

Abstract
A new fluorogenic fluorescein derivative containing an α,β-unsaturated aldehyde moiety produces a selective fluorescent signal enhancement in the presence of cysteine or peptides containing N-terminal cysteine residues. The mechanism is based on synergistic covalent and supramolecular interactions.
An excess or deficiency of specific biological thiols serves as evidence of disease states. The investigation of fluorescent probes for thiol detection is an active area of study.1 Aldehyde-functionalized fluorophores and chromophores can be used for highly selective detection of homocysteine (Hcy) and/or cysteine (Cys) over all other sulfhydryls due to characteristic thiazinane or thiazolidine formation, respectively.2 Moreover, selectivity for Cys3 has been directly achieved at room temperature via visible detection using α,β-unsaturated aldehydes initially in 2005.4 Unlike our previous work in which the response toward thiols resulted in fluorescence quenching, we report herein a new fluorogenic fluorescein derivative (1) exhibiting a combination of covalent and non-covalent interactions between the fluorophore and bound analyte which generates a turn-on response. These unique interactions result in heightened selectivity, emission, site-specific peptide and protein residue detection and amplified selectivity as a function of bound analyte complexity.
Compound 1 is synthesized via a Reimer–Tiemann formylation reaction of fluorescein4 followed by olefination (see ESI†). The reaction of 1 with excess Cys involves conjugate addition and thiazolidine formation (Scheme 1). The disappearance of the aldehyde and olefin resonances of 1 along with the concomitant appearance of thiazolidine resonances in the 1H NMR centered at ca. 6.72 ppm (Fig. S4, ESI†), along with ESI-TOF HRMS evidence (Fig. S5, ESI†), show that 2 equiv. Cys react with 1.
Scheme 1.

The reaction of 1 with Cys (excess) in 0.1 M phosphate buffer pH 7.4 at rt.
The absorption properties of 1 (ε501 = 46 981 cm−1 M−1 in 0.1 M phosphate buffer pH 7.4, Fig. S6, ESI†) are similar to that of fluorescein5 with a slight red shift. The absorption spectra of 1 as a function of pH (Fig. S7, ESI†) reveal an isosbestic point at 463 nm. The pKa of the phenolic hydroxyl of 1 is 6.22. Emission of 1 is suppressed and red shifted as compared to fluorescein as a result of the extended conjugation and electron withdrawing aldehyde. The addition of a large excess of Cys (10 mM) to a 2 μM solution of 1 restores a large portion of the suppressed xanthene emission with essentially no change in absorption observed at the 463 nm wavelength chosen for excitation (Fig. S8, ESI†). Therefore, the emission intensities are directly proportional to the relative quantum yields of the system. The quantum yield of 1 as referenced to fluorescein at the same conditions (λex = 463 nm, A463nm = 0.042, 0.1Mphosphate buffer pH 7.4, φfluorescein pH 7.4=0.876 based upon Sjoback et al.)5 is calculated to be 0.054. The quantum yield of the 1-Cys reaction product is calculated to be 0.289.
We optimized the fluorogenic response of compound 1 to Cys through investigation of the absorption and excitation properties of 1 and of its reaction product with Cys as it formed over time. In the absence of Cys, the excitation spectrum of 1 is blue shifted as compared to the corresponding absorption spectrum (inset Fig. 1A, λmax abs = 501 nm vs. λmax ex = 485 nm) with a shape similar to that of the absorption spectrum of 1 in acidic buffer (see pH titration in Fig. S7, ESI†). Interestingly, under these conditions, excitation near the absorption maximum resulted in little emission from 1 indicating that the major component, 1 with the phenol ionized (pKa = 6.22), is strongly absorbing but with limited or no emission. The weak emission of 1 at these conditions is largely from the minor species.
Fig. 1.

Spectral properties of compound 1 (9.38 ×10−7 M) in the presence of Cys (10.0 mM). (A) Absorption spectra as a function of time (60 min). Inset: normalized absorption and excitation spectra of 1. (B) Excitation spectra monitored at the 525 nm emission peak of the reaction product as a function of time (60 min). Inset: fluorescence enhancement factor of compound 1 as a function of excitation wavelength. Vertical lines represent the optimized wavelength of 508 nm chosen for excitation of all subsequent emission spectra. All spectra were collected at rt. Excitation and emission bandpass = 5 nm.
Fluorescence intensity of the reaction product monitored over time at its 525 nm emission maximum increased by ~41-fold as the excitation spectra shifted to more closely resemble the corresponding absorption spectra (reaction product λmax abs and λmax ex from Fig. 1 are both 499 nm). This indicates that emission of the dianion (carboxylate and phenolate both ionized) of the reaction product is not suppressed as it was for 1 in the absence of thiol. Differences between the absorption and excitation spectra of 1, combined with opposite shifts in the absorption and excitation spectra (blue vs. red, respectively) as the Cys reaction product formed afforded a window of excitation wavelengths wherein the fluorogenic response could be maximized. The enhancement factor increased from as little as a few fold when excited near 400 nm to >40-fold when excited near 510 nm (inset, Fig. 1B). The optimized enhancement is much greater than the ~5-fold difference between the quantum yield of 1 (0.054) and the reaction product (0.289).
Interestingly, excitation wavelengths at (i) the λmax ex of 1, (ii) the λmax abs of 1, or (iii) within the spectral region which displayed the greatest increase in absorbance in response to Cys (~495 nm) did not result in the greatest fluorescence intensity enhancements. Therefore, these are not the best choices for excitation wavelengths as might initially be assumed. A well-defined isosbestic point in the absorption spectra was observed at 508 nm. Constant absorbance combined with this wavelength being (i) near the maximum in the excitation spectrum of the reaction product and (ii) in the region where the fluorescence enhancement was the largest was chosen as the excitation wavelength for all further experiments.
The fluorogenic response of 1 towards other thiols was investigated following optimization using Cys. Emission of 1 in the presence of various thiols (λem = 524 nm upon excitation at 508 nm) demonstrated that compound 1 displayed selectivity towards specific biothiols of interest including Cys and Hcy (Fig. 2, see also complete emission and absorption spectra in Fig. S9, ESI†). After complete reaction, 1 displayed 1.43-fold selectivity for Cys over Hcy as a result of different quantum yields of the reaction products (φ1–Cys = 0.289 vs. φ1–Hcy = 0.202). The Cys reaction product forms at a faster rate resulting in selectivity over Hcy as great as ~2.9-fold early in the course of the reaction (Fig. 2A, Fig. S10 and S11, ESI†). Importantly, response of 1 to Cys and Hcy was found to be linear with concentration (100 μM–1.0 mM). Selectivity towards Cys of greater than 2-fold was observed for concentrations as low as 100 μM (Fig. S11, ESI†) and although not yet optimized, the limit of detection for Cys was found to be ~3-fold lower than that of Hcy (39 μM vs. 114 μM following 20 min of reaction, see Fig. S12, ESI†).
Fig. 2.
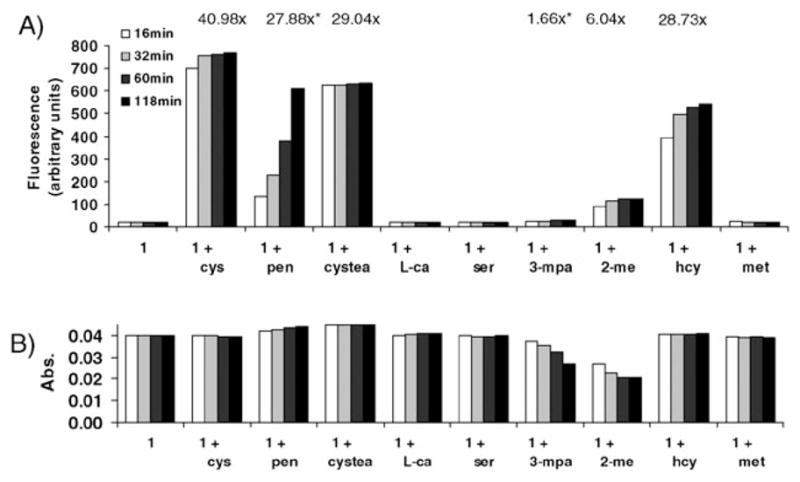
Response of compound 1 to thiols (10.0 mM) of interest and their structural analogs. (A) Fluorescence emission at 524 nm. (B) Absorbance at the 508 nm excitation wavelength. *Signal still increasing after 2 h. Conditions are the same as in Fig. 1. Abbreviations: pen = penicillamine, cystea = cysteamine, L-ca = L-cysteic acid, 3-mpa = 3-mercaptopropionic acid, 2-me = 2-mercaptoethanol.
Close inspection of Fig. 2 reveals that significant fluorogenic response resulted only from compounds containing both a free thiol and amine in which a thiazinane or thiazolidine hetero-cycle could form. The mechanism behind this selectivity was investigated and confirmed through a series of structurally related analogs containing various functional groups and combinations thereof and through computer-assisted molecular modeling (vide infra).
Thiols attack the β carbon of aldehyde 1, thereby restoring xanthene emission. Once formed, the covalent complexes can undergo further interaction with the chromophore thereby affording increased selectivity by affecting the ionization state of the phenolic hydroxyl. Energy-minimized simulations (Sybyl 8.0, Tripos Inc.) indicate that for amino thiols in general at neutral pH, depending on the length of the alkyl chain, electrostatic interactions between the NH3+ group with both the phenolate and the carboxylate moieties of 1 occur (Fig. 3).
Fig. 3.

Energy-minimized structures of the reaction products of 1 with Cys (left) and Hcy (right) showing the distance between the NH3+ and the phenolate groups. The electrostatic interactions between NH3+ and the carboxylate and/or phenolate groups of 1 are influenced by increased ionic strength (addition of 1 M NaCl) resulting in a corresponding decrease in selectivity toward Cys (see Fig. S13–S15, ESI†).
In the case of Cys and Hcy there is a greater fluorescence response for the Cys-adduct (Fig. 2, Fig. S9, ESI†). The Cys side chain is shorter than that of Hcy, allowing the NH3+ group to more tightly bind the xanthene phenolate oxygen rather than the carboxylate, thereby directly enhancing fluorophore ionization and emission. In contrast, the alkyl chain in Hcy is longer allowing the NH3+ group to instead favor formation of a salt bridge with the carboxylate moiety of 1, which has a relatively minor effect on fluorophore moiety ionization (Fig. 3). In the case of thiols without cationic character, such as mercaptoethanol and mercaptopropionate, emission is relatively lower as electrostatic interactions with the phenolate oxygen are relatively diminished. The observed trend in emission enhancement of 1 is thus Cys (40.98-fold) > Hcy (28.73-fold) > mercaptoethanol (6.04-fold) > mercaptopropionate (1.66-fold) after complete reaction with excess thiol (1 μM compound 1: 10.0 mM thiol).
The reaction of N-terminal cysteine residues with aldehydes has been applied to the site-specific modification, ligation, and labeling of peptides and proteins.6 The selectivity of 1 for N-terminal cysteine-containing peptides is evidenced by observing the fluorescence emission in the presence of N- and C-terminal dipeptides (Fig. 4).
Fig. 4.

Response of compound 1 to thiols (0.5 mM) and peptides (0.5 mM) of interest. (A) Fluorescence emission at 524 nm. (B) Absorbance at the 508 nm excitation wavelength. *Signal still increasing after 2 h.
In solutions containing 1 and Cys-Gly (an N-terminal cysteine dipeptide) or Glu-Cys (a C-terminal cysteine dipeptide), only the solution containing Cys-Gly exhibits significant fluorescence emission enhancement. A similar result is obtained for a solution of 1 and laminin-11 (a synthetic nonapeptide containing an N-terminal cysteine residue, also known as peptide 11). Laminin-11 has been used for the development of anti-metastatic drugs.7 Interestingly, the reaction of 1 with N-terminal Cys-containing peptides affords greater signal enhancement compared to simple thiols. The greatest signal enhancement of all analytes resulted from the nonapeptide (Fig. 4, see also complete emission and absorption spectra in Fig. S16, ESI†). Previously reported functional xanthenes8 that exhibit analyte-dependent emission based on an analogous combination of supramolecular and covalent interactions between sugar-derived boronate ester complexes and a rhodamine fluorophore also exhibit significantly enhanced emission in the presence of polysaccharides as compared to mono- and disaccharides.9
We propose that this increased fluorescence from larger analytes is due to more complex non-covalent interactions and/or better shielding from solvent molecules, other fluorophores, or other potential sources of non-radiative decay including dynamic quenching by dissolved oxygen as compared to smaller analyte complexes. Further studies of the impact of probe-bound analytes on the spectral properties of fluorophores are currently underway in our laboratory.
In conclusion, we have synthesized a new fluorogenic dye that displays signal amplification in the presence of biothiols. Fluorescence signaling in the presence of analytes of interest including Cys, Hcy, and N-terminal cysteine peptides was observed to be in excess of 40 times that of compound 1 alone. The fluorescence intensity was directly proportional to the analyte concentration and a greater than 2-fold selectivity towards cysteine over homocysteine was achieved. Importantly, compound 1 did not respond to structurally similar analogs. Additionally, fluorescence resulting from the reaction of 1 and N-terminal cysteine dipeptides and larger peptides was observed to be greater than upon reaction with simple thiols. The design of new dyes to attain specific signal transduction in the presence of bioactive analytes is ongoing.
Supplementary Material
Acknowledgments
Support from the National Institutes of Health via award RO1 EB002044 is gratefully acknowledged.
Footnotes
Electronic supplementary information (ESI) available: Experimental details. Synthesis and characterization of compound 1 plus additional spectral data for compound 1 and its reaction products with thiols and peptides. See DOI: 10.1039/c0cc01398f
Notes and references
- 1.Beija M, Afonso CAM, Martinho JMG. Chem Soc Rev. 2009;38:2410. doi: 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]; Chen X, Zhou Y, Peng XJ, Yoon J. Chem Soc Rev. 2010;39:2120. doi: 10.1039/b925092a. [DOI] [PubMed] [Google Scholar]
- 2.Rusin O, St Luce NN, Agbaria RA, Escobedo JO, Jiang S, Warner IM, Dawan FB, Lian K, Strongin RM. J Am Chem Soc. 2004;126:438. doi: 10.1021/ja036297t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esterbauer H, Ertl A, Scholz N. Tetrahedron. 1976;32:285. [Google Scholar]; Ohmori S, Ikeda M, Hattori H, Hagiwara K, Iwase C. J Clin Chem Clin Biochem. 1983;21:851. doi: 10.1515/cclm.1983.21.12.851. [DOI] [PubMed] [Google Scholar]
- 4.Wang WH, Rusin O, Xu XY, Kim KK, Escobedo JO, Fakayode SO, Fletcher KA, Lowry M, Schowalter CM, Lawrence CM, Fronczek FR, Warner IM, Strongin RM. J Am Chem Soc. 2005;127:15949. doi: 10.1021/ja054962n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sjoback R, Nygren J, Kubista M. Spectrochim Acta, Part A. 1995;51:L7. [Google Scholar]
- 6.Zhang LS, Tam JP. Anal Biochem. 1996;233:87. doi: 10.1006/abio.1996.0011. [DOI] [PubMed] [Google Scholar]; Ren HJ, Xiao F, Zhan K, Kim YP, Xie HX, Xia ZY, Rao J. Angew Chem, Int Ed. 2009;48:9658. doi: 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starkey JR, Dai S, Dratz EA. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1998;1429:187. doi: 10.1016/s0167-4838(98)00236-2. [DOI] [PubMed] [Google Scholar]
- 8.Jiang S, Escobedo JO, Kim KK, Alpturk O, Samoei GK, Fakayode SO, Warner IM, Rusin O, Strongin RM. J Am Chem Soc. 2006;128:12221. doi: 10.1021/ja063651p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halo TL, Appelbaum J, Hobert EM, Balkin DM, Schepartz A. J Am Chem Soc. 2009;131:438. doi: 10.1021/ja807872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.