 was 4.210 for co-inheritance of KPTN allele with the deafness phenotype, but <3 for family 1030, though segregation of alleles was consistent with linkage.
Genomic structure
We determined the genomic structure of KPTN by using exonic primers at approximately 200-bp overlapping intervals on genomic DNA from peripheral blood lymphocytes of unaffected family members as template. PCR conditions were those described in the mapping section above, and annealing temperatures for each primer set are listed in Table 1. PCR products of predicted and longer-than-predicted size were directly sequenced and compared to the cDNA sequence to identify intron-exon boundaries. Analysis was facilitated by Sequencher 3.1 (Ann Arbor, MI).
Mutation screening
To determine whether mutations in KPTN caused hearing loss in DFNA4, direct sequencing and SSCP were used to screen for disease-causing mutations in the coding and the splice-site regions of 2E4-kaptin in family 1070; data from affected and non-affected family members and unrelated controls were compared. DNA sequencing reactions were conducted by using a dye terminator sequencing reaction with AmpliTaq DNA polymerase (PE Applied Biosystems). The reactions were run and analyzed with an automated sequencer (Applied Biosystems Model 377) at the University of Iowa DNA Facility. SSCP screening was performed by generating primer pairs that amplified approximately 150 bp segments of KPTN. PCR reaction components and conditions were the same as employed in determining the genomic structure of KPTN. SSCP was performed on 6% nondenaturing PAGE at room temperature. Bands were visualized with silver staining.
RESULTS
Immunofluorescence
Cochlear hair cells project clusters of stereocilia from their apical surface. Each stereocilium contains a unipolar cross-linked bundle of actin filaments which stain with phalloidin (Fig. 1A, green). KPTN can be detected with the polyclonal antibody at the tips of these stereocilia by double-label immunofluorescence (Fig. 1A, top panel, red). At the very tip, only red staining is seen, demonstrating that the KPTN extends beyond the end of the actin filaments. A region of overlap containing both actin and KPTN stains yellow and extends down each stereocilium approximately 20–30% of its length. This staining is similar to that obtained with the anti-KPTN monoclonal IgM, Mab2E4 (Fig. 1A, bottom panel). This double-label experiment significantly advances our previous results of staining obtained by staining with anti-KPTN Mab2E4 only (Bearer & Abraham, 1999). First, the double labelling provides definitive evidence that KPTN is not only co-localized with actin, but extends beyond the ends of the actin filaments, the site predicted to contain the polymerization machine; and second, polyclonal antibodies raised against cloned protein give the same pattern as MAb2E4 originally raised against endogenous protein from human blood platelets. Thus, we can have more confidence that the cloned cDNA encodes the protein present at the tips of these stereocilia in the cochlea.
Fig. 1.
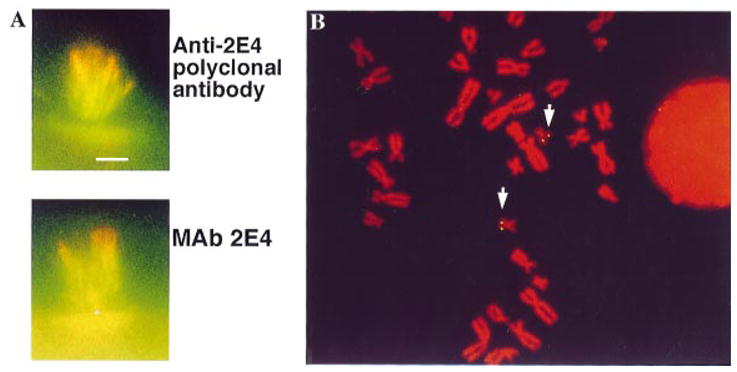
(A) Stereocilia projecting from single hair cells stained with polyclonal anti-KPTN antibody (top panel, red) or monoclonal anti-KPTN, Mab 2E4 (bottom panel, red) and double labeled for actin (both panels, green). Bar = 5 μm. (B) KPTN is encoded on chromosome 19 by fluorescence in situ hybridization. Arrows indicate the duplicated gene on both replicated chromatids of the two chromosome 19s.
Fluorescence in situ hybridization
The KPTN cDNA identified two matching sites on the replicated chromatids of both homologous chromosome 19s from this diploid nucleus (Fig. 1B). No signals were seen on other chromosomes. This cDNA is the same plasmid that produced the recombinant protein used to generate the polyclonal antibody used for the staining shown in Fig. 1A.
PCR mapping
Human cells containing the full complement of chromosomes produced the expected 240-bp product. No product was observed in either of the two host cells, mouse and hamster. The only hybrid cell line to produce a PCR of the correct size contained chromosome 19. When the same PCR reaction was carried out on DNA extracted from radiation hybrid cell lines containing fragments of chromosome 19 (unpublished, C. L. Jackson) the presence of a KPTN product showed the highest concordance (91%–95%) with 3 markers – APOC2, HRC (histidine-rich Ca-binding protein), and TNNT1 (troponin T1) – located at 19q13.2, 19q13.3, and 19p13.4, respectively. These data regionally place the location of KPTN as chromosome 19q13.2–13.4.
Additional radiation hybrid mapping with Genebridge 4, a different radiation hybrid panel, localized KPTN to the interval flanked by WI-9028 and NIB1805 in 19q13.4. The gene was further localized by screening a complete YAC library and sub-cloning to identify a single positive YAC clone, 810F7, which contains the marker D19S606. This marker has been mapped on the genetic linkage map to 76.2 cM from the top linkage group (Gyapay et al. 1994), and falls between D19S412 and D19S571 on the YAC Contig map. This set of results places KPTN between D19S412 and NIB1805, a ~2.8 cR span on the Whitehead radiation hybrid map.
We had hoped to sub-localize KPTN within the Whitehead contigs anchored on chromosome 19. The gene should fall in the interval between WI-7903 and RP S9 on the WC19.5 contig but our screen did not detect KPTN on either of two YACs reported to span this region. This contig is linked at this point by only one marker (RP S9), verified on the distal YAC but ambiguous on the proximal YAC. Our negative result suggests the contig has a gap at this point which contains KPTN and the other markers found on 810F7.
Co-segregation with DFNA4 families, genomic structure and mutation screening
In one family (1070), an autosomal dominant progressive sensorineural hearing loss which begins in the second decade and leads to profound impairment by age 40 maps to this region of chromosome 19q13 (Chen et al. 1995). Here, we describe a second family with a different phenotypic hearing impairment, a congenital sensorineural hearing deficit, which also co-segregates with polymorphic markers in this locus (Family 1030, Fig. 2).
Fig. 2.

Pedigree of a second family, with sensorineural hearing loss, that has been mapped to an interval which overlaps with the DFNA4 locus. Black and yellow chromosomes are inherited from the affected father, solid blue and white from the unaffected mother, hatched green and hatched blue contributed by unaffected spouse in the second generation. Obligate crossovers in individuals 10 and 11 define the flanking markers. (squares, male; circles, female; black filling indicates affected individual, and black chromosome co-segregates with defect). Numbers beside each chromosome indicate alleles for each marker.
Using PCR-based linkage analysis, we determined the markers that flank the co-segregating interval inherited by affected members in each of these families and compared it with the mapped location of KPTN (diagrammed in Fig. 3). There was complete overlap between the interval containing the mutation in the first family (1070) and KPTN. While the two families overlap, the interval inherited in family 1030 does not include the KPTN interval. KPTN thus remains a candidate for the gene responsible for deafness only in the 1070 family.
Fig. 3.
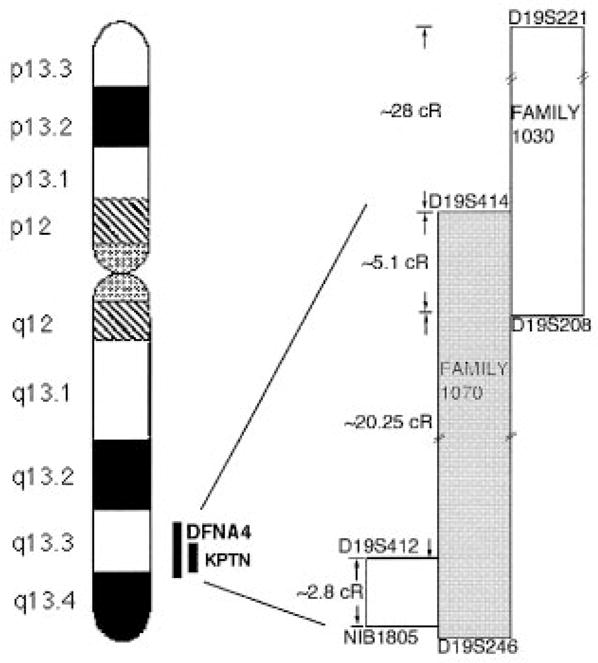
Region of interest within chromosome 19 showing the common intervals between KPTN and DFNA4 (Family 1070) and between DFNA4 and Family 1030. Drawing is not to scale. Distances are indicated in cRays according to the Whitehead RH map.
The genomic structure of KPTN consists of 11 exons with splice acceptor and donor sites following the consensus GT-AG rule (Fig. 4 and Table 1). The smallest exon is 74 bases, and the largest is greater than 300 bases. Intronic sequences vary from less than 100 bases to more than 900 bases. Sequence analysis of coding regions and splice sites in hearing-impaired persons in either the DFNA4 family (1070) or family 1030 failed to demonstrate any deafness-causing mutations.
Fig. 4.

Genomic structure of KPTN. Dark areas of boxes indicate the coding sequence. The dashed area of exon 1 indicates that the 5′ end of the gene was not resolved here, although the full length cDNA including the untranslated 5′ end for KPTN has been obtained (Bearer & Abraham, 1999). The indicated lengths of exons correspond to the coding sequence.
DISCUSSION
In this report, we show that KPTN, an actin-binding protein which we have characterized functionally in vitro (Bearer, 1992, 1995; Bearer & Abraham, 1999), extends beyond the ends of actin filaments within the stereocilia. Since the length of stereocilia is determined by their actin filaments and is a crucial aspect of their frequency specificity, it will be interesting to determine whether KPTN plays an integral role in this process.
Localization of KPTN to chromosome 19q13.4 suggested that it might be involved in non-syndromic deafness associated with the DFNA4 locus (Chen et al. 1995). Other actin-binding proteins that are associated with deafness include myosin VIIA (USH1B, DFNB2 and DFNA11) (Weil et al. 1995; Tamagawa et al. 1996) and diaphanous (DFNA1) (Lynch et al. 1997). Although we failed to demonstrate deafness-causing mutations in the coding region of KPTN in two families segregating for DFNA4 deafness, KPTN remains a candidate gene for non-syndromic deafness if other families are mapped to this genomic region. That two families with different clinical features of inherited deafness both map to this region of chromosome 19 suggests that there is either a cluster of several genes in sequence that contribute to hearing, or that different mutations in the same gene produce different clinical phenotypes of hearing deficit. There is precedence for either scenario (Keats & Berlin, 1999). An example of a gene cluster associated with deafness is the two gap junction proteins encoded by GJB2 and GJB6 on chromosome 13; and there are two different deafness-causing genes in the DFNA2 interval on chromosome 1. The lack of mutations in the coding region does not preclude abnormalities in expression levels caused by mutations in transcriptional regulatory regions of KPTN. Syntenic comparison with the murine genome further supports a role for KPTN in hearing, as it places the murine orthologue of KPTN within the Nijmegan waltzer deletion. These mice have sensorineuro-vestibular defects and a variable hearing deficiency, but little work has been done on them in the past 30 years (Van Abeelen & Van der Kroon, 1967; Van Abeelen & Kalthoven, 1970).
Acknowledgments
We thank Rosemaria Preparata and Kathleen Santoro for technical assistance. We are indebted to Linda Ashworth at Lawrence Livermore National Laboratories on the chromosome 19 project for her help in selecting markers and correlating our findings with existing maps. This work was supported by NIH GM47368 (E. L.B), grant NIH-DC03544 (RJHS) and the Solomon Faculty Research Award from Brown University (E.L.B.).
References
- Bearer EL. An actin-associated protein present in the microtubule organizing center and the growth cones of PC-12 cells. J Neurosci. 1992;12(3):750–761. doi: 10.1523/JNEUROSCI.12-03-00750.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL. Role of actin polymerization in cell locomotion: molecules and models. Am J Resp Cell and Molecular Biol. 1993;8(6):582–591. doi: 10.1165/ajrcmb/8.6.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL. Cytoskeletal domains in the activated platelet. Cell Motil Cytoskel. 1995;30:50–66. doi: 10.1002/cm.970300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, Abraham M. 2E4: A novel actin-associated protein from human blood platelets found in lamellipodia and the tips of the stereocilia of the inner ear. Euro J Cell Biol. 1999;78:117–126. doi: 10.1016/S0171-9335(99)80013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AH, et al. Linkage of a gene for dominant non-syndromic deafness to chromosome 19. Hum Mol Genet. 1995;4:1073–6. doi: 10.1093/hmg/4.6.1073. [DOI] [PubMed] [Google Scholar]
- Gyapay G, et al. The 1993–94 Genethon human genetic linkage map. Nat Genet. 1994;7(2 Spec):246–339. doi: 10.1038/ng0694supp-246. [DOI] [PubMed] [Google Scholar]
- Gyapay G, et al. A radiation hybrid map of the human genome. Hum Mol Genet. 1996;5(3):339–46. doi: 10.1093/hmg/5.3.339. [DOI] [PubMed] [Google Scholar]
- Hudspeth AJ. How hearing happens. Neuron. 1997;19:947–950. doi: 10.1016/s0896-6273(00)80385-2. [DOI] [PubMed] [Google Scholar]
- Jackson CL, et al. Construction and characterization of radiation hybrids for chromosome 9, and their use in mapping cosmid probes on the chromosome. Som Cell Molec Gen. 1992;18:285–301. doi: 10.1007/BF01233864. [DOI] [PubMed] [Google Scholar]
- Keats BJ, Berlin CI. Genomics and hearing impairment. Genome Res. 1999;9(1):7–16. [PubMed] [Google Scholar]
- Lynch ED, et al. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science. 1997;278(5341):1315–8. [PubMed] [Google Scholar]
- Mark HFL, et al. Fluorescent in situ hybridization as an adjunct to conventional cyto-genetics. Ann Clin Lab Sci. 1994;24:153–163. [PubMed] [Google Scholar]
- Steel KP. Inherited hearing defects in mice. Ann Rev Genet. 1995;29:675–701. doi: 10.1146/annurev.ge.29.120195.003331. [DOI] [PubMed] [Google Scholar]
- Steel KP, et al. Unravelling the genetics of deafness. Ann Otol Rhinol Laryngol Suppl. 1997;168:59–62. [PubMed] [Google Scholar]
- Tamagawa Y, et al. A gene for a dominant form of non-syndromic sensorineural deafness (DFNA11) maps within the region containing the DFNB2 recessive deafness gene. Hum Mol Genet. 1996;5(6):849–52. doi: 10.1093/hmg/5.6.849. [DOI] [PubMed] [Google Scholar]
- Tilney LG, et al. Actin filaments, stereocilia, and hair cells of the bird cochlea V. How the staircase pattern of stereociliary length is generated. J Cell Biol. 1988;107:2563–2574. doi: 10.1083/jcb.106.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, et al. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu Rev Cell Biol. 1992;8:257–74. doi: 10.1146/annurev.cb.08.110192.001353. [DOI] [PubMed] [Google Scholar]
- Van Abeelen J, Kalthoven J. Behavioral ontogeny of the Nijmegan waltzer, a neurological mutant in the mouse. Anim Behav. 1970;18:711–718. doi: 10.1016/0003-3472(70)90016-3. [DOI] [PubMed] [Google Scholar]
- Van Abeelen J, Van der Kroon P. Nijmegan walzer – a new neurological mutant in the mouse. Genet Res Camb. 1967;10:117–118. doi: 10.1017/s0016672300010818. [DOI] [PubMed] [Google Scholar]
- Weil D, et al. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16:191–3. doi: 10.1038/ng0697-191. [DOI] [PubMed] [Google Scholar]