

Abstract
Protein methylation is predominantly found on lysine and arginine residues, and carries many important biological functions, including gene regulation and signal transduction. Given their important involvement in gene expression, protein methylation and their regulatory enzymes are implicated in a variety of human disease states such as cancer, coronary heart disease and neurodegenerative disorders. Thus, identification of methylation sites can be very helpful for the drug designs of various related diseases. In this study, we developed a method called PMeS to improve the prediction of protein methylation sites based on an enhanced feature encoding scheme and support vector machine. The enhanced feature encoding scheme was composed of the sparse property coding, normalized van der Waals volume, position weight amino acid composition and accessible surface area. The PMeS achieved a promising performance with a sensitivity of 92.45%, a specificity of 93.18%, an accuracy of 92.82% and a Matthew’s correlation coefficient of 85.69% for arginine as well as a sensitivity of 84.38%, a specificity of 93.94%, an accuracy of 89.16% and a Matthew’s correlation coefficient of 78.68% for lysine in 10-fold cross validation. Compared with other existing methods, the PMeS provides better predictive performance and greater robustness. It can be anticipated that the PMeS might be useful to guide future experiments needed to identify potential methylation sites in proteins of interest. The online service is available at http://bioinfo.ncu.edu.cn/inquiries_PMeS.aspx.
Introduction
Protein methylation, which was discovered more than 40 years ago [1], is an important and reversible protein post-translational modification (PTM). This PTM includes N-methylation [2], [3], [4] of either the backbone or side-chain of arginine, lysine, histidine, proline, alanine and asparagine, O-methylation [5] of either internal carboxyl groups of glutamate or isoaspartate residues and COOH-terminal lipidated cysteine residues, and S-methylation [6] of either cysteine or methionine residues. Among them, arginine and lysine are the most frequently methylated residues. Arginine methylation is catalyzed by a family of enzymes called protein arginine methyltransferases (PRMTs) [7]. PRMTs are classified into two groups, type I PRMTs catalyze the formation of NG-monomethylarginine (MMA) and asymmetric ω-NG, NG-dimethylarginine (aDMA), type II enzymes form MMA and symmetric ω-NG, N′G-dimethylarginine (sDMA) [8]. Similarly, lysine methylation involves the addition of one to three methyl groups on the amino acid’s ε-amine group, to form mono-, di- or tri-methyllysine by lysine methyltransferases (KMTs) [2]. Lysine specific demethylases (KDMs) work in coordination with histone lysine methylases to maintain global histone methylation patterns [9].
It has now been shown that protein arginine methylation has an important role in gene regulation and signal transduction, and lysine methylation is correlated with either gene activation or repression depending on the site and degree of methylation [10]. Given their important involvement in gene regulation, arginine methylation, lysine methylation and their regulatory enzymes are implicated in a variety of human disease states such as cancer [9], [11], coronary heart disease [12], multiple sclerosis [13], rheumatoid arthritis [14] and neurodegenerative disorders [15]. Thus, understanding the mechanisms governing these basic epigenetic phenomena will surely represent a very attractive target for drug discovery to prevent the onset of various related diseases. Furthermore, identification of protein methylation sites is of fundamental importance to understand the methylation dynamics and molecular mechanism. Unfortunately, it is often laborious, time intensive and expensive to determine protein methylation sites using conventional experiments including methylation-specific antibodies, Chip-Chip and mass spectrometry [16]–[18]. Therefore, a robust computational prediction tool is desirable to reduce the number of experiments needed to identify potential methylation sites in proteins of interest.
Actually, several computational methods have been developed to handle these methylation sites prediction problems from primary protein sequences. Plewczynski et al. [19] designed the first methylation sites predictor within their AutoMotif Server using regular expression technique. Subsequently, Daily et al. [20] developed a method for arginine and lysine methylation prediction, using support vector machine (SVM) based on the hypothesis that PTMs preferentially occur in intrinsically disordered regions. Chen et al. [21] built a web server MeMo for identifying methylation sites by utilizing orthogonal binary coding scheme to represent protein sequence fragment. Further, Shao et al. [22] combined Bi-profile Bayes feature extraction with SVM to predict arginine and lysine methylation. MASA was constructed by Shien et al. [23] for methylation sites prediction, where considered both sequence information and structural characteristics such as accessible surface area (ASA) and secondary structure of residues surrounding methylation sites. Recently, Hu et al. [24] presented a method for predicting protein methylarginine and methyllysine based on multi-sequence features and nearest neighbor algorithm.
However, most existing prediction methods applied orthogonal encoding scheme to characterize protein sequence information. The orthogonal encoding uses a 20 dimensional vector of binary values 0 or 1 to represent each residue. Each bit in this vector means the occurrence of one kind of amino acid. Thus, there is one 1 and nineteen 0 in each vector. It is obvious that orthogonal representation doesn’t contain preferences on amino acids or position information and physicochemical properties of residues. Additionally, the highest prediction sensitivity was 82.1% for methylarginine [23], only 79.73% for methyllysine among the existing methods [24]. Hence it has become a crucial issue to improve the quality of predicting protein methylation sites by selecting more informative feature descriptors.
In view of this, a novel approach called PMeS was developed to identify methylation sites based on an enhanced feature encoding scheme for extracting the most informative amino acids features. Here, the enhanced feature encoding scheme was composed of sparse property coding (SPC), normalized van der Waals volume (VDWV), position weight amino acid composition (PWAA) and solvent accessible surface area (ASA). SPC and VDWV were utilized to characterize protein sequence information and physicochemical properties of amino acids surrounding methylation sites. PWAA and ASA were applied to represent sequence-order information and structural characteristic around methylation sites, respectively. Our current work contained the following contents: (1) four types of features and feature analysis were considered; (2) SVM was employed to deal with the problem of binary classification; (3) ten-fold cross-validation method was chosen to evaluate the performance of SVM classifier; (4) the effect of window length was discussed; (5) the ratio of positive to negative samples was investigated; (6) the robustness of PMeS was considered; and (7) the predictive performance of PMeS was compared with that of the existing models.
Materials and Methods
Data Collection
All training data were extracted from UniProtKB/Swiss-Prot database (version 2011_05, www.uniprot.org) and PhosphoSitePlus (2011_05, www.phosphosite.org). Firstly, we obtained 98 proteins covering 246 experimental methylarginine sites by searching information containing “Omega-N-methylarginine”, “symmetric dimethylarginine” and “asymmetric dimethylarginine”, and 137 proteins covering 367 experimental methyllysine sites through the keywords “N6, N6, N6-trimethyllysine”, “N6, N6-dimethyllysine” and “N6-methyllysine” from UniProtKB/Swiss-Prot database (see Tables S1 and S2). PhosphoSitePlus is an online systems biology resource providing an extensive, manually curated phosphorylation site database and other commonly studied PTMs including acetylation, methylation, ubiquitination, and O-glycosylation. We obtained 68 non-redundant proteins covering 155 experimental methylarginine sites and 78 non-redundant proteins covering 147 experimental methyllysine sites from PhosphoSitePlus (see Tables S3 and S4). However, the dataset may contain several high sequence identity proteins. To avoid such overestimation of predictive performance, we clustered the protein sequences with a threshold of 40% identity by CD-HIT program [25] to remove the highly homologous sequences.
Secondly, the sliding window strategy was utilized to extract positive and negative data from protein sequences as training data, which were represented by peptide sequences with arginine and lysine symmetrically surrounded by flanking residues. Experimentally validated methylarginine and methyllysine were defined as positive datasets, excluding those annotated by “potential”, “probable” or “by similarity” in the description field. Negative datasets included all arginines and lysines that were not marked by any methylation information on the same proteins. Although not all of these sites are necessarily true negatives, it is reasonable to believe that a large majority of them are [26]. Moreover, the redundancy reducing process was also carried out on training data. For example, for two methylated arginine peptide sequences with 100% identity, when the methylarginine sites in the two proteins were in the same positions, only one was kept. After strictly following the above procedures, we attained 355 high quality positive sites and 3960 negative sites for methylarginine, and 322 positive sites and 4126 negative sites for methyllysine. Here, the feasible window size for both arginine and lysine was 15 after several trials of 9, 11, 13, 15, 17 and 19.
Finally, to ensure unbiased and objective results, five negative training sets were obtained by randomly extracting from the negative datasets. The average predictive performance obtained using the five sets of training data was calculated by the following cross-validation.
The Enhanced Feature Encoding Scheme
Sparse property coding
The specificity and diversity of protein structure and function are largely attributed to the composition of various properties of each of the 20 amino acids [27]. Physicochemical encoding is particularly suited for peptides since it exploits the fixed length of the sequence [28]. Peptide sequences have been coded using physicochemical properties in three ways: sparse property coding, continuous property coding and property projection coding [29]. Methylation on lysine and arginine residues does not alter their charge, but it does increase their hydrophobicity [30], [31]. Thus, we adopted a sparse property coding based on the hydrophobicity and charged character of amino acid residue. The sparse property coding (SPC) divided the 20 amino acid residues into four different groups according to their hydrophobicity and charged character: the hydrophobic group G1 = {A,F,G,I,L,M,P,V,W}, the polar group G2 = {C,N,Q,S,T,Y}, the positively charged group G3 = {H,K,R} and the negatively charged group G4 = {D,E} [32]. Then each amino acid residue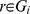 was encoded as follows:
was encoded as follows:
| (1) |
where 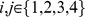 and
and 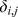 is the Kronecker delta symbol. Consequently, a peptide sequence p with sliding window size N can be mapped to a 4N-dimension vector
is the Kronecker delta symbol. Consequently, a peptide sequence p with sliding window size N can be mapped to a 4N-dimension vector
| (2) |
within the feature space by concatenating the encoded amino acids, where 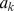 is the kth position residue in peptide sequence p.
is the kth position residue in peptide sequence p.
The SPC reflects the distribution of residues with the same unique characteristic and portrays the essence of protein sequence. It can effectively overcome the defect of orthogonal encoding which doesn’t contain physicochemical properties of amino acids. On the other hand, the SPC reduces the dimension of the input space, so the computational complexity is largely decreased.
Van der Waals volume (VDWV)
Van der Waals volume of side groups is a determinant for binding sites [33]. Therefore, we took into account the normalized van der Waals volume (VDWV) of the amino acid side chain as a feature to code the peptides. The normalized van der Waals volume of 20 kinds of amino acids is presented in Supplementary Table S5 [34].
Position weight amino acid composition
To avoid losing the sequence-order information, we presented position weight amino acids composition (PWAA) to extract the sequence position information of amino acid residues around the methylation sites and non-methylation sites. Given an amino acid residue ai (i = 1,2,…,20), we can express the position information of amino acid ai in the protein sequence fragment p with 2L +1 amino acids by following formula:
| (3) |
where L denotes the number of upstream residues or downstream residues from the central site in the protein sequence fragment p, 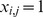 if ai is the jth position residue in protein sequence fragment p, otherwise
if ai is the jth position residue in protein sequence fragment p, otherwise 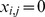 . In general, the closer residue ai is to the central site (0 position), the absolute value of
. In general, the closer residue ai is to the central site (0 position), the absolute value of 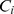 is smaller. Finally, a protein sequence fragment p is defined as 20 dimension feature vectors.
is smaller. Finally, a protein sequence fragment p is defined as 20 dimension feature vectors.
| (4) |
Solvent accessible surface area
A side-chain of amino acid that undergoes post-translational modification (PTM) prefers to be accessible on the surface of a protein [35]. Pang et al. [35] investigated the structural environment of 8378 incidences of 44 types of post-translational modifications (PTMs). It has been observed that protein methylation prefers to occur in regions that are intrinsically disorder and easily accessible. Therefore, the solvent accessibility of amino acid residues surrounding the methylation sites may be adapted to evaluate the classifying performance when distinguishes between the methylation site and non-methylation sites.
Most of the experimental methylation proteins do not have corresponding protein tertiary structures in the protein data bank (PDB). Consequently, we used RVP-Net [36], [37] to calculate the solvent accessible surface area (ASA) for each residue of a protein sequence. RVP-net applied a neural network to predict real value of ASA of residues based on neighborhood information, with 18.0–19.5% mean absolute error, defined as per residue absolute difference between the predicted and experimental values of relative ASA [36]. The computed ASA value was the percentage of the solvent-accessible area of each amino acid on the protein sequence. The ASA values of amino acids surrounding the methylation site were extracted and normalized.
Support Vector Machine
SVM is a supervised learning method for classification and regression designed by Vapnik [38]. The principle of the SVM method is to transform the samples into a high dimension Hilbert space and seek an optimal separating hyperplane which maximizes the margin in feature space. SVM has shown successful ability to classify complex data sets without over-fitting issues, thus it’s considered as a machine learning tool for methylation prediction. For actual implementation we used the LIBSVM package (version 3.0) [39]. Here, a radial basis function was chosen as the kernel function, the penalty parameter and the kernel width parameter were tuned based on the training set using the grid search strategy in LIBSVM.
Evaluation Methods
Ten-fold cross-validation was applied to evaluate the powers of the prediction method proposed in this study. The training data are divided into 10 groups by splitting each dataset into 10 approximately equal-sized subgroups. Then 9 subgroups are merged into a training data set while the remnant subgroup is taken as a testing data set. This process is repeated 10 times and the average performance of 10-fold cross-validation is used to estimate the performance. We adopted four major parameters for performance assessment: sensitivity (Sn), specificity (Sp), accuracy (Acc) and Matthews Correlation Coefficient (MCC). All of the above measurements are defined as follows:
| (5) |
| (6) |
| (7) |
| (8) |
where TP,TN,FP,FN denote the number of true positives, true negatives, false positives and false negatives, respectively. Sensitivity and specificity illustrate the correct prediction ratios of positive (methylation) samples and negative (non-methylation) samples respectively, while accuracy represents the correct ratio among both positive and negative data sets. The MCC takes into account true and false positives and negatives, and it is generally regarded as a balanced measure which can be used even if the classes are of very different sizes, for these reasons the MCC is more reliable than the accuracy. The value of MCC ranges from −1 to 1, and a larger MCC stands for better prediction performance.
Results and Discussion
Investigation of Different Features
As described in the Materials and Methods section, the enhanced feature encoding scheme included four types of features: sparse property coding (SPC), normalized van der Waals volume (VDWV), position weight amino acids composition (PWAA) and solvent accessible surface area (ASA). Here we constructed ten prediction models composed by SPC, VDWV, PWAA and ASA to investigate the influences of different features.
SPC Feature Analysis
As mentioned above, the SPC feature is mainly based on the hydrophobicity and charged character of amino acid residue. To determine whether methylation and non-methylation sites have distinct physiochemical properties, we calculated statistically significant differences in the distribution of physicochemical properties of amino acid residues surrounding methylation and non-methylation sites based on the paired Welch's t-test. As shown in Figure 1, from −7 to +7 positions, the ratios of hydrophobic amino acids around methylarginine were 2.3% to 29.2% higher than those of non-methylarginine with P-value ≤3.59e-02 (see Table S6). Especially for the +1 position, hydrophobic residues around methylarginine account for 77.3%, about 29.2% higher than those of non-methylarginine (P = 3.84e-09). From −7 to +7 positions, polar and negatively charged residues surrounding non-methylarginine were 1.15% to 7.12% higher than those of methylarginine (P<0.05). This analysis reveals that methylarginine and non-methylarginine have distinct physiochemical properties. In fact, some studies suggested that the arginine residue becomes more hydrophobic due to addition of methyl groups and may engage in more van der Waal interactions [8].
Figure 1. The distribution of physicochemical properties of residues around methylarginine and non-methylarginine.

G1 is hydrophobic residue, G2 is polar residue, G3 is positively charged residue, and G4 is negatively charged residue.
While compared with non-methyllysine, the ratios of four different attributive residues around methyllysine have not changed much, as shown in Figure 2, which indicates that the incorporation of methyl groups to the lysine side chain changes the physicochemical properties of the affected residues only slightly. It is worth noting that the ratios of polar residues surrounding methyllysine were 2.81% to 6.03% higher than those of non-methyllysine from −5 to −2 positions (P≤8.46e-04). Most enzymes bind the methyllysine in a polar environment, which resembles the ‘carbonyl cage’ of SET domains rather than the hydrophobic pockets of chromo domain-related motifs [40]. The methyl groups are coordinated by a set of electrostatic interactions between polar residues of the protein and the trimethylammonium. CH…O-H bonds form between oxygen on the enzyme's sidechains and methyl groups of the methyllysine [41]. These interactions cumulatively position one of the methyl groups in the vicinity of the iron for hydroxylation to occur [24]. All these researches strengthen the role of surrounding sites in the enzymes' reorganization.
Figure 2. The distribution of physicochemical properties of residues around methyllysine and non-methyllysine.

G1 is hydrophobic residue, G2 is polar residue, G3 is positively charged residue, and G4 is negatively charged residue.
VDWV feature analysis
Figure 3 gives the mean values of normalized van der Waals volume (VDWV) of residues around methylation sites and non-methylation sites based on training data. From −7 to +7 positions, the mean values of VDWV of residues surrounding methylarginine were lower than those of non-methylarginine, especially for the -1 and +1 position. Most of P-values were less than 0.05 (see Table S7), indicating that there was significant difference between the VDWV surrounding methylarginine and that surrounding non-methylarginine. From −7 to −1 positions, there was obvious difference between the VDWV surrounding methyllysine and that surrounding non-methyllysine (P≤1.24e-05). This reveals that the upstream residues may have a significant influence on methyllysine.
Figure 3. The mean value of normalized van der Waals volume (VDWV) of residues around methylation sites and non-methylation sites.

PWAA feature analysis
PWAA feature reflects the position information of residues surrounding methylation sites and non-methylation sites. In order to analyze position specific properties, we adopted WebLogo [42] to generate the graphical sequence logo for the relative frequency of the corresponding amino acid at each position around methylation and non-methylation sites. As we can see from Figure 4, the methylated arginines (R) are enriched in arginine-glycine (R–G) regions which are much different from non-methylated arginines. Indeed, motif analysis reveals many arginine methylation are associated with RGG/RXG/RGX [43] or GXXR [20] motifs. The conserved residues at specific sequence sites are under strong selective pressure and therefore are always functional relevant. The type I PRMTs is known to methylate a number of proteins that contain an RGG-motif [44]. The repeated RGG-motif is known as a RNA-binding motif [45], and this also supports the role of arginine methylation in the regulation of mRNA binding [46]. In contrast, no amino acids surrounding methylated lysines (K) are obviously conserved in the current available data (Fig. 4). Therefore, sequence profiles of the flanking regions of methylarginine are more conservative with higher specificity than those of methyllysine.
Figure 4. Sequence logo plots of methylation sites and non-methylation sites represent normalized amino acid frequencies for ±7 amino acids.

ASA feature analysis
Figure 5 summarizes the average accessible surface area (ASA) formed from the 15-mer methylation sites and the 15-mer non-methylation sites in the constructed data set. Most of the methylation or non-methylation sites (0 position) were located in the highly ASA, which was consistent with those data reported in the literature [35]. The average ASA of neighborhood residues were 23.09% to 39.01% and 25.54% to 49.90% for methylarginine and methyllysine, respectively. The fluctuant range of ASA of residues surrounding methylation sites was bigger than that of non-methylation sites. This implies that the methylation processing might have occurred where the structural surroundings are relatively large variation range. The mean ASA that surrounds the methylarginine exceeded that around non-methylarginine, especially in the −6, −3, 0, +1, +4, +5 and +6 positions (P≤5.21e-03, see Table S8). Interestingly, the mean ASA around the methyllysine was slightly below that around non-methyllysine, especially in the −6, −3, −1, +2 and +3 positions (P≤3.06e-02). Generally speaking, the ASA of residues around the methylation sites and non-methylation sites have a little difference.
Figure 5. The average accessible surface area (ASA) of residues around methylation sites and non-methylation sites.

There were two possible reasons for limiting the ASA analysis in the methylation: first, the negative sites were obtained as not being previously experimentally identified; second, the ASA values were predicted by RVP-Net server. Table S9 gives the predicted ASA and experimental ASA of methylation sites with known tertiary structure of protein data bank. There are some differences between the predicted ASA and experimental ASA of methylation sites. The experimental ASA of most methyllysine are more than 30%. In the RVP-Net, the residue is exposed when its ASA is more than 16%. Thus, it seems that it may be important that the methyllysine need be solvent exposed. While the experimental ASA of several methylarginine (eg. P53674, R230 and R231) are lower than 12%. If the experimental ASA of most methylation sites were obtained, we could get a more reliable conclusion.
Optimal feature set
When the window size was 15 and the ratio between positive and negative samples was 1∶3, the predictive performance of models trained with various features for methylarginine and methyllysine are shown in Tables 1 and 2, respectively. According to statistical comparison of sensitivity (see Table S10), the model trained with SPC outperformed that trained with PWAA, VDWV or ASA (P≤2.21e-03), which was in agreement with the results of above feature analysis. But in general, the models trained with individual features could not effectively distinguish methylation sites from non-methylation sites. However, the predictive performance of the methyllysine model trained with the combination of SPC and PWAA (SPC+PWAA) or SPC, PWAA and ASA (SPC+PWAA+ASA) had some improvements (P≤2.48e-02). The predictive performance of the methylarginine model trained with the combination of SPC, PWAA and ASA (SPC+PWAA+ASA) also had some improvements (P≤6.92e-04). Furthermore, both methylarginine and methyllysine, the performance of the model trained with SPC+PWAA+ASA+VDWV had been remarkably enhanced (P<0.05). This demonstrated that all four types of features contributed to distinguishing between methylation sites and non-methylation sites. There was a strong complementary effect among these features. Henceforth, the combination of SPC, PWAA, ASA and VDWV was selected as an optimal feature set to learn the predictive model.
Table 1. The performance of models trained with various features for methylarginine.
| Training features | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
| SPC | 68.96±1.52 | 92.71±1.09 | 86.78±0.56 | 63.78±1.66 |
| PWAA | 60.85±0.57 | 95.51±0.50 | 86.85±0.41 | 62.76±1.16 |
| ASA | 51.94±0.15 | 99.81±0.15 | 87.84±0.12 | 66.37±0.45 |
| VDWV | 56.34±1.22 | 98.91±0.34 | 88.24±0.54 | 67.08±1.81 |
| SPC+PWAA | 71.66±2.14 | 91.87±0.60 | 86.82±0.75 | 64.40±2.55 |
| SPC+ASA | 65.69±1.22 | 95.46±0.69 | 88.01±0.68 | 66.42±1.93 |
| PWAA+ASA | 66.42±2.37 | 92.13±0.90 | 85.70±1.02 | 60.72±2.78 |
| SPC+PWAA+ASA | 74.09±1.44 | 93.35±1.45 | 88.54±0.64 | 68.93±1.88 |
| SPC+PWAA+VDWV | 74.54±3.56 | 94.31±1.17 | 89.37±0.83 | 71.69±3.79 |
| SPC+PWAA+ASA+VDWV | 80.73±1.58 | 92.28±1.24 | 89.39±1.35 | 72.45±1.73 |
The corresponding measurement was represented as the average value±standard deviation. The window size was 15 and the ratio between positive and negative samples was 1∶3.
Table 2. The performance of models trained with various features for methyllysine.
| Training features | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
| SPC | 61.69±1.00 | 99.79±0.16 | 90.31±0.21 | 73.44±0.60 |
| PWAA | 58.88±0.65 | 99.46±0.20 | 89.36±0.27 | 70.53±0.86 |
| ASA | 53.31±3.04 | 98.34±0.33 | 87.14±0.82 | 63.40±2.62 |
| VDWV | 54.38±2.49 | 99.48±0.35 | 88.26±0.42 | 67.27±1.16 |
| SPC+PWAA | 65.88±1.77 | 99.79±0.16 | 91.35±0.43 | 76.40±1.23 |
| SPC+ASA | 64.44±3.32 | 99.01±0.44 | 90.40±0.76 | 73.41±2.23 |
| PWAA+ASA | 63.00±3.29 | 99.34±0.35 | 90.30±0.76 | 73.20±2.21 |
| SPC+PWAA+ASA | 69.94±2.78 | 99.11±0.40 | 91.85±0.64 | 77.59±1.84 |
| SPC+PWAA+VDWV | 68.88±3.74 | 99.83±0.15 | 92.13±1.19 | 78.59±2.54 |
| SPC+PWAA+ASA+VDWV | 73.56±2.08 | 99.11±0.39 | 92.75±0.25 | 80.15±0.64 |
The corresponding measurement was represented as the average value±standard deviation. The window size was 15 and the ratio between positive and negative samples was 1∶3.
Moreover, we noticed that the performance of the predictive models on arginine was much better than on lysine in Table 1 and 2. This observation agrees with the above feature analysis, which the difference of the physiochemical properties between methylarginine and non-methylarginine is more obvious than that of methyllysine and non-methyllysine, and the sequence pattern of methylarginine is more conservative with higher specificity than that of methyllysine.
Investigation of Window Sizes
For each methylation or non-methylation sites, its profile feature and ASA feature were taken from a sequence fragment containing the n nearest residues (spatially); thus, it is crucial to confirm the appropriate window size and to realize its effects on the prediction performance. The predictive performance of models trained with different window sizes (9 to 19) are illustrated in Tables S11 and S12, where training feature was SPC+PWAA+ASA+VDWV and the ratio between positive and negative samples was 1∶3. The results showed that the window size had much more impact on the Sn and MCC than on the Sp and Acc, especially for methylarginine. Based on statistical comparison of sensitivity (see Table S13), there were significant differences between the methylarginine model with window size of 15 and those of 9, 13, 17, 19 (P≤3.29e-03). The methyllysine model with window size of 15 outperformed that with window sizes of 9, 11, and 19 (P≤1.25e-02). There was no statistical difference among the methyllysine model with window sizes of 13, 15 and 17 (P>1.44e-01). Based on the computational efficiency and overall performance of the models trained with different window length, 15-mer was adopted as the feasible window size for the two methylation residues in this study.
Investigation of the Ratios between Positive Samples and Negative Samples
As we can see from the Table 1 and 2, the Sp and Acc were relatively stable on different features, whereas the Sn and MCC fluctuated wildly, and it was relatively hard to get a higher sensitivity when the ratio of positive samples to negative samples was 1∶3. This is because the positive examples are extremely few and one incorrect prediction leads to a large decrease on sensitivity, and a larger negative set would cause the trained model preferentially to predict negative data correctly, driven by the requirement to maximize accuracy. Thus, it is very important to use a suitable ratio between positive samples and negative samples to construct the prediction model. As shown in Tables S14 and S15, after the ratio between positive and negative samples arrived at 1∶5, the MCC of the predictive models using different ratios of positive and negative samples decreased with increasing the size of the negative set (P≤2.38e-02, see Table S16). The best performance of methylarginine models was obtained when the ratio between positive and negative samples was 1∶1 (P≤1.42e-02). The corresponding Sn, Sp, Acc and MCC were 92.45%, 93.18%, 92.82% and 85.69%, respectively. For methyllysine, when the ratio between positive and negative samples were 1∶1 and 1∶3, there was no statistical difference based on MCC comparison (P = 5.43e-02). Except for 1∶3, when the ratio between positive and negative samples was 1∶1 the best performance of methyllysine models was obtained (P≤1.85e-02), the Sn, Sp, Acc and MCC were 84.38%, 93.94%, 89.16% and 78.68%, respectively. Given the narrowing of the gap between the sensitivity and the specificity, 1∶1 was as the suitable ratio between positive samples and negative samples to construct the optimal predictive model PMeS.
Investigation of the Robustness of PMeS
To test the robustness of our predictive model PMeS, the self-consistency validation, leave-one-out validation and K-fold cross-validation were calculated. Table S17 presents the three test performances of methylarginine model. Based on MCC comparison (see Table S18), there was no statistical difference among different cross-validation (P≥6.29e-01). Importantly, it is proposed that the leave-one-out test might overfit in small samples, whereas the K-fold cross-validation should do better [47]. However, we observed that the leave-one-out test results were quite similar with 4-, 6-, 8-, 10-fold cross-validations, which demonstrated the robustness and stability of the PMeS. One vital factor that could result in misleadingly high prediction performance and possibly influence prediction stability is sequence homology in training dataset [48]. As described in the Data collection section, we carried out homology reducing process on training dataset. This data preprocessing might be helpful to enhance the robustness of the PMeS.
Independent Test
Moreover, to validate our algorithm against other sources of methylation data from experimental papers, we collected 46 experimental methyllysine sites and 27 experimental methylarginine sites from scientific literatures to construct the independent test sets (see Tables S19 and S20). None of independent test proteins was included in the training dataset. As shown in Table 3, besides the Sn, the other three measurements of independent test for methylarginine were quite similar with those of training test (P≥1.12e-01, see Table S21). For methyllysine, the Sp of the independent test was slightly higher than that of training test (P = 2.40e-03), the other three measurements of independent test were 3.29% to 8.29% lower than those of training test (P≤5.50e-03). If the performance of the independent test is much worse than that of training test, then the trained model may be over-fitting for the training data. Generally, the performance of the independent test was just a little lower than those of training test, which was also acceptable. Moreover, the negative sites were obtained as not being previously experimentally identified, which might be a possible reason for influencing the predictive results.
Table 3. Independent test results of PMeS.
| Residue type | Number of positivetest data | Number of negativetest data | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
| Arginine | 27 | 27 | 85.19±3.64 | 96.30±3.25 | 90.74±2.18 | 81.99±3.95 |
| Lysine | 46 | 46 | 76.09±1.90 | 95.65±3.03 | 85.87±2.49 | 73.15±3.41 |
The corresponding measurement was represented as the average value±standard deviation.
Comparisons with Existing Methods
In order to further evaluate the prediction performance of the PMeS method objectively, we made comparisons with other methylation predictor. Here the performance of the PMeS on the dataset adopted in MASA [23] and Hu's method [24] were evaluated as shown in Tables 4 and 5, respectively. For methyllysine, the four measurements in PMeS were 7.99% to 34.24% higher than those in MASA (P≤4.18e-04, see Table S22). For methylarginine, besides the Sp (P = 2.21e-01), the other three measurements in PMeS were 3.41% to 7.01% higher than those in MASA (P≤3.52e-02). Compared with the training features (AA+ASA) in MASA, our significant improvements can be attributed to the adoption of the physicochemical properties of residues, as elucidated in the above feature analysis, the physicochemical properties are effective in identifying methylation status. Similarly, for methyllysine, except the Sn (P = 4.70e-01, see Table S23), the other three measurements in PMeS were 6.76% to 14.26% higher than those in Hu's method (P<4.67e-02). For methylarginine, the four measurements in PMeS were so much better than those in Hu's method (P<0.05). Compared with Hu's method, our improvements may come from SPC feature. In some problems (e.g. HIV protease), where the training set could be not completely representative of the test set, the sparse orthonormal representation works very well [49]. In summary, the PMeS outperformed MASA and Hu's method, which justified the effectiveness of SPC+PWAA+ASA+VDWV as feature for methylation sites prediction.
Table 4. Comparison of PMeS with MASA on the dataset adopted in MASA method.
| Prediction methods | Residue type | Training features | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
| MASA | Arginine | AA+ASA | 82.1 | 87.4 | 84.8 | 69.6(a) |
| Lysine | AA+ASA | 75.1 | 74.0 | 74.6 | 49.2(b) | |
| PMeS(c) | Arginine | SPC+PWAA+ASA+VDWV | 86.18±2.43 | 90.24±2.33 | 88.21±1.29 | 76.61±4.02 |
| Lysine | SPC+PWAA+ASA+VDWV | 83.09±3.14 | 99.23±0.84 | 91.16±1.69 | 83.44±3.07 |
The MCC for methylarginine in MASA [23] was 79.6%, which was the author’s mistake in calculation. We corrected it for 69.6% by the calculating formula of MCC. (b)The MCC for methyllysine in MASA was 56.1%, which was the author’s mistake in calculation. We corrected it for 49.2% by the calculating formula of MCC. (c) The corresponding measurement was represented as the average value±standard deviation. Abbreviation: AA, amino acid.
Table 5. Comparison of PMeS with Hu’s method on the dataset adopted in Hu's method.
| Prediction methods | Residue type | Training features | Sn (%) | Sp (%) | Acc (%) | MCC (%) |
| Hu's method | Arginine | AAF+PSSM+SD | 74.39±2.21 | 74.11±3.27 | 74.25±1.46 | 48.52±2.85 |
| Lysine | AAF+PSSM+SD | 79.73±1.66 | 74.54±3.61 | 77.02±1.95 | 54.28±3.74 | |
| PMeS | Arginine | SPC+PWAA+ASA+VDWV | 82.03±2.53 | 84.41±3.82 | 83.22±3.06 | 66.57±4.53 |
| Lysine | SPC+PWAA+ASA+VDWV | 79.11±2.98 | 88.44±2.52 | 83.78±1.48 | 68.54±4.79 |
The corresponding measurement was represented as the average value±standard deviation. Abbreviation: AAF, amino acid factors; PSSM, position specific scoring matrix; SD, structural disorder.
Conclusion
Methylation prediction methods in previous studies, such as MeMo [21], BPB-PPMS [22] and MASA [23], have focused only on orthogonal encoding scheme to represent protein sequence information, where do not contain preferences on amino acids or position information and physicochemical properties of residues. However, the enhanced feature encoding scheme PMeS in this study incorporated the amino acid sequence, position information, physicochemical properties of residues with structural characteristic to improve the prediction of protein methylation sites. Feature analysis showed that methylation and non-methylation sites had distinct physiochemical properties, and the SPC, VDWV, PWAA and ASA features all contributed to the methylation prediction. The cross-validation results demonstrated that PMeS achieved a promising performance and outperformed other methylation prediction tools. In addition, the PMeS had a greater robustness. It can be anticipated that the PMeS might be useful to guide future experiments needed to identify potential methylation sites in proteins of interest. Datasets and Matlab code can be downloaded from our website (http://bioinfo.ncu.edu.cn/inquiries_PMeS.aspx).
Supporting Information
246 experimentally identified methylarginine sites in 98 proteins were extracted from UniProtKB/Swiss-Prot database.
(DOC)
367 experimentally identified methyllysine sites in 137 proteins were extracted from UniProtKB/Swiss-Prot database.
(DOC)
155 methylarginine sites in 68 proteins were extracted from PhosphoSitePlus.
(DOC)
147 methyllysine sites in 78 proteins were extracted from PhosphoSitePlus.
(DOC)
The normalized van der Waals volume of 20 kinds of amino acids.
(DOC)
The distribution of physicochemical properties of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
Average van der Waals volume (VDWV) of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
Average accessible surface area (ASA) of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
The list of proteins containing experimental methylation sites which are located in the protein regions with known tertiary structure of protein data bank (PDB).
(DOC)
The predictive performance of model trained with different features was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of models trained with different window sizes for methylarginine.
(DOC)
The performance of models trained with different window sizes for methyllysine.
(DOC)
The predictive result of models with different window sizes was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of models trained with different ratio of positive to negative samples for methylarginine.
(DOC)
The performance of models trained with different ratio of positive to negative samples for methyllysine.
(DOC)
The predictive result of models with different ratios of positive to negative samples was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of the methylarginine model based on self-consistency, K-fold (4-, 6-, 8- and 10-fold) cross-validation and leave-one-out validation.
(DOC)
MCC of methylarginine model by different cross-validation was compared via P -values on the paired Welch's t-test.
(DOC)
We collected 46 experimentally identified methyllysine sites in 39 unique proteins from the scientific literature (PubMed).
(DOC)
We collected 27 experimentally identified methylarginine sites in 24 unique proteins from the scientific literature (PubMed).
(DOC)
Statistical comparison of training test with independent test based on the paired Welch's t-test.
(DOC)
Statistical comparison of PMeS with MASA on the dataset adopted in MASA method.
(DOC)
Statistical comparison of PMeS with Hu's method on the dataset adopted in Hu's method.
(DOC)
Acknowledgments
We are thankful for academic editor, Professor Niall James Haslam, and two anonymous reviewers, whose suggestions have greatly improved the quality of this manuscript. We also would like to thank UniProtKB/Swiss-Prot and PhosphoSitePlus for supplying methylation data on proteins.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the National Natural Science Foundation of China (21175064, 21163014 and 21065006). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Paik WK, Kim S. Enzymatic methylation of protein fractions from calf thymus nuclei. Biochem Bioph Res Co. 1967;29:14–20. doi: 10.1016/0006-291x(67)90533-5. [DOI] [PubMed] [Google Scholar]
- 2.Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436:1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- 3.Bedford MT, Richard S. Arginine methylation: An emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Sayegh J, Webb K, Cheng DH, Bedford MT, Clarke SG. Regulation of protein arginine methyltransferase 8 (PRMT8) activity by its N-terminal domain. J Biol Chem. 2007;282:36444–36453. doi: 10.1074/jbc.M704650200. [DOI] [PubMed] [Google Scholar]
- 5.Predel R, Brandt W, Kellner R, Rapus J, Nachman RJ, et al. Post-translational modifications of the insect sulfakinins-Sulfation, pyroglutamate-formation and O-methylation of glutamic acid. Eur J Biochem. 1999;263:552–560. doi: 10.1046/j.1432-1327.1999.00532.x. [DOI] [PubMed] [Google Scholar]
- 6.Lapko VN, Cerny RL, Smith DL, Smith JB. Modifications of human beta A1/beta A3-crystallins include S-methylation, glutathiolation, and truncation. Protein Sci. 2005;14:45–54. doi: 10.1110/ps.04738505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aleta JM, Cimato TR, Ettinger MJ. Protein methylation: a signal event in post-translational modification. Trends Biochem Sci. 1998;23:89–91. doi: 10.1016/s0968-0004(98)01185-2. [DOI] [PubMed] [Google Scholar]
- 8.Pahlich S, Zakaryan RP, Gehring H. Protein arginine methylation: Cellular functions and methods of analysis. Biochim Biophys Acta. 2006;1764:1890–1903. doi: 10.1016/j.bbapap.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Varier RA, Timmers HTM. Histone lysine methylation and demethylation pathways in cancer. BBA-Rev Cancer. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Paik WK, Paik DC, Kim S. Historical review: the field of protein methylation. Trends Biochem Sci. 2007;32:146–152. doi: 10.1016/j.tibs.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Shukla A, Chaurasia P, Bhaumik SR. Histone methylation and ubiquitination with their cross-talk and roles in gene expression and stability. Cell Mol Life Sci. 2009;66:1419–1433. doi: 10.1007/s00018-008-8605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, et al. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 13.Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: A role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci. 2006;26:11387–11396. doi: 10.1523/JNEUROSCI.3349-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki A, Yamada R, Yamamoto K. Shoenfeld Y, Gershwin ME, editors. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. 2007. pp. 323–339. Autoimmunity, Pt D - Autoimmune Disease, Annus Mirabilis. Oxford: Blackwell Publishing. [DOI] [PubMed]
- 15.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Turner BM. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 17.Snijders APL, Hung ML, Wilson SA, Dickmana MJ. Analysis of arginine and lysine methylation utilizing peptide separations at neutral pH and electron transfer dissociation mass spectrometry. J Am Soc Mass Spectrom. 2010;21:88–96. doi: 10.1016/j.jasms.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Johnson DS, Li W, Gordon DB, Bhattacharjee A, Curry B, et al. Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets. Genome Res. 2008;18:393–403. doi: 10.1101/gr.7080508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plewczynski D, Tkacz A, Wyrwicz LS, Rychlewski L. AutoMotif server: prediction of single residue post-translational modifications in proteins. Bioinformatics. 2005;21:2525–2527. doi: 10.1093/bioinformatics/bti333. [DOI] [PubMed] [Google Scholar]
- 20.Daily KM, Radivojac P, Dunker AK. Intrinsic disorder and protein modifications: building an SVM predictor for methylation. IEEE Symposium on CIBCB, San Diego, California, 475–481. 2005.
- 21.Chen H, Xue Y, Huang N, Yao XB, Sun ZR. MeMo: a web tool for prediction of protein methylation modifications. Nucleic Acids Res. 2006;34:W249–W253. doi: 10.1093/nar/gkl233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao JL, Xu D, Tsai SN, Wang YF, Ngai SM. Computational identification of protein methylation sites through bi-profile bayes feature extraction. PLoS ONE. 2009;4:e4920. doi: 10.1371/journal.pone.0004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shien DM, Lee TY, Chang WC, Hsu JBK, Horng JT, et al. Incorporating structural characteristics for identification of protein methylation sites. J Comput Chem. 2009;30:1532–1543. doi: 10.1002/jcc.21232. [DOI] [PubMed] [Google Scholar]
- 24.Hu LL, Li Z, Wang K, Niu S, Shi X-H, et al. Prediction and analysis of protein methylarginine and methyllysine based on multi sequence features. Biopolymers. 2011;95:763–771. doi: 10.1002/bip.21645. [DOI] [PubMed] [Google Scholar]
- 25.Li WZ, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 26.Gao JJ, Thelen JJ, Dunker AK, Xu D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol Cell Proteomics. 2010;9:2586–2600. doi: 10.1074/mcp.M110.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niu S, Huang T, Feng KY, Cai YD, Li YX. Prediction of tyrosine sulfation with mRMR feature selection and analysis. J Proteome Res. 2010;9:6490–6497. doi: 10.1021/pr1007152. [DOI] [PubMed] [Google Scholar]
- 28.Nanni L, Lumini A. A new encoding technique for peptide classification. Expert Syst Appl. 2011;38:3185–3191. [Google Scholar]
- 29.Rögnvaldsson T, You L, Garwicz D. Bioinformatic approaches for modeling the substrate specificity of HIV-1 protease: an overview. Expert Rev Mol Diagn. 2007;7:435–451. doi: 10.1586/14737159.7.4.435. [DOI] [PubMed] [Google Scholar]
- 30.Stallcup MR. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene. 2001;20:3014–3020. doi: 10.1038/sj.onc.1204325. [DOI] [PubMed] [Google Scholar]
- 31.Teyssier C, Le Romancer M, Sentis S, Jalaguier S, Corbo L, et al. Protein arginine methylation in estrogen signaling and estrogen-related cancers. Trends Endocrin Met. 2010;21:181–189. doi: 10.1016/j.tem.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Zhang ZH, Wang ZH, Zhang ZR, Wang YX. A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine. Febs Letters. 2006;580:6169–6174. doi: 10.1016/j.febslet.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Rudbeck ME, Nilsson Lill SO, Barth A. Influence of the molecular environment on phosphorylated amino acid models: a density functional theory study. J Phys Chem B. 2012;116:2751–2757. doi: 10.1021/jp206414d. [DOI] [PubMed] [Google Scholar]
- 34.Fauchere JL, Charton M, Kier LB, Verloop A, Pliska V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int J Peptide Protein Res. 1988;32:269–278. doi: 10.1111/j.1399-3011.1988.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 35.Pang CNI, Hayen A, Wilkins MR. Surface accessibility of protein post-translational modifications. J Proteome Res. 2007;6:1833–1845. doi: 10.1021/pr060674u. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad S, Gromiha MM, Sarai A. Real value prediction of solvent accessibility from amino acid sequence. Proteins. 2003;50:629–635. doi: 10.1002/prot.10328. [DOI] [PubMed] [Google Scholar]
- 37.Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 38.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–297. [Google Scholar]
- 39.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. Software available at. 2001. http://www.csie.ntu.edu.tw/~cjlin/libsvm.
- 40.Xiao B, Wilson JR, Gamblin SJ. SET domains and histone methylation. Curr Opin Struct Biol. 2003;13:699–705. doi: 10.1016/j.sbi.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 42.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wooderchak WL, Zang TZ, Zhou ZS, Acuna M, Tahara SM, et al. Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the "RGG" paradigm. Biochemistry. 2008;47:9456–9466. doi: 10.1021/bi800984s. [DOI] [PubMed] [Google Scholar]
- 44.Pang C, Gasteiger E, Wilkins MR. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genomics. 2010;11:92. doi: 10.1186/1471-2164-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiledjian M, Dreyfuss G. Primary structure and binding activity of the hnRNPU protein: binding RNA through RGG box. EMBO J. 1992;11:2655–2664. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dolzhanskaya N, Merz G, Aletta JM, Denman RB. Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. J Cell Sci. 2006;119:1933–1946. doi: 10.1242/jcs.02882. [DOI] [PubMed] [Google Scholar]
- 47.Dong LH, Yuan Y, Cai YD. Using bagging classifier to predict protein domain structural class. J Biomol Struct Dyn. 2006;24:239–242. [PubMed] [Google Scholar]
- 48.Xu JL, He Y, Qiang BQ, Yuan JG, Peng XZ, et al. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinformatics. 2008;9:8. doi: 10.1186/1471-2105-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rögnvaldsson TS, Etchells TA, You LW, Garwicz D, Jarman IH, et al. How to find simple and accurate rules for viral protease cleavage specificities. BMC Bioinformatics. 2009;10:149. doi: 10.1186/1471-2105-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
246 experimentally identified methylarginine sites in 98 proteins were extracted from UniProtKB/Swiss-Prot database.
(DOC)
367 experimentally identified methyllysine sites in 137 proteins were extracted from UniProtKB/Swiss-Prot database.
(DOC)
155 methylarginine sites in 68 proteins were extracted from PhosphoSitePlus.
(DOC)
147 methyllysine sites in 78 proteins were extracted from PhosphoSitePlus.
(DOC)
The normalized van der Waals volume of 20 kinds of amino acids.
(DOC)
The distribution of physicochemical properties of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
Average van der Waals volume (VDWV) of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
Average accessible surface area (ASA) of residues around methylation sites and non-methylation sites was compared via P -values on the paired Welch's t-test.
(DOC)
The list of proteins containing experimental methylation sites which are located in the protein regions with known tertiary structure of protein data bank (PDB).
(DOC)
The predictive performance of model trained with different features was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of models trained with different window sizes for methylarginine.
(DOC)
The performance of models trained with different window sizes for methyllysine.
(DOC)
The predictive result of models with different window sizes was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of models trained with different ratio of positive to negative samples for methylarginine.
(DOC)
The performance of models trained with different ratio of positive to negative samples for methyllysine.
(DOC)
The predictive result of models with different ratios of positive to negative samples was compared via P -values on the paired Welch's t-test.
(DOC)
The performance of the methylarginine model based on self-consistency, K-fold (4-, 6-, 8- and 10-fold) cross-validation and leave-one-out validation.
(DOC)
MCC of methylarginine model by different cross-validation was compared via P -values on the paired Welch's t-test.
(DOC)
We collected 46 experimentally identified methyllysine sites in 39 unique proteins from the scientific literature (PubMed).
(DOC)
We collected 27 experimentally identified methylarginine sites in 24 unique proteins from the scientific literature (PubMed).
(DOC)
Statistical comparison of training test with independent test based on the paired Welch's t-test.
(DOC)
Statistical comparison of PMeS with MASA on the dataset adopted in MASA method.
(DOC)
Statistical comparison of PMeS with Hu's method on the dataset adopted in Hu's method.
(DOC)