

Abstract
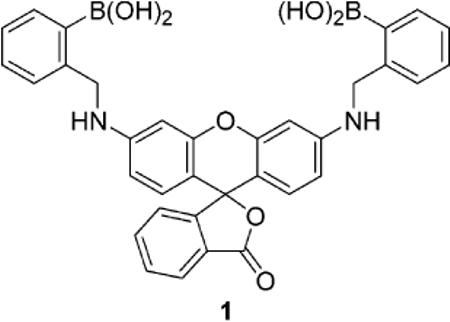
Novel chromophoric compound 1 promotes the HPLC postcolumn detection of mono- and oligosaccharides. The detection of chromatographic peaks in the visible region for glucose, fructose, maltodextrins, sialic acid, and a ganglioside can be accomplished with a standard UV–vis detector. The use of selective, reversible binding agents in automated HPLC assays should allow for improved monitoring of specific analytes as well as material recovery.
The analysis of saccharides by HPLC has played a major role in the field of glycobiology. The method of detection is an important issue because carbohydrates do not absorb in the visible region.1 Sugars can be detected by refractivity, but this method is not highly sensitive and is typically limited to isocratic chromatography. Direct UV detection of saccharides below 210 nm limits solvent choice and requires ultra-pure solvents. Electrochemical detection by pulsed ampero-metric detection (PAD) is often performed at ca. pH 13. Mass spectrometry, coupled with chromatographic separations, requires specialized, expensive equipment.1 Evaporative light scattering detection (ELSD) has attracted great recent attention for the chromatographic detection of carbohydrates; however, molecules with a lower MW range that have the potential to evaporate along with the mobile phase may require advanced detector design. Buffer choice is also limited to only a few salts because of evaporation with the mobile phase. The system may also require relatively high maintenance.2
Carbohydrates may be derivatized with chromopohores or fluorophores prior to separation; however, this procedure can hamper separation via the appendage of moieties with intrinsically similar properties. Postcolumn derivatization techniques have thus attracted great attention in carbohydrate analysis.1–3 Many older automated detection systems based on a specific, postcolumn reaction of sugars require specially designed acid-resistant reagent delivery and detection systems, cause excessive peak broadening, and are incompatible with certain solvents used for separations.2,3 More recently, milder reactions with fluorogens have been reported.4 The reagents currently used in postcolumn derivatization are typically selective for a family of compounds (for instance, aldoses, ketoses, uronic acids, aminosugars, etc.). The reactions are irreversible. The use of more selective synthetic chromogenic and/or fluorogenic receptors as postcolumn detection agents could significantly improve the analysis of a component of interest. Moreover, if binding is via non- or reversible covalent interactions, recovery of expensive or scarce biomolecules should be possible. Herein we report the use of new rhodamine-derived boronic acid 1 as a postcolumn derivatization agent in an automated HPLC method toward the detection of a variety of sugars and related biomolecules.
Recently, we reported that xanthene dye functionalized boronic acids can be formed in situ from resorcinarenes.5 These compounds were used toward the detection of several mono- and oligosaccharides.5,6 In the current work, we functionalize a related rhodamine scaffold as a result of its high molar absortivity in the visible region7 as well as the potential for favorable boron–nitrogen interactions to promote sugar-boronate formation.8 Compound 1 is readily synthesized via a reductive amination reaction between commercially available rhodamine 110, 2-formylphenylboronic acid, and NaBH4 (Scheme 1).9 It exhibits selectivity for fructose over glucose in solution, in keeping with the behavior of most boronic acid derivatives.8 We observe a blue shift of the λmax from 550 to 530 nm upon addition of d-fructose or d-glucose (Figure 1). The apparent equilibrium constant for the interaction of 1 and d-fructose is 3806 M−1; for d-glucose the value is 375 M−1.
Scheme 1.
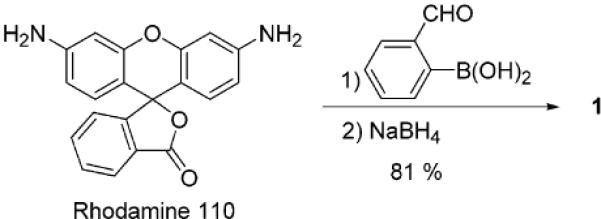
Synthesis of 1
Figure 1.
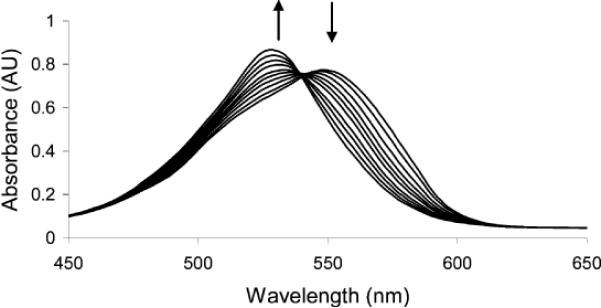
UV–vis spectra of solutions of 1 (1.64 × 10−5 M, 0.16 M pH 9.5 carbonate buffer in 1:2 MeOH/H2O) upon titration with d-fructose. The final concentration of fructose was 8.33 × 10−4 M.
Fructose is a common energy source and sweetener metabolized at a high rate in animals and humans. High D-fructose intake is implicated in the pathogenesis of hyper-triglyceridaemia, atherosclerosis,10 and insulin resistance.11 Nonenzymatic glycosidation products form more rapidly from fructose than from glucose.12 Further elucidation of the biochemical role of d-fructose requires better methods of analysis. For instance, the determination of the relatively low fructose levels in human plasma is hampered in large part as the result of an excess (ca. 100-fold) of glucose.13 Levels of fructose reported vary among laboratories and on the technique employed, including enzymatic detection.14 Many methods are based on GC or HPLC determinations but often involve tedious sample preparation and derivatization.14
We can monitor mixtures of d-glucose and d-fructose via automated postcolumn HPLC detection with 1.15 We clearly observe a peak for d-fructose even in the presence of a 100-fold excess of d-glucose (Figure 2).
Figure 2.
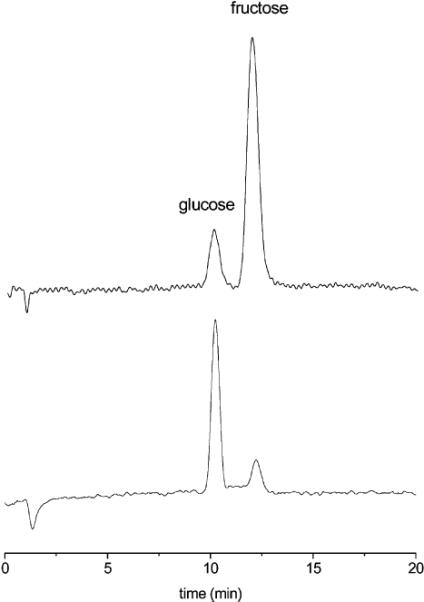
(Top) Chromatogram of a 1:1 mixture of d-fructose and d-glucose (20.0 μg, λ = 560 nm). (Bottom) Chromatogram of a mixture of d-fructose (4.5 μg) in the presence of a 100-fold excess of d-glucose.
The new postcolumn detection method is also applicable to the monitoring of oligosaccharides. Current oligosaccharide colorimetric HPLC detection methods typically require prior complete hydrolysis to monosaccharides or precolumn covalent attachment to a chromophore.16,17 The classical color tests for monosaccharides cannot be used to directly detect oligosaccharides containing more than three residues.16 The colorimetric response is only related to the molar concentration of oligosaccharide, not the concentration by weight.16 Recently, we reported that boronic acid functionalized xanthenes allowed us to generate strong colorimetric responses for larger oligosaccharides.5 We can apply these findings to the colorimetric HPLC detection of oligosaccharides. Under conditions (reactor temperature set to 94 °C) similar to those used for the monosaccharides above, mixtures of maltotriose and maltohexaose (80 μg) can be monitored (Figure 3).
Figure 3.
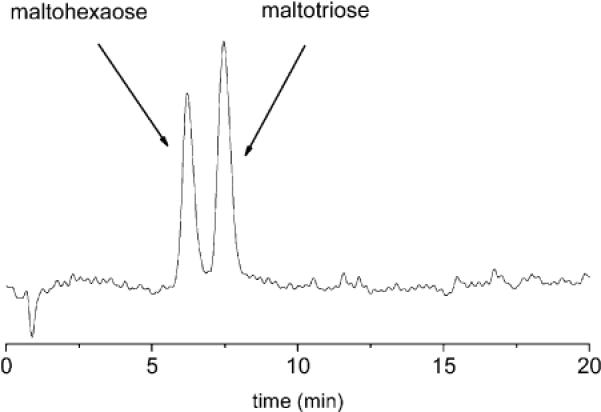
Chromatogram of a 1:1 mixture of maltohexaose and maltotriose (80 μg, λ = 560 nm).
Sialic acids are found in glycoproteins, glycopeptides, and glycolipids. Unbalanced sialic acid levels can lead to alterations in cell adhesion, which is implicated in certain cancers and graft rejection. An increase in the levels of both soluble and cellular sialic acid can be a marker for cancer diagnosis.18 Gangliosides are a natural source of sialic acids. The function of sialic acids in gangliosides is presently not completely understood.19 Improved methods of analysis would aid in the elucidation of their biochemistry. The analysis of the sialic acid content of gangliosides is challenging. The common Warren20 and Svennerholm21 color tests (the most commonly used assays for sialic acid22) employ high temperatures and harsh reagents and suffer from interference with other carbohydrates. In addition, many assays require prior liberation of the sialic acids, which often results in decomposition.23 In the case of enzymatic hydrolysis incomplete sialic acid liberation can be a problem.21
Compound 1 can be used for the postcolumn HPLC detection of N-acetylneuraminic acid, the most commonly occurring sialic acid, and GM1 (Figure 4), a commercially available ganglioside. The retention times and detection limits for N-acetylneuraminic acid, GM1, and the other analytes described above are summarized in Table 1.
Figure 4.
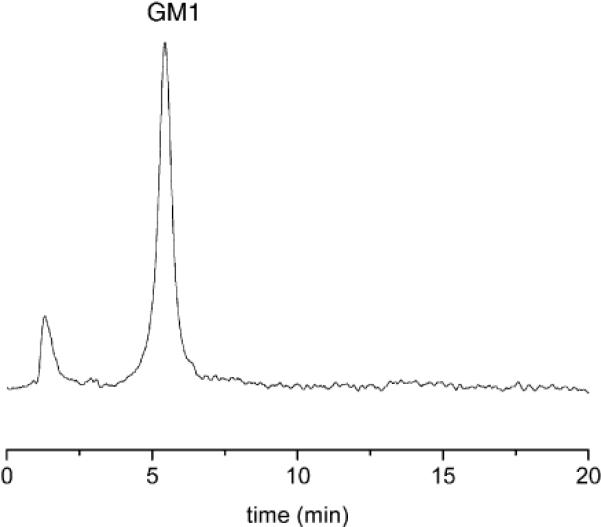
Chromatogram of GM1 (220 μg, λ = 520 nm).
Table 1.
Retention Times and Limits of Detection (LOD) for Various Carbohydrates
| carbohydrate | retention time (min) | LOD (μg) |
|---|---|---|
| d-fructose | 10.1 | 2.3 |
| d-glucose | 12.0 | 7.1 |
| maltohexaose | 6.0 | 30.1 |
| maltotriose | 7.3 | 35.8 |
| N-acetylneuraminic acid | 7.2 | 45.4 |
| GM1 | 5.5 | 26.8 |
In conclusion, we have synthesized new boronic acid probe 1 and demonstrated its use as a detection agent in an automated postcolumn HPLC system. The selectivity of 1 for fructose promotes fructose monitoring in the presence of a large excess of glucose. The affinity of 1 for oligosaccharides and sugar-containing biomolecules such as GM1 allows for colorimetric monitoring upon chromatographic elution. We are continuing to study the synthesis and properties of chromophoric and fluorophoric chemosensors and supramolecular materials as HPLC postcolumn reagents.
Acknowledgment
We gratefully acknowledge the National Institutes of Health (EB002044) for support of this research.
References
- (1).Chaplin MF, Monosaccharides . In: Carbohydrate Analysis. A Practical Approach. Chaplin MF, Kennedy JF, editors. Oxford University Press; Oxford: 1994. pp. 24–25. [Google Scholar]
- (2).Review: Herbreteau B. Analusis. 1992;10:355.
- (3).Reviews: Honda S. J. Chromatogr. A. 1996;720:183. doi: 10.1016/0021-9673(95)00022-4.. Honda S. Anal. Biochem. 1984;140:1. doi: 10.1016/0003-2697(84)90130-1.
- (4).For example: Mopper K, Dawson R, Liebezeit G, Hansen H-P. Anal. Chem. 1980;52:2018.
- (5).He M, Johnson R, Escobedo JO, Beck JA, Kim KK, St. Luce NN, Davis CJ, Lewis PT, Fronczek FR, Melancon BJ, Mrse AA, Treleaven WD, Strongin RM. J. Am. Chem. Soc. 2002;124:5000. doi: 10.1021/ja017713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Davis CJ, Lewis PT, McCarroll ME, Read MW, Cueto R, Strongin RM. Org. Lett. 1999;1:331. doi: 10.1021/ol990105a. [DOI] [PubMed] [Google Scholar]; (b) Lewis PT, Davis CJ, Cabell LA, He M, Read MW, McCarroll ME, Strongin RM. Org. Lett. 2000;2:589. doi: 10.1021/ol9903990. [DOI] [PubMed] [Google Scholar]
- (7).Miljanic S, Cimerman Z, Frkanec L, Žinic M. Anal. Chim. Acta. 2002;468:13. [Google Scholar]
- (8).Recent reviews describing sugar detection with boronic acid compounds: James TD, Shinkai S. Top. Curr. Chem. 2002;218:159.. Wang W, Gao X, Wang B. Curr. Org. Chem. 2002;6:1285.
- (9).Rhodamine 110 (0.1 g, 0.27 mmol) and 2-formylphenylboronic acid (0.082 g, 0.54 mmol) are mixed in absolute EtOH (20 mL) and PhCH3 (3.1 mL). A Dean–Stark trap is fitted, and the reaction mixture is heated at reflux overnight (18–24 h). After cooling, the solvent is removed in vacuo to afford a yellow oil. Anhydrous MeOH (25 mL) is added, and NaBH4 (0.041 g, 1.08 mmol, 4 equiv) is added over 5 min. The reaction is stirred at room temperature for 2 h and poured into ice–water (10 mL). A small amount of saturated NaHCO3 (5 mg) is added. The aqueous solution is extracted with CH2Cl2 (3 × 50 mL). The aqueous layer is collected, dried, and filtered. The solvent is removed in vacuo to afford 0.13 g (81%) of product. To obtain an analytical standard for sensing work, further purification is performed by reversed-phase HPLC using a Dynamax 60 Å C18 (21.4 mm i.d. × 25 cm) column with a flow rate of 5 mL/min and a gradient of 70/30 MeOH/H2O to 100% MeOH in 30 min. Data for 1: 1H NMR (300 MHz, DMSO-d6) δ 4.53 (s, 4H), 6.74–6.86 (m, 4H), 7.11–7.23 (m, 8H), 7.58–7.63 (m, 2H), 8.05 (d, J = 7.2 Hz, 2H), 8.54 (s, 2H); 13C NMR (500 MHz, DMSO-d6) δ 22.07, 28.66, 28.95, 46.45, 97.17, 105.85, 114.91, 118.83, 122.48, 124.05, 124.33, 125.24, 126.38, 128.13, 128.83, 129.73, 150.87, 152.33, 154.98, 168.87; UV λmax 545; FAB-MS m/z (glycerol matrix) calcd for C34H28B2N2O7 598.22 M+, found 710.1 [M + 2 C2H6O2 − 4H2O]+
- (10).MacDonald I. Adv. Exp. Med. Biol. 1975;60:57. doi: 10.1007/978-1-4615-9029-3_4. [DOI] [PubMed] [Google Scholar]
- (11).Thorburn AW, Storlein LH, Jenkins AB, Khouri S, Kreagen EW. Am. J. Clin. Nutr. 1989;49:1155. doi: 10.1093/ajcn/49.6.1155. [DOI] [PubMed] [Google Scholar]
- (12).(a) Bunn HF, Higgins PJ. Science. 1981;213:222. doi: 10.1126/science.12192669. [DOI] [PubMed] [Google Scholar]; (b) Burden AC. Lancet. 1984;11:986. doi: 10.1016/s0140-6736(84)91206-6. [DOI] [PubMed] [Google Scholar]; (c) Suárez G, Rajaram R, Oronski AL, Gawinowicz MA. J. Biol. Chem. 1989;264:3674. [PubMed] [Google Scholar]
- (13).Pettit BR, King GS, Blau K. Biomed. Mass Spectrom. 1980;7:309. doi: 10.1002/bms.1200070708. [DOI] [PubMed] [Google Scholar]
- (14).Pitkaänen E, Kanninen T. Biol. Mass Spectrom. 1994;23:590. doi: 10.1002/bms.1200230909. [DOI] [PubMed] [Google Scholar]
- (15).HPLC experiments are performed on a CM4000 multiple solvent delivery system (LDC/Milton Roy) and a SpectroMonitor 3100 UV–vis detector (LDC/Milton Roy) using a 700CH carbohydrate column (6.5 mm × 300 mm, Alltech Associates Inc.). The column is maintained at constant temperature using a CH-30 column heater (Eppendorf). The postcolumn detection system consists of a He cylinder connected to a RDR-1 reagent delivery/reaction module (Timberline). The RDR-1 unit contains a pressurized reagent reservoir, a mixing tee, and a thermostated reaction block with a Teflon reaction coil (0.02 in. i.d. × 1 m) with a nominal volume of 0.2 mL. The HPLC column is connected to the RDR-1. The RDR-1 is attached to a SpectroMonitor 3100 UV–vis detector (LDC/Milton Roy). The column temperature is set to 85 °C, and the temperature of the reaction block is set to 50 °C. Absorbance is monitored at 560 nm except for sialic acid and GM1, which are monitored at 520 nm. The reagent consists of a solution of 1 in 0.05 M buffer (pH = 10.5, carbonates). The final concentration of 1 is 4.0 × 10−6 M. The reagent is introduced at a flow rate of 0.5 mL/min, and the reactor temperature is kept at 50 °C. The mobile phase is 100% deionized H2O
- (16).Kennedy JF, Pagliuca G, Oligosaccharides . In: Carbohydrate Analysis. A Practical Approach. Chaplin MF, Kennedy JF, editors. Oxford University Press; Oxford: 1994. pp. 46–48. [Google Scholar]
- (17).Reviews: Bardelmeijer HA, Waterval JCM, Lingeman H, van't Hof R, Bult A, Underberg WJM. Electrophoresis. 1997;18:2214. doi: 10.1002/elps.1150181212.. LoGuidice JM, Lhermitte M. Biomed. Chromatogr. 1996;10:290. doi: 10.1002/(SICI)1099-0801(199611)10:6<290::AID-BMC623>3.0.CO;2-H.
- (18).Reviews: Schutter EMJ, Visser JJ, Vankamp GJ, Mensdorffpouilly S, Vandijk W, Hilgers J, Kenemans P. Tumor Biol. 1992;13:121.. Roth J. Histochem. J. 1993;25:687. doi: 10.1007/BF00211765.. Kobata A. Acc. Chem. Res. 1993;26:319.. Reuter G, Gabius H-J. Biol. Chem. Hoppe-Seyler. 1996;377:325. doi: 10.1515/bchm3.1996.377.6.325.
- (19).(a) Schauer R, Kelm S, Rerter G, Roggentin P, Shaw L. In: Biology of the Sialic Acids. Rosenberg A, editor. Plenum; New York: 1995. p. 7. [Google Scholar]; (b) Nagai Y, Iwamori M. In: Biology of the Sialic Acids. Rosenberg A, editor. Plenum; New York: 1995. p. 197. [Google Scholar]
- (20).Warren L. J. Biol. Chem. 1959;234:1971. [PubMed] [Google Scholar]
- (21).Svennerholm L. Biochim. Biophys. Acta. 1957;24:604. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- (22).Hikita T, Tadano-Aritomi K, Iida-Tanaka N, Toyoda H, Suzuki A, Toida T, Imanari T, Abe T, Yanagawa Y, Ishizuka I. Anal. Biochem. 2000;281:193. doi: 10.1006/abio.2000.4561. [DOI] [PubMed] [Google Scholar]
- (23).Mattoo R, Roseman S. Anal. Biochem. 1997;246:30. doi: 10.1006/abio.1996.9987. [DOI] [PubMed] [Google Scholar]