

Abstract
Genistein is a bioflavonoid enriched in soy products. However, high levels of maternal soy consumption have been linked to the development of infant leukemia ALL and AML. The majority of infant leukemia is linked to mixed lineage leukemia gene (MLL) translocations. Previous studies have implicated topoisomerase II (Top2) in genistein-induced infant leukemia. In order to understand the roles of the two Top2 isozymes in and the molecular mechanism for genistein-induced infant leukemia, we carried out studies in vitro using purified recombinant human Top2 isozymes, as well as studies in cultured mouse myeloid progenitor cells (32Dc13) and Top2β knockout mouse embryonic fibroblasts (MEFs). First, we showed that genistein efficiently induced both Top2α and Top2β cleavage complexes in the purified system as well as in cultured mouse cells. Second, genistein induced proteasomal degradation of Top2β in 32Dc13 cells. Third, the genistein-induced DNA double-strand break (DSB) signal, γ-H2AX, was dependent on the Top2β isozyme and proteasome activity. Fourth, the requirement for Top2β and proteasome activity was mirrored in genistein-induced DNA sequence rearrangements, as monitored by a DNA integration assay. Together, our results suggest a model in which genistein-induced Top2β cleavage complexes are processed by proteasome, leading to the exposure of otherwise Top2β-concealed DSBs and subsequent chromosome rearrangements, and implicate a major role of Top2β and proteasome in genistein-induced infant leukemia.
Keywords: Genistein, topoisomerase IIbeta, infant leukemia, MLL translocation, proteasome
Introduction
Genistein is a natural bioflavonoid (isoflavone) mostly found in soy-based foods. Genistein exhibits cancer-chemopreventive and antitumor activities, as well as anti-oxidant, anti-inflammatory and anti-in vitro angiogenesis effects [1–3]. However, clinical studies have suggested a strong link between a prior exposure to dietary flavonoids including genistein and infant leukemia. It was demonstrated that maternal consumption of bioflavonoid-rich foods led to an approximately 10-fold higher risk of infant acute myelogenous leukemia (AML) [4, 5].
Infant leukemia is frequently associated with chromosome translocations involving the mixed lineage leukemia 1 (MLL) gene [6]. It has been estimated that up to 80% of infant acute lymphoblastic leukemia (ALL) and 65% of infant acute myelogenous leukemia (AML) are linked to MLL translocations [7]. These translocations can take place in utero and are associated with poor prognosis, especially in infant ALL [5]. Interestingly, MLL translocations are also hallmarks of more than 70% of t-AML (therapy-related acute myelogenous leukemia) associated with topoisomerase II (Top2)-based chemotherapy in cancer patients [8]. Mapping of chromosomal breakpoints of MLL translocations has revealed the clustering of breakpoints within an 8.3 kb region of the human MLL gene, known as the breakpoint cluster region (BCR) [8]. The genomic breakpoints in infant leukemia and t-AML tend to co-localize with Top2 cleavage sites, suggesting a possible link between infant leukemia and Top2 [8, 9].
Genistein is known to induce DNA topoisomerase II-linked DNA breaks (Top2 cleavage complexes) [3]. There are two human Top2 isozymes, Top2α and Top2β, that share 70% sequence identity [10]. Top2α has been suggested to function in cell cycle events such as DNA replication and chromosome condensation/segregation [11], whereas Top2β has been shown to be involved in transcription[12–15]. Recent studies have shown that cancer chemotherapeutic drugs that target Top2 poison both Top2α and Top2β [10]. It has been suggested that Top2α targeting (poisoning) is primarily responsible for the antitumor activity of these drugs while Top2β targeting could lead to secondary malignancies such as (Top2 drug) therapy-related acute myelogenous leukemia (t-AML) [16]. Since genistein is a Top2-tartgeting compound, we envision that genistein may induce infant leukemia through poisoning of the Top2β isozyme.
Previous studies have demonstrated that the induction of DNA double-strand breaks (DSBs) by Top2-targeting drugs characteristically requires the proteasome activity [17]. It has been shown that Top2-targeting drugs induce preferential degradation of the Top2β isozyme (Top2β down-regulation) in various cells, leading to the exposure of the otherwise Top2-concealed DSBs [16, 17]. It has been proposed that Top2β down-regulation is the underlying mechanism for Top2 drug-induced DNA sequence rearrangements and carcinogenesis [16].
In the present study, we have tested the role of Top2β and proteasome in genistein-induced DSBs and chromosome rearrangements. Our results suggest that proteasomal processing of genistein-induced Top2β cleavage complexes results in DSB formation and DNA sequence rearrangements, thus implicating an important role of Top2β and proteasome in genistein-induced MLL translocations and infant leukemia.
MATERIALS AND METHODS
shRNA-mediated knockdown of Top2β in 32Dc13 mouse myeloid progenitor cells
The Control (Ctrl) or Top2β shRNA sequences were selected using the Whitehead Institute siRNA selection program (http://jura.wi.mit.edu/bioc/siRNAext/) and the corresponding oligo duplex DNAs were cloned into a LentiLox 3.7 vector with an inserted neomycin-resistant gene. Standard procedures were then followed to generate stable Top2β- or control-knockdown 32Dc13 cells lines.
Top2-mediated DNA cleavage assay
The Top2 cleavage assay was performed as described [18].
Measurement of plasmid integration frequency
For measuring the plasmid integration frequency in MEFs, procedures were followed as previously described [19]. For measuring the plasmid integration frequency in 32Dc13 cells, 2 × 106 cells were seeded 24 hours prior to the experiment. Treatment and transfection were performed as described [19]. 6 hrs post-transfection, cells were washed three times, replenished with fresh medium and incubated for additional 24 hrs to allow the expression of the blasticidin resistance gene. Cells were then trypsinized and cultured in methylcellulose (Methocult® 3134 medium) in RPMI/IL3 medium supplemented with blasticidin (3 μg/ml). A small aliquot of cells were also cultured in the absence of blasticidin for survival determination. After ten days, blasticidin-resistant colonies were counted by the IPI MiniCount colony counter. Plasmid integration frequency was determined as the ratio of the number of blasticidin-resistant colonies and the number of surviving cells (number of colonies without blasticidin selection times the dilution factor). The average and standard error of mean were then calculated and plotted.
Top2β down-regulation
To determine genistein- or VP-16-induced Top2β down-regulation, procedures were followed as described previously [17, 20].
Band depletion assay
To determine the level of drug-induced intracellular Top2 cleavage complexes by the band depletion assay as previously described [16].
Results
Genistein induces Top2 cleavage complexes and proteasomal degradation of Top2β (Top2β down-regulation)
As shown in Fig. 1A, genistein, like VP-16 (a prototypic Top2 poison), induced both Top2α and Top2β-mediated DNA cleavage in a concentration-dependent manner, most certainly reflecting the formation of cleavage complexes with the two Top2 isozymes (Fig. 1A, note the disappearance of the full length DNA (labeled **) and the appearance of the cleaved DNA fragments (labeled *)). Relative to VP-16, genistein appeared to be slightly more effective in inducing Top2β- than Top2α-mediated DNA cleavages. While VP-16 induced comparable levels of Top2α- and Top2β-mediated DNA cleavages (lanes 3–8 and lanes 17–22), genistein could induce more extensive Top2β-mediated DNA cleavages as compared to Top2α-mediated DNA cleavages at all concentrations of genistein being tested (lanes 9–14 and lanes 23–28). We also tested whether genistein could induce Top2 cleavage complexes in cells. As shown in Fig. 1B, genistein, like VP-16, caused depletion of both Top2α and Top2β immunobands within 15 min of treatment as evidenced by a band depletion assay [16], suggestive of the formation of Top2 cleavage complexes (too large to enter the gel due to the large size of the Top2-DNA covalent complexes). As expected for reversible Top2 cleavage complexes [21], genistein- (as well as VP-16-) induced depletion of both Top2α and Top2β immunobands was largely abolished following replenishing the cells with drug-free medium and further incubation for 30 min (Fig. 1B, labeled Rev + genistein and Rev + VP-16), demonstrating the reversibility of genistein-induced Top2 cleavage complexes.
Figure 1. Induction of Top2 cleavage complexes and Top2β down-regulation by genistein.

A. Genistein induces Top2α- and Top2β-mediated DSBs in vitro. VP-16 (0 μM (lanes 2 & 16), 3.13 μM (lanes 3 & 17), 6.25 μM (lanes 4 & 18), 12.5 μM (lanes 5 & 19), 25.0 μM (lanes 6 & 20), 50.0 μM (lanes 7 & 21), 100 μM (lanes 8 & 22)) and genistein (0 μM (lanes 2 & 16), 3.13 μM (lanes 9 & 23), 6.25 μM (lanes 10 & 24), 12.5 μM (lanes 11 & 25), 25.0 μM (lanes 12 & 26), 50.0 μM (lanes 13 & 27), 100 μM (lanes 14 & 28)) were incubated with 32P-labeled linearized plasmid DNA in the presence of purified recombinant hTop2α (lanes 1–14) or hTop2β (lanes 15–28) as described in Materials and Methods (DNA cleavage assay). **, full length 32P-labeled linearized plasmid DNA; *, cleaved DNA fragments. DNA cleavage products were then analyzed by agarose gel electrophoresis, followed by autoradiography. B. Genistein traps both Top2α and Top2β cleavage complexes in mouse 32Dc13 progenitor cells. The amounts of genistein-induced Top2α and the Top2β cleavage complexes were measured by the band depletion assay. 32Dc13 cells, expressing both Top2 isozymes were treated with genistein (100, 250 or 500 μM) or VP-16 (250 μM) for 15 min, followed by lysis in 6X sample buffer. Cell lysate were then analyzed by SDS-PAGE and immunoblotted with isozyme-specific antibodies. This assay measures the levels of free Top2 isozymes in the lysate as trapping on DNA leads to the decreased free Top2 levels. To demonstrate the reversibility of genistein- and VP-16-induced Top2 cleavage complexes, drug-treated cells were further incubated in drug-free medium for an additional 30 min, followed by immunoblotting analysis. C. Mouse 32Dc13 progenitor cells, expressing both Top2 isozymes, were treated with genistein (500 μM) or VP-16 (100 μM) for 0 and 2 hrs in the absence or presence of the proteasome inhibitor MG132 (10 μM). Cells were then lysed in alkaline lysis buffer followed by neutralization and S7 nuclease digestion. After SDS-PAGE, the amount of the Top2 isozymes was measured by immunoblotting.
The formation of Top2 cleavage complexes (e.g. induced by VP-16) is known to trigger degradation of Top2β through a proteasome-dependent pathway (Top2β down-regulation), resulting in the exposure of otherwise Top2β-concealed DSBs [17]. To test whether genistein can also induce Top2β down-regulation, the Top2β protein levels were monitored in genistein-treated 32Dc13 hematopoietic progenitor cells. As shown in Fig. 1C, genistein (500 μM) treatment reduced the Top2β level by about 50% in 2 hrs. By contrast, the level of Top2α did not change significantly upon genistein treatment. In addition, genistein-induced Top2β degradation was prevented by co-treatment with the proteasome inhibitor MG132 (Fig. 1C). These results suggest that similar to VP-16, genistein induces Top2β down-regulation through a proteasome-dependent pathway.
Genistein-induced DNA double-strand breaks (DSBs) require Top2β and proteasome
Previous studies have linked VP-16-induced Top2β down-regulation to the formation of DSBs [16, 17]. Since our studies have demonstrated that genistein induces Top2β down-regulation, we tested the possibility that genistein could induce DSBs in a proteasome-dependent manner. As shown in Fig. 1C, genistein, like VP-16, induced γ-H2AX (phosphorylation of histone H2AX at Ser-139, a prototypic DSB marker) in 32Dc13 cells, suggesting that genistein can induce DSBs. Interestingly, genistein-induced DSBs, like VP-16-induced DSBs, were significantly reduced in the presence of the proteasome inhibitor MG132, suggesting that genistein-induced DSBs requires proteasome activity. We also tested the possibility that genistein-induced DSBs require Top2β. As shown in Fig. 2A, genistein induced γ-H2AX in Top2β+/+, but not in top2β−/−, MEFs. As a positive control, we showed that VP-16-induced γ-H2AX also required Top2β (Fig. 2A). By contrast, camptothecin (CPT), a Top1 (topoisomerase I)-targeting drug, induced γ-H2AX independent of Top2β (Fig. 2A). Together, these results suggest that genistein-induced DSBs, like VP-16-induced DSBs, require both proteasome and Top2β.
Figure 2. Genistein-induced DNA damage signal is Top2β- and proteasome-dependent in primary MEFs.
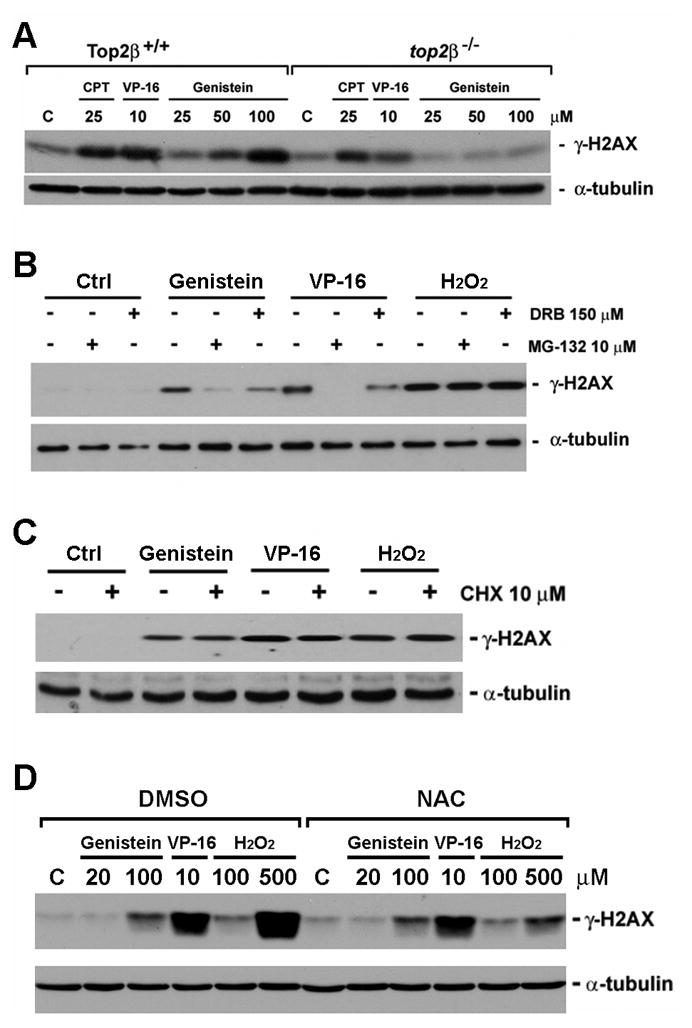
A. Primary Top2β+/+ and top2β−/− MEFs were treated with 25 μM CPT, 10 μM VP-16 and or increasing concentrations of genistein (25, 50, 100 μM) for 1 hour, followed by lysis in 6X SDS sample buffer and immunoblotted with antibodies against γ-H2AX and α-tubulin. B. The genistein-induced γ-H2AX signal requires proteasome activity and transcription. Primary Top2β+/+ MEFs were treated with MG132 (10 μM) or DRB (150 μM) for 30 min, followed by co-treatment with VP-16 (10 μM), genistein (100 μM) or hydrogen peroxide (H2O2, 400 μM) for 1 hr. Western blotting was performed as described in A. C. Genistein-induced γ-H2AX does not require protein synthesis. Primary Top2β+/+ MEFs were treated with cycloheximide (CHX, 10 μM) for 30 min, followed by co-treatment with VP-16 (10 μM), genistein (100 μM) or hydrogen peroxide (H2O2, 400 μM) for 1 hr. Western blotting was performed as described in A. D. N-acetyl cysteine had no effect on the genistein-induced DNA damage. Primary Top2β+/+ MEFs were treated with 0.1% DMSO or 1 mM N-acetyl cysteine (NAC, 1 mM) for 30 min, followed by co-treatment with genistein (20 μM, 100 μM), VP-16 (10 μM) or H2O2 (100 μM, 500 μM) for 2 hrs. Western blotting was performed as described in A.
VP-16-induced Top2β cleavage complexes have been suggested to arrest transcription, triggering proteasomal degradation of Top2β and concomitant exposure of the Top2β-concealed DSBs [17]. To determine whether genistein-induced DSBs also specifically involves active transcription, we tested the effect of the transcription inhibitor DRB and the protein synthesis inhibitor cycloheximide (CHX) on the induction of genistein-induced DSB signal, γ-H2AX. As shown in Fig. 2B, genistein-induced γ-H2AX in Top2β+/+ MEFs was also largely prevented by the transcription inhibitor DRB (Fig. 2B), but not by the protein synthesis inhibitor cycloheximide (Fig. 2C). Additionally, we showed that the requirement for active transcription and proteasome in DSB induction by genistein and VP-16 is specific since the induction of γ-H2AX by hydrogen peroxide (H2O2) was not affected by neither the proteasome inhibitor MG132 nor the transcription inhibitor DRB (Fig. 2B). It indicated that in contrast to Top2-mediated DNA damage, reactive oxygen species (ROS)-induced DNA damage did not involve transcription or proteasome-mediated processing. Furthermore, we have showed that while N- acetyl cysteine (NAC, 1 mM) could almost completely block the H2O2-induced γ-H2AX signal, it had minimal effect on the genistein- or VP-16-induced γ-H2AX signal (Fig. 2D). These results further suggested that genistein could induce DNA damage through a mechanism that did not involve ROS.
Genistein-induced DNA sequence rearrangements are Top2β- and proteasome-dependent
Genistein is known to induce chromosome translocations involving the MLL locus in CD34+ hematopoietic progenitors [22]. However, the molecular basis for genistein-induced rearrangement, other than the involvement of DSBs, is not clear. Since genistein induces Top2β- and proteasome-dependent DSBs, we hypothesize that genistein-induced chromosome translocations could result from DSBs generated from proteasomal processing of Top2β cleavage complexes. To test the possible involvement of Top2β and proteasome in genistein-induced chromosome translocations, we employed the plasmid integration assay that measures the integration frequency of a plasmid-born resistance gene into chromosomal DNAs in order to score DNA sequence rearrangement events induced by genistein. To test the involvement of Top2β, we have generated stable Top2β knockdown and control knockdown 32Dc13 lines expressing Top2β shRNA (shTop2β) and control shRNA (shCtrl), respectively. Immunoblotting analysis showed that 32Dc13-shTop2β cells expressed much reduced level of Top2β as compared to 32Dc13-shCtrl cells (Fig. 3A, insert). As shown in Fig. 3A, genistein (100 μM) stimulated plasmid integration at a much higher frequency in 32Dc13-shCtrl cells than in 32Dc13-shTop2β cells. A similar trend of decrease in plasmid integration frequency was observed for VP-16 (0.5 μM). We have also employed plasmid integration assay using SV-40-transformed Top2β+/+ and top2β−/− MEFs. As shown in Fig. 3B, genistein-stimulated plasmid integration was much less frequent in top2β−/− cells as compared to Top2β+/+ cells. Together, these results suggest strongly that Top2β plays an important role in genistein-, and VP-16-, induced DNA sequence rearrangements.
Figure 3. Genistein-induced plasmid integration is Top2β and proteasome-mediated.
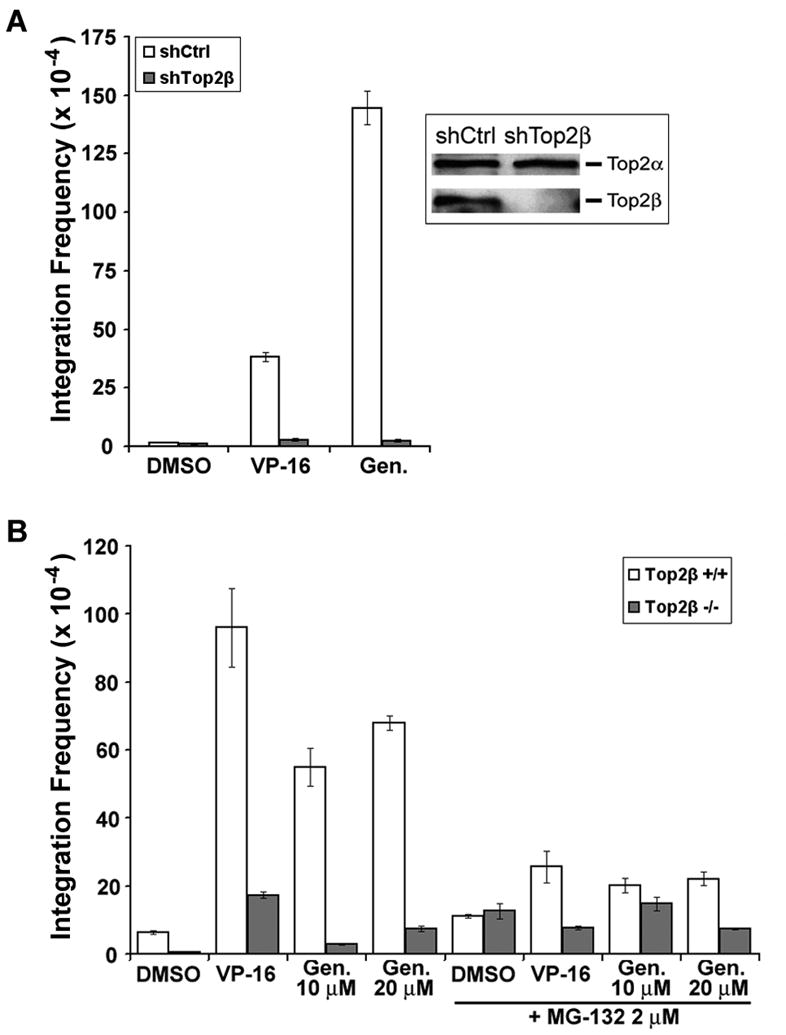
A. Mouse 32Dc13 progenitor cells expressing control shRNA (shCtrl) or Top2β shRNA (shTop2β) were transfected with linearized pUCSV-BSD plasmid in the presence of DMSO (0.1%), VP-16 (0.5 μM) or genistein (100 μM) as indicated. Plasmid integration frequency was then determined and plotted as histograms. B. SV40-transformed Top2β+/+ and top2β−/− MEFs were transfected with the linearized pUCSV-BSD plasmid in the presence of DMSO (0.1%), VP-16 (0.5 μM) or genistein (10 or 20 μM) as indicated. In addition, similar transfections were also performed in the presence of the proteasome inhibitor MG132 (2 μM). Plasmid integration frequencies were then determined.
We next tested the role of proteasome in genistein-induced chromosome rearrangements using the plasmid integration assay. As shown in Fig. 4B, co-treatment with the proteasome inhibitor MG132 during transfection led to a greater than 2-fold decrease in genistein-stimulated plasmid integration frequency, similar to that observed for VP-16 (this study and also see ref. [16]). These findings are in agreement with the model that proteasomal processing of Top2β cleavage complexes exposes Top2β-concealed DSBs, leading to chromosome rearrangements.
Figure 4. A proposed model for genistein-induced DNA sequence rearrangements and infant leukemia.

In this model, genistein stabilizes Top2β-DNA covalent adducts (Top2β cleavage complex) on chromosomal DNA within the transcribed regions. Top2β cleavage complexes arrest transcription elongation, triggering Top2β degradation through a proteasome-dependent pathway. Proteasomal degradation of Top2β cleavage complexes expose Top2β-concealed DSBs. Subsequent repair of the DSBs through the non-homologous end joining pathway (NHEJ) leads to DNA sequence rearrangements. DSBs located within MLL BCR can undergo NHEJ leading to MLL translocations and hence infant leukemias.
Discussion
Previous studies have demonstrated that genistein-induced chromosomal DNA cleavages as well as chromosome translocations involve the MLL BCR [9, 22]. These genistein-induced DNA cleavages and chromosome translocations have been suggested to result from genistein-trapped Top2 cleavage complexes within the MLL BCR [8, 23, 24]. However, the relative contribution of the two Top2 isozymes (i.e. Top2α and Top2β) as well as the mechanism for processing Top2 cleavage complexes into DSBs, in genistein-induced chromosome translocations is unknown. Studies of etoposide (VP-16)-induced carcinogenesis in the classical mouse skin carcinogenesis model have suggested that etoposide-induced skin carcinogenesis is primarily Top2β-mediated [16]. It has been suggested that etoposide behaves as a stage II tumor promoter, presumably by promoting DNA sequence rearrangements [17]. The predominant role of the Top2β isozyme in etoposide-induced carcinogenesis could suggest that the Top2β isozyme may be involved in genistein-induced MLL translocations and infant leukemia.
In the current studies, we show that genistein-induced DNA sequence rearrangements in murine myeloid progenitor D32c13 cells and MEFs require both Top2β and the proteasome activity. Although genistein can induce the formation of both Top2α and Top2β cleavage complexes in these cells, the strong dependence on Top2β rather than Top2α for genistein-induced DNA sequence rearrangements is most likely related to the differential cellular processing of Top2β and Top2α cleavage complexes. It is known that Top2β cleavage complexes are more readily converted into DSBs than Top2α cleavage complexes through a proteasome-dependent pathway [16, 17]. Indeed, we have shown that genistein-induced DSBs are both Top2β- and proteasome-dependent. Consequently, while genistein induces both Top2α and Top2β cleavage complexes to a similar extent, genistein-induced DNA sequence rearrangements are Top2β-dependent, presumably due to the propensity of Top2β cleavage complexes to undergo proteasomal processing into DSBs (see Fig. 4 for a schematic model).
The difference between the two Top2 isozymes in mediating cellular responses to the Top2-targeting drugs is probably related to their differential regulation and distinct roles in various cellular functions. Top2α is known to be a proliferation marker and participate in cell cycle events such as DNA replication, chromosome condensation and sister-chromatid segregation [11]. It seems possible that the chemopreventive and antitumor activities of genistein could at least in part result from its targeting of the Top2α isozyme. Top2α cleavage complexes induced by genistein could selectively kill tumor cells due to elevated levels of Top2α and hence elevated Top2α cleavage complexes in tumor cells. By contrast, the Top2β isozyme is expressed more or less at a constant level in both proliferating and quiescent cells [11], and has been implicated in regulation of gene expression, presumably by controlling regional chromatin condensation/decondensation [13, 25]. Top2β has been shown to localize in the transcribed regions of human rDNA repeats [26], as well as the promoter and transcribed regions of protein-coding genes [13]. As genistein-induced MLL translocations occur within the transcribed region of the MLL gene (i.e. the MLL BCR) and the transcribed regions of the various MLL partner genes, it seems plausible that the initial step may involve trapping of Top2β into cleavage complexes within the transcribed regions of both MLL (e.g. BCR) and its partner genes. The Top2β-DNA covalent adducts (cleavage complexes) then arrest the elongating RNA polymerase complexes, triggering proteasomal degradation of Topβ-DNA covalent adducts and concomitant exposure of otherwise Top2β-concealed DSBs. The initial formation of genistein-induced DSBs could have at least two outcomes; first, these DSBs may directly undergo NHEJ resulting in chromosomal translocations (e.g. MLL translocations). Second, these DSBs could induce apoptosis, resulting in activation of apoptotic nucleases (e.g. CAD) and the formation of secondary DSBs at nuclease hypersensitive sites (e.g. the MLL BCR). These DSBs formed at nuclease hypersensitive sites, which are reflected in the formation of high-molecular-weight DNA fragmentations [27–29], can also undergo NHEJ causing chromosomal translocations (e.g. MLL translocations at MLL BCR). Clearly, the possible involvement of CAD in genistein-induced MLL translocations and infant leukemia needs further investigation.
Although the concentration of genistein used in our studies seems to be much higher than the peak plasma concentration as determined previously (8 μmol/L, [30]), it is possible that the intracellular accumulation of genistein may reach much higher levels. In addition, our in vitro studies have shown that genistein could induce Top2β-mediated DNA cleavages at concentrations as low as 3.13, 6.25 and 12.5 μM (Fig. 1A, lanes 23–25). These results could suggest that genistein may induce low levels of chromosome breaks at physiologically achievable concentrations in vivo. Although undetectable by biochemical methods, these low levels of chromosome breaks at specific chromosomal regions (e.g. the MLL BCR) could contribute to chromosome translocations and infant leukemia.
Conclusions
In the present study, we have tested the role of Top2β and proteasome in genistein-induced DSBs and chromosome rearrangements. We show that genistein, like other Top2 drugs, induces proteasomal degradation of Top2β. In addition, we show that genistein-induced DNA damage and sequence rearrangements are Top2β-mediated and can be prevented by co-treatment with the proteasome inhibitor MG132. These results suggest that proteasomal processing of genistein-induced Top2β cleavage complexes results in DSB formation and DNA sequence rearrangements, thus implicating an important role of Top2β and proteasome in genistein-induced MLL translocations and infant leukemia.
Supplementary Material
Acknowledgments
We thank Dr. Makoto Kimura (Laboratory for Remediation Research, Plant Science Center, Saitama, Japan) for providing the pUSCV-BSD plasmid. This work was supported in part by a NIH grant CA102463, a New Jersey Commission on Cancer Research Grant 06-2419-CCR-EO, a Department of Defense Idea Award W81XWH-07-1-0407 and a Department of Defense grant W81XWH06-1-0514.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fotsis T, Pepper M, Adlercreutz H, Hase T, Montesano R, Schweigerer L. Genistein, a dietary ingested isoflavonoid, inhibits cell proliferation and in vitro angiogenesis. J Nutr. 1995;125:790S–797S. doi: 10.1093/jn/125.suppl_3.790S. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch Biochem Biophys. 2008;476:107–112. doi: 10.1016/j.abb.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 3.Peterson G. Evaluation of the biochemical targets of genistein in tumor cells. J Nutr. 1995;125:784S–789S. doi: 10.1093/jn/125.suppl_3.784S. [DOI] [PubMed] [Google Scholar]
- 4.Ross JA. Maternal diet and infant leukemia: a role for DNA topoisomerase II inhibitors? Int J Cancer Suppl. 1998;11:26–28. [PubMed] [Google Scholar]
- 5.Felix CA, Lange BJ. Leukemia in infants. Oncologist. 1999;4:225–240. [PubMed] [Google Scholar]
- 6.Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, Blair CK, Reaman GH, Ross JA. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children's oncology group. Cancer Epidemiol Biomarkers Prev. 2005;14:651–655. doi: 10.1158/1055-9965.EPI-04-0602. [DOI] [PubMed] [Google Scholar]
- 7.Canaani E, Nakamura T, Rozovskaia T, Smith ST, Mori T, Croce CM, Mazo A. ALL-1/MLL1, a homologue of Drosophila TRITHORAX, modifies chromatin and is directly involved in infant acute leukaemia. Br J Cancer. 2004;90:756–760. doi: 10.1038/sj.bjc.6601639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Rowley JD. Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) 2006;5:1282–1297. doi: 10.1016/j.dnarep.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci USA. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Austin CA, Marsh KL. Eukaryotic DNA topoisomerase II beta. Bioessays. 1998;20:215–226. doi: 10.1002/(SICI)1521-1878(199803)20:3<215::AID-BIES5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 11.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 12.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 13.Lyu YL, Lin CP, Azarova AM, Cai L, Wang JC, Liu LF. Role of topoisomerase IIbeta in the expression of developmentally regulated genes. Mol Cell Biol. 2006;26:7929–7941. doi: 10.1128/MCB.00617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyu YL, Wang JC. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIbeta. Proc Natl Acad Sci USA. 2003;100:7123–7128. doi: 10.1073/pnas.1232376100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsutsui K, Sano K, Kikuchi A, Tokunaga A. Involvement of DNA topoisomerase IIbeta in neuronal differentiation. J Biol Chem. 2001;276:5769–5778. doi: 10.1074/jbc.M008517200. [DOI] [PubMed] [Google Scholar]
- 16.Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF. From the Cover: Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci USA. 2007;104:11014–11019. doi: 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang A, Lyu YL, Lin CP, Zhou N, Azarova AM, Wood LM, Liu LF. A Protease Pathway for the Repair of Topoisomerase II-DNA Covalent Complexes. J Biol Chem. 2006;281:35997–36003. doi: 10.1074/jbc.M604149200. [DOI] [PubMed] [Google Scholar]
- 18.Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 19.Hars ES, Lyu YL, Lin CP, Liu LF. Role of apoptotic nuclease caspase-activated DNase in etoposide-induced treatment-related acute myelogenous leukemia. Cancer Res. 2006;66:8975–8979. doi: 10.1158/0008-5472.CAN-06-1724. [DOI] [PubMed] [Google Scholar]
- 20.Mao Y, Desai SD, Ting CY, Hwang J, Liu LF. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J Biol Chem. 2001;276:40652–10658. doi: 10.1074/jbc.M104009200. [DOI] [PubMed] [Google Scholar]
- 21.Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 22.Barjesteh van Waalwijk van Doorn-Khosrovani S, Janssen J, Maas LM, Godschalk RW, Nijhuis JG, van Schooten FJ. Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis. 2007;28:1703–1709. doi: 10.1093/carcin/bgm102. [DOI] [PubMed] [Google Scholar]
- 23.Kantidze OL, Razin SV. Chemotherapy-related secondary leukemias: A role for DNA repair by error-prone non-homologous end joining in topoisomerase II - Induced chromosomal rearrangements. Gene. 2007;391:76–79. doi: 10.1016/j.gene.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Malik M, Nitiss KC, Enriquez-Rios V, Nitiss JL. Roles of nonhomologous end-joining pathways in surviving topoisomerase II-mediated DNA damage. Mol Cancer Ther. 2006;5:1405–1414. doi: 10.1158/1535-7163.MCT-05-0263. [DOI] [PubMed] [Google Scholar]
- 25.Sano K, Miyaji-Yamaguchi M, Tsutsui KM, Tsutsui K. Topoisomerase IIbeta activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS One. 2008;3:e4103. doi: 10.1371/journal.pone.0004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Govoni M, Neri S, Labella T, Sylvester JE, Novello F, Pession A. Topoisomerase-II-mediated DNA cleavage within the human ribosomal genes. Biochem Biophys Res Commun. 1995;213:282–288. doi: 10.1006/bbrc.1995.2127. [DOI] [PubMed] [Google Scholar]
- 27.Stanulla M, Wang J, Chervinsky DS, Thandla S, Aplan PD. DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Mol Cell Biol. 1997;17:4070–4079. doi: 10.1128/mcb.17.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sim SP, Liu LF. Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. J Biol Chem. 2001;276:31590–31595. doi: 10.1074/jbc.M103962200. [DOI] [PubMed] [Google Scholar]
- 29.Betti CJ, Villalobos MJ, Jiang Q, Cline E, Diaz MO, Loredo G, Vaughan AT. Cleavage of the MLL gene by activators of apoptosis is independent of topoisomerase II activity. Leukemia. 2005;19:2289–2295. doi: 10.1038/sj.leu.2403966. [DOI] [PubMed] [Google Scholar]
- 30.King RA, Bursill DB. Plasma and urinary kinetics of the isoflavones daidzein and genistein after a single soy meal in humans. Am J Clin Nutr. 1998;67:867–872. doi: 10.1093/ajcn/67.5.867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.