

Abstract
Objectives
Aspirin, a major anti-platelet and cancer preventing drug, irreversibly blocks the cyclooxygenase activity of prostaglandin H synthase-1 (PGHS-1). Considerable differences in aspirin effectiveness are observed between individuals, and some of this variability may be due to PGHS-1 protein variants. Our overall aim is to determine which, if any, of the known variants in the mature PGHS-1 protein lead to functional alterations in cyclooxygenase catalysis or inhibition by aspirin. The present study targeted four PGHS-1 variants: R53H, R108Q, L237M and V481I.
Methods
Wildtype human PGHS-1 and the four polymorphic variants were expressed as histidine-tagged, homodimeric proteins in insect cells using baculovirus vectors, solubilized with detergent, and purified by affinity chromatography. The purified proteins were characterized in vitro to evaluate cyclooxygenase and peroxidase catalytic parameters and the kinetics of cyclooxygenase inhibition by aspirin and NS-398.
Results
Compared to wildtype, several variants exhibited a higher COX/POX ratio (up to 1.5-fold, for R108Q), an elevated arachidonate Km (up to 1.9-fold, for R108Q), and/or a lower aspirin reactivity (up to 60% less, for R108Q). The decreased aspirin reactivity in R108Q reflected both a 70% increase in the Ki for aspirin and a 30% decrease in the rate constant for acetyl group transfer to the protein. Computational modeling of the brief aspirin pulses experienced by PGHS-1 in circulating platelets during daily aspirin dosing predicted that the 60% lower aspirin reactivity in R108Q gives a 15-fold increase in surviving cyclooxygenase activity; smaller, ~2-fold increases in surviving cyclooxygenase activity were predicted for L237M and V481I. NS-398 competitively inhibited cyclooxygenase catalysis of the wildtype (Ki = 6 μM) and inhibited cyclooxygenase inactivation by 1.0 mM aspirin in both wildtype (IC50 = 0.8 μM) and R108Q (IC50 = 2.1 μM).
Conclusions
Of the four PGHS-1 variants examined, R108Q has the largest functional effects, with evidence for impaired interactions with cyclooxygenase substrate and inhibitors. As Arg108 is located on the protein surface and not in the active site, the effects of R108Q suggest a novel, unsuspected mechanism for modulation of the PGHS-1 active site structure. The lower intrinsic aspirin reactivity of R108Q, V481I and L237M, combined with the rapid hydrolysis of aspirin in the blood, suggests that these variants decrease the anti-platelet effectiveness of the drug. These PGHS-1 variants are uncommon but aspirin is very widely used, so a considerable number of individuals could b e affected. Further examination of these and other PGHS-1 variants will be needed to determine whether PGHS-1 genotyping can be used to personalize anti-cyclooxygenase therapy.
Keywords: prostaglandin H synthase-1, polymorphisms, cyclooxygenase, aspirin
INTRODUCTION
Prostanoids originating from arachidonate via the cyclooxygenase (COX) activity of prostaglandin H synthase-1 (PGHS-1) have important pathophysiological functions [1]. COX-1 inhibitors, principally aspirin, are part of current therapy for ischemic stroke and myocardial infarction; over one third of adults in the US take aspirin for prevention of cardiovascular disease [2, 3]. In addition, aspirin is emerging as an effective cancer prevention agent [4, 5]. PGHS-1 has very weak affinity for aspirin (Ki ~20 mM) [6, 7] but the acetyl group of aspirin can transfer to Ser529 (human PGHS-1 numbering) in the COX active site [8]. This covalent modification blocks access to the catalytic Tyr384 radical, permanently inhibiting COX activity in affected platelets [9, 10].
A substantial fraction of patients do not respond to aspirin therapy for cardiovascular disease, and some of this “aspirin resistance” has been attributed to genetic variation [11]. For example, our group has shown that individuals with genetic variants in PTGS1, the human gene for PGHS-1, may benefit less from aspirin with respect to cancer prevention [12]. A number of sequence variants, mostly single nucleotide polymorphisms (SNPs), have been reported in the dbSNP database for PTGS1 [13]. Of particular interest are coding region SNPs that lead to structural changes in the mature PGHS-1 protein (residues 24-599 [14]), as these may directly impact COX-1 catalysis or pharmacology. There are over thirty such SNPs listed in dbSNP [13]. Of these, five have been characterized in vitro as recombinant proteins, but the possibility of altered aspirin inhibition kinetics in the variants was not examined [15]. COX inhibition by aspirin follows a two-step mechanism (Eq. 1; E, PGHS-1), characterized by a dissociation constant (Ki) for the first step and a first order rate constant (k2) for the second step [6]:
| (Eq. 1) |
As a consequence, aspirin shows “time dependent” kinetics, with exponential decay of COX activity over time when PGHS-1 is preincubated with the drug [16].
The covalent and essentially irreversible modification of PGHS-1 by aspirin is unique among anti-COX drugs, compensating for weak aspirin binding; the IC50 is 2 μM in blood ex vivo [17]. Aspirin is particularly potent in anucleate platelets, which cannot replace inactive, acetylated PGHS-1 [9].
To begin evaluation of the functional impact of PGHS-1 structural variants, we selected a group of four variants that were among the most prevalent and/or were positioned in the vicinity of structural and functional landmarks in crystallographic models: R53H, R108Q, L237M and V481I. We expressed wildtype human PGHS-1 and these four variants in an insect cell system and used the purified proteins to evaluate the effects of the structural changes on COX catalysis and aspirin reactivity.
METHODS
Materials
Aspirin (ASA) and NS-398 were from Cayman Chemical Company (Ann Arbor, MI), fatty acids were purchased from NuChek Preps (Elysian, MN) and Tween-20 (10% solution) was obtained from Anatrace (Maumee, OH). Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs (Beverly, MA), oligonucleotides were from Integrated DNA Technologies (Coralville, IA), and reagents for DNA manipulation were from Promega (Madison, WI). The plasmid transfer vector pAcSG2 and BaculoGold linearized baculovirus DNA were from PharMingen (San Diego, CA). QuikChange site-directed mutagenesis kit and E. coli strain XL-10 were from Stratagene (La Jolla, CA). Sf9 cells, E. coli strain DH5α, Grace’s supplemented medium, and fetal bovine serum were from Invitrogen (Carlsbad, CA). Ni-NTA agarose was purchased from Qiagen (Valencia, CA). All other reagents were obtained from Sigma (St. Louis, MO).
Construction of plasmid for recombinant wildtype and variant PGHS-1
The cDNA we originally cloned as “wildtype” PGHS-1 [18] was later found to actually code for the minor allele at position 237, i.e., L237M [19]. Consequently, introduction of codons for a 6×His tag sequence downstream of the signal peptide cleavage site near the amino terminus [20] produced a plasmid with the coding sequence for the L237M variant. To generate coding sequence for true PGHS-1 wildtype (i.e., carrying the major allele at all targeted positions), the codon for methionine at position 237 was mutated to a codon for leucine, using the QuikChange kit and the following primer pairs (base changes underlined): M237L-f: 5′-CATTTATGGAGACAATCTGGAGCGTCAGTATC-3′ M237L-r: 5′-GATACTGACGCTCCAGATTGTCTCCATAAATG-3′ The resulting plasmid coding for wildtype PGHS-1 was then used as the template for introducing point mutations corresponding to the R53H, R108Q and V481I variants of PGHS-1, using the primer pairs below (base changes underlined), again with the QuikChange mutagenesis kit. R53H-f: 5′-CTTCGGCCTTGACCACTACCAGTGTGACTG-3′ R53H-r: 5′-CAGTCACACTGGTAGTGGTCAAGGCCGAAG-3′ R108Q-f: 5′-GCCACCTTCATCCAAGAGATGCTCATGCGC-3′ R108Q-r: 5′-GCGCATGAGCATCTCTTGGATGAAGGTGGC-3′ V481I-f: 5′-CCTTCCAGGAGCTCATAGGAGAGAAGGAG-3′ V481I-r: 5′-CTCCTTCTCTCCTATGAGCTCCTGGAAGG-3′ The integrity of each plasmid described below was verified by restriction enzyme digestion and by DNA sequencing at the Microbiology and Molecular Genetics Core Facility, UT Health Science Center at Houston.
Baculovirus generation and expression of recombinant proteins
Generation, amplification, and titer determination of recombinant baculovirus containing the desired PGHS-1 cDNA, and expression of the recombinant proteins used previously published techniques [20]. Solubilization of recombinant PGHS-1 with detergent (Tween 20) and purification by affinity chromatography on Ni-NTA agarose followed the procedures described for recombinant PGHS-2 [21].
Protein characterization
Total protein concentrations were determined by a modified Lowry assay [22]. PGHS-1 variants were analyzed by polyacrylamide gel electrophoresis under denaturing conditions [23], visualization with Coomassie staining, and densitometry using ImageJ [24] to determine the fraction of total protein present as recombinant PGHS-1.
COX activity
Oxygen uptake was measured polarographically in standard assays at 30 or 37 °C in 3.0 ml of 100 mM KPi, pH 7.2 with 1.0 mM phenol, 1.0 μM heme and 0.025% Tween-20, using a “Standard” electrode membrane (YSI, Yellow Springs, OH) [25]. One unit of COX activity gives a rate of 1 nmol O2 consumed/min. Specific activities were calculated by dividing the observed COX activity measured at 30 °C by the concentration of recombinant PGHS-1 present. COX kinetic parameters were determined at 37 °C by measuring the activity in the reaction mixture described above, but with 2-100 μM arachidonate in the presence of 100 μM HOOH (to ensure full COX activation); in some cases the Tween concentration was raised to 0.165%. Arachidonate was suspended at 30 mM in 0.1 M Tris HCl, pH 8.5. Following our earlier approach [26, 27], the velocities were normalized to those for the same batch of enzyme with 100 μM arachidonate without added peroxide, and then fitted to the Michaelis-Menten equation using Kaleidagraph (Synergy Software) to estimate the Vmax (Relative) and Km values.
COX inhibition
COX inhibition kinetics were assessed at 37 °C with a standard electrode membrane in the standard reaction mixture. Inhibitors were dissolved in DMSO; the final DMSO concentration was below 1% and did not affect activity. Two experimental designs were used. The first was used for reversible PGHS-1 inhibitors, such as NS-398. Enzyme (~10−8 M) was added last to start the reaction after equilibration of the reaction mixture with arachidonate (2-100 μM) and a fixed level of inhibitor. Data for activity as a function of arachidonate concentration at each inhibitor level were analyzed by a 1/v vs. 1/S plot to determine the type of inhibition; these data were also fitted to the Michaelis-Menten equation to estimate the apparent Vmax and Km values.
The second inhibitor assay focused on the progressive, irreversible inhibition by aspirin and involved timed preincubation of enzyme (~10−8 M) with inhibitor before starting the COX reaction by addition of 100 μM arachidonate. This assay measures the amount of surviving COX activity, i.e., enzyme that has not progressed to tight inhibitor-protein complexes [28]. Data for surviving COX activity as a function of preincubation time at a fixed inhibitor level were fitted to the equation: v = V0 (exp(−k2’t)), where t is the preincubation time, V0 is the COX activity at t = 0 and k2’ is the apparent rate constant. The k2’ value is expected to be a saturable function of inhibitor concentration for time-dependent inhibitors [6]: k2’ = k2/(1 + Ki/I). Note that if I << Ki, this equation simplifies to k2’ = k2(I)/Ki. When NS-398 was examined as an antagonist of time-dependent inhibition by 1 mM aspirin, an IC50 value for NS-398 was estimated by fitting the k2’ vs. NS-398 data to a three-parameter logistic function: k2’ = k2max’/(1 + (I/IC50)n), where k2 max’ is the k2’ value without NS-398 and n is the Hill slope. IC50 values were converted to Ki values using the Cheng-Prusoff equation [29].
The timed preincubation assay described above was modified to measure COX inhibition kinetics at the higher aspirin levels needed to observe saturable behavior and thus to quantify Ki and k2 values individually. First, the reaction of aspirin with PGHS-1 was slowed by decreasing the reaction temperature from 37 to 20°C. The second modification was to use the potassium salt of aspirin instead of the acidic form. Aspirin was coverted to the salt by treatment with potassium carbonate, adapting an old procedure [30]; the stable aspirin salt was dried under vacuum and ground to a powder. The aspirin content in the salt was determined from the increase in A530 after mixing with ferric chloride (with and without prior hydrolysis by alkali) [31]. For the COX reactions, a portion of the aspirin salt was weighed out and dissolved at the desired concentration in reaction buffer within 45 min of use (thereby keeping aspirin hydrolysis below 2%). Even at 50 mM, the potassium salt of aspirin lowered the pH of the reaction buffer by only 0.03 units. Use of the potassium salt thus greatly extended the practical upper limit of aspirin concentration.
Peroxidase (POX) activity
The stirred assay mixture contained 2.5 ml of 100 mM Tris HCl, pH 8.0 with 0.50 mM guaiacol, 1.0 μM heme, and 0.40 mM HOOH at room temperature [32]. Reaction was started by injection of enzyme and the initial velocity calculated from the rate of increase in A436 due to oxidized guaiacol (ε436 = 6.39 (mM oxidized guaiacol)−1 cm−1). One unit of POX activity gives a velocity of 1 nmol guaiacol oxidized/min.
Statistical analysis
The results of replicate experiments are presented as mean ± standard deviation of the mean. In most cases an unpaired, two-sided t test was used to assess statistical differences between individual variants and wildtype PGHS-1 (GraphPad Software, La Jolla, CA). Differences with a P-value below 0.05 were considered to be statistically significant.
Analysis of COX product profiles
Duplicate reactions at room temperature included purified recombinant PGHS-1 wildtype or variant (1-9 μl, adjusted to consume ~10 nmol of arachidonate) in 0.30 ml of 25 mM KPi pH 7.2 containing 1 mM phenol, 80 μM [1-14C]arachidonate, 0.2 mM SnCl2, and 1 μM heme. The SnCl2 stock solution (20 mM in 10 mM HCl) was prepared immediately before use and kept on ice. Reactions were started by injection of enzyme and quenched after 90 s by adding 0.60 ml of ice-cold toluene-ethyl acetate (1:1), 7 μl of 1.2 M HCl and 200 mg of NaCl, followed by vigorous vortexing. After brief centrifugation, the organic phase was transferred to a vial containing 0.1 g of anhydrous Na2SO4 and the aqueous phase was extracted again. The organic extracts were combined and stored at −20 °C until analyzed. For chromatographic analysis, 300 μl of each extract was spiked with 50 μg each of unlabeled arachidonate and PGF2α (and other standards as desired) and the volume was reduced to ~30 μl under vacuum before application to the preabsorbent zone of a Whatman LK6D TLC plate. The plate was developed with the upper phase of a mixture of ethyl acetate -2,2,4-trimethylpentane - acetic acid - water (110:50:20:100; v/v) [33]. Radioactive bands were visualized by exposure to a storage phosphor sheet and analyzed quantitatively on a Molecular Dynamics Storm model 860 imager. The positions of unlabeled standards were visualized by exposure to iodine vapor.
Aspirin hydrolysis
The decomposition kinetics of aspirin were determined by monitoring the generation of SA [31] when 5.33 mM ASA was incubated at 37 °C in the standard reaction mixture without arachidonate (0.10 M KPi, pH 7.2 containing 1.0 mM phenol, 1.0 μM heme and 0.025% Tween 20). Aliquots (200 μl) were withdrawn periodically and mixed with 1.0 mL of ferric nitrate reagent (4.0 g of Fe(NO3)3·9H2O dissolved in 12 ml of 1 M HCl and diluted with water to 100 ml). SA levels were calculated from the A529 values using a standard curve with 0 - 0.70 μmol of standard SA dissolved in 200 μl of the incubation buffer. The half-life for ASA observed under these conditions was 18 hr, quite comparable to the value of 15.4 hr reported at the same temperature for 0.1 M phosphate buffer, pH 7.4 [34]. As a consequence of this long half-life for ASA and the relatively short incubations used in the present study (< 7 min) it was not necessary to correct for ASA hydrolysis.
Computational modeling of COX-1 inhibition by pulsatile aspirin exposure
A computational model was devised to predict the impact of differences in aspirin reactivity on COX inhibition kinetics during a transient pulse of the drug, such as that seen in the blood after oral ingestion of aspirin. The model, implemented in Berkeley Madonna X v.8.3.22, consisted of the following reactions and rate constant values. E, enzyme (PGHS-1); ASA, aspirin; SA, salicylate; AcOH, acetate.
| 1) E + ASA ⇔ E(ASA) | reversible binding of aspirin to PGHS-1 |
| k1: 1 × 105 M−1s−1 k−1: 1 × 103 s−1 | |
| 2) E(ASA) ⇒ E-Ac + SA | acetylation of Ser529 of PGHS-1, release of salicylic acid |
| k2: 0.33 s−1 (wildtype); k2: 0.133 s-1 (R108Q) | |
| 3) E + SA ⇔ E(SA) | reversible binding of salicylate to PGHS-1 |
| k3: 1 × 105 M−1s−1 k-3: 1 × 103 s−1 | |
| 4) ASA (in gut) ⇒ ASA (in blood) | diffusion of aspirin from gut into blood |
| k4: 1 × 10−3 s−1 | |
| 5) ASA ⇒ SA + AcOH | hydrolysis of aspirin (written as first order reaction) |
| k5: 7.7 × 10−4 s−1 | |
| 6) SA (in blood) ⇒ SA (in urine) | transport of salicylate from blood to urine |
| k6: 5 × 10−5 s−1 | |
Reactions 4-6 generate a rapid aspirin pulse and a slower pulse in salicylate. Reactions 1-3 predict how the aspirin and salicylate pulses affect COX activity, with reversible inhibition by aspirin (Reaction 1) and salicylate (reaction 3) and irreversible inactivation by aspirin (Reaction 2). Note that the very high Ki values for aspirin and salicylate [6] mean that only a negligibly small fraction of enzyme is reversibly inhibited at any time by these agents. Initial concentrations were set at zero, except for E (1 × 10−8 M) and ASA (in gut; 1 × 10−4 M). The model was run for 200 minutes to simulate the effects of a single aspirin pulse. To simulate the effects of a second aspirin pulse, the concentrations of all enzyme intermediates were decreased by 15% and the resulting level of free enzyme was supplemented with 15% of its initial value (reflecting daily replacement of 15% of existing platelets), ASA and SA in blood were set to zero (reflecting complete clearance after 24 hours), ASA in the gut was set to its initial value (relecting fresh oral aspirin ingestion), and the model run for another 200 minutes. This process was repeated to predict the effects of further cycles of daily aspirin ingestion.
RESULTS
Catalytic properties of recombinant PGHS-1 proteins
The wildtype and each of the four PGHS-1 variants were expressed in catalytically active form. Purification of the recombinant proteins removed host cell lipids, small molecule reductants and endogenous proteins, avoiding potential sources of interference for reliable characterization of COX and peroxidase (POX) catalytic activities. The COX specific activities of the purified proteins ranged from 16 to 34 k units/mg PGHS protein, comparable to previous reports for recombinant PGHS-1 and -2 [35]. We analyzed the COX/POX ratio in the purified proteins to control for specific activity differences due to variations in expression and purification [27]. The COX/POX ratio for the V481I variant was indistinguishable from the wildtype value, but the ratio was significantly elevated in the other three variants (13-53%; p < 0.05; Table 1). This indicates that the structural changes in R108Q, L237M, and perhaps R53H increase the efficiency of COX catalysis relative to POX catalysis.
Table 1.
Kinetic parametersa for wildtype (WT) and variant PGHS-1 proteins
| PGHS-1 construct |
VCOX/VPOX (Rel)b |
COX Km (μM arachidonate) |
|
|---|---|---|---|
| 0.025% Tween 20 |
0.165% Tween 20 |
||
| WT | 1.00 ± 0.07 | 1.5 ± 0.4 | 19.8 ± 3.7 |
| R53H | 1.13 ±0 .03 p = 0.042 |
1.9 ± 0.2 | Not measured |
| R108Q | 1.53 ± 0.12 p = 0.003 |
2.8 ± 0.4 p = 0.016 |
29.7 ± 3.3 p = 0.026 |
| L237M | 1.27 ±0 .09 p = 0.015 |
1.8 ± 0.3 | Not measured |
| V481I | 1.07 ±0 .14 | 1.8 ± 0.2 | Not measured |
Averages ± SD for n = 3 unless indicated otherwise; p-value for difference with wildtype shown if < 0.05.
Ratio of COX rate to POX rate, normalized to wildtype value.
Observed rate of COX decay (k’) in a preincubation experiment divided by the fixed aspirin level.
A saturable response of COX to arachidonate level was observed with each of the purified recombinant proteins (data from one experiment for wildtype and for the R108Q variant are shown in Fig. 1). The calculated Km values for the variants were indistinguishable from the wildtype value, except for R108Q, where the Km was almost doubled (Table 1). This difference in Km value between R108Q and wildtype was small in absolute terms, so the substrate dependence for wildtype and R108Q was also examined in reactions at a higher Tween 20 level (0.165%), which is known to increase the apparent Km value [36]. Saturable behavior was also seen at the higher detergent level (Fig. 1), and the resulting Km value for R108Q was 50% above the wildtype value (Table 1), confirming the elevated Km value observed under standard assay conditions. Thus, the interaction with arachidonate was considerably more difficult to saturate in R108Q than in wildtype PGHS-1.
Fig. 1.
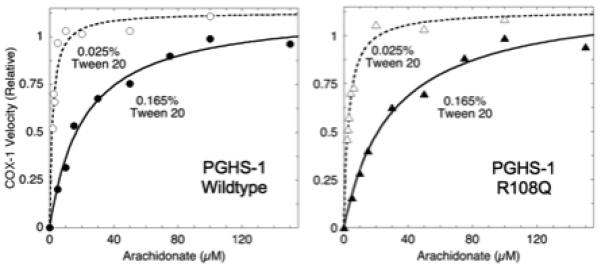
Substrate dependence of COX activity in wildtype PGHS-1 and the R108Q variant. COX activity was determined by oxygen electrode measurements at 37 °C with the indicated fatty acid concentrations and 0.025 or 0.165% Tween 20. The lines represent least-squares fits to the Michaelis-Menten equation. Data shown are for one of three similar experiments.
COX product profiles
Each of the purified recombinant proteins was reacted with radiolabeled arachidonate and the products analyzed by thin layer chromatography (Fig. 2). The wildtype enzyme produced PGH2 and several secondary prostanoids derived from it (PGD2, PGE2 and PGF2α), 12-HHT from decomposition of PGH2, and some non-cyclized COX side products (11- and 15-HETEs). Each of the PGHS-1 variants had a product profile quite similar to that of the wildtype enzyme (Fig. 2). Although there were differences in the relative proportions of individual products, there was no indication that any of the variants produced a novel arachidonate metabolite. Inclusion of 1 mM glutathione in the buffer for reactions with wildtype PGHS-1 and the R108Q variant resulted in a decreased amount of PGH2 and a comparable increase in the 12-HHT (data not shown); this effect of GSH was reported earlier for the wildtype enzyme [37].
Fig. 2.
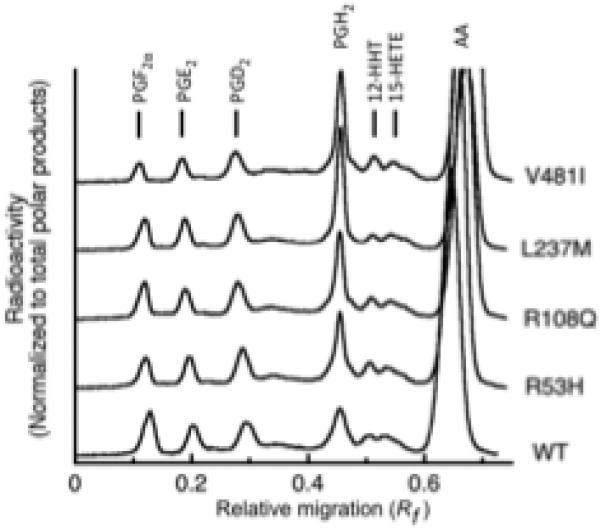
Thin layer chromatographic analysis of arachidonate reaction products from PGHS-1 wildtype (WT) and variants. The positions of standards are indicated. Details are described in Methods.
Inhibition of COX activity by aspirin
The response of the PGHS-1 variants to aspirin was first examined by monitoring the loss in COX activity during timed preincubation with 1.0 mM aspirin (Fig. 3A). Loss of COX activity followed first order kinetics for wildtype PGHS-1 and all variants. The decay rates in the R53H, L237M and V481I variants were comparable to that for wildtype, but COX decay was markedly slower in the R108Q variant. The COX activities decayed at similar, very slow rates (0.02-0.03 min−1) in the controls without aspirin (data not shown).
Fig. 3.
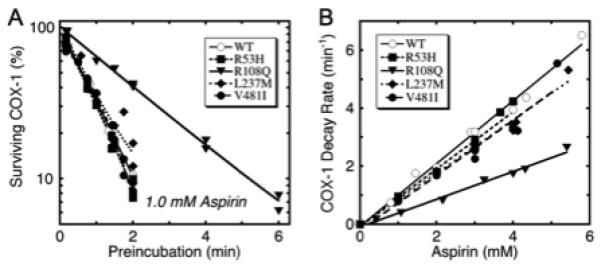
Kinetics of COX inhibition by aspirin in wildtype PGHS-1 and variants. A) Decay of COX activity during preincubation with 1.0 mM aspirin. Lines represent fits for single exponential decay. B) Effect of aspirin level on COX decay rate during preincubation. Each data point represents the fitted decay rate for a separate experiment with the indicated aspirin level and at least five different lengths of preincubation. Rate constants were corrected for control COX decay rates in absence of aspirin. Lines represent linear fits.
For more detailed comparison of acetylation kinetics, the individual COX decay rate constants determined from preincubation experiments at 0.4-5.3 mM aspirin, similar to those in Fig. 3A, were plotted as a function of the aspirin level during preincubation (Fig. 3B). For each protein the decay rate was a linear function of the aspirin concentration, indicating that the aspirin levels tested were well below the Ki value. This is consistent with the reported Ki of 14-20 mM for ovine PGHS-1 [6, 7]. The time- and concentration-dependence of COX activity loss for wildtype PGHS-1 and for the variants (Fig. 3) is consistent with the mechanism in Eq. 1.
The linear relationship between the observed COX decay rate and aspirin level (Fig. 3B) was exploited to obtain a single parameter characterizing aspirin reactivity for each experiment at a given level of aspirin, calculated as k2’/[aspirin]; averages of these aspirin reactivity values are shown in Table 2. The aspirin reactivity for wildtype human PGHS-1 was 1.04 mM−1 min−1, in reasonable accord with the value of 0.3 mM−1 min−1 determined for ovine PGHS-1 at a lower temperature of 30°C [6]. The aspirin reactivity values for the variants were 40-91% of the wildtype value, with the differences being statistically significant (Table 1). Thus, the structural changes in each variant decreased the reactivity toward aspirin, with the difference most marked in the R108Q variant.
Table 2.
Aspirin reactivity of wildtype (WT) and variant PGHS-1 proteins
| PGHS-1 construct |
Aspirin reactivitya (relative) |
|---|---|
| WT | 1.00 ± 0.10 (n = 9) |
| R53H | 0.91 ± 0.07 (n = 9) p = 0.042 |
| R108Q | 0.40 ± 0.05 (n = 7) p = 0.0001 |
| L237M | 0.86 ± 0.05 (n = 9) p = 0.001 |
| V481I | 0.82 ± 0.10 (n = 9) p = 0.001 |
Average ± SD of k2’/[aspirin], normalized to wildtype value (1.04 mM−1 min−1).
Analysis of Ki and k2 values for aspirin
We used the potassium salt of aspirin and reduced temperature to analyze COX inhibition kinetics of wildtype PGHS-1 and the R108Q variant over a greatly expanded range of aspirin levels (Fig. 4). Clearly saturable and very reproducible behavior was evident for both enzymes. The fitted values of Ki and k2 from triplicate experiments are shown in Fig. 4. R108Q has a 70% higher Ki and a 30% lower k2, with both differences being statistically significant. These results indicate that both the aspirin binding and the acetyl transfer steps are adversely affected in R108Q, as might be expected if the structure of the aspirin binding site is perturbed in the variant. Note that the value of k2’/aspirin (≈ k2/Ki) determined at 37°C (Fig. 3 and Table 2) for R108Q is 40% of the wildtype value, while the value of k2/Ki determined at 20 °C (Fig. 4) for R108Q is 42% that for wildtype. This agreement between measurements at the two temperatures confirms that the temperature change affects wildtype and R108Q similarly, validating the use of low temperature for detailed aspirin kinetic analysis.
Fig. 4.
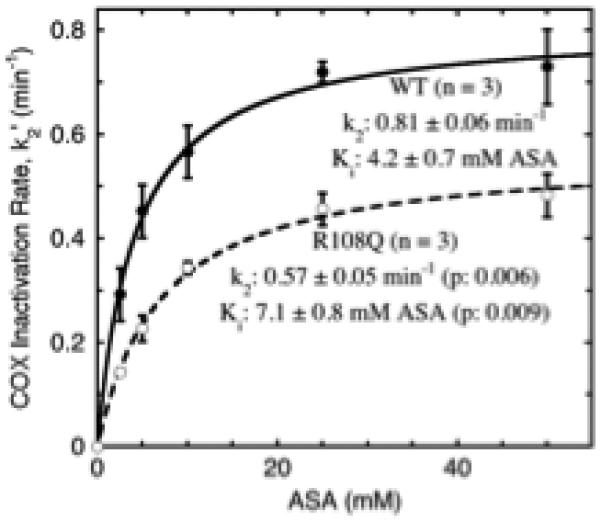
Analysis of Ki and k2 values for aspirin in wildtype PGHS-1 (WT) and the R108Q variant. Apparent COX decay rates during preincubation (k2’ values) were determined at 20 °C at each of the aspirin (potassium salt) levels indicated. Each point and error bars represent the average ± SD at a given aspirin level in three separate k2’ vs. [aspirin] experiments. The parameters shown are the averages ± SD for k2 and Ki values from fits to the Michaelis-Menten equation in the three individual experiments. The lines shown are the fits of the pooled data from all three experiments to the Michaelis-Menten equation. Details are described in Methods.
Effects of NS-398 on COX-1 inhibition by aspirin
We analyzed wildtype PGHS-1 and the R108Q variant for the actions of a coxib, NS-398, on the kinetics of COX inhibition with 1.0 mM aspirin. As shown in Fig. 5, adding NS-398 to the preincubation with aspirin slowed the rate of acetylation of both wildtype and the R108Q variant. The attenuating effect on acetylation rates appeared to increase as a saturable function of the NS-398 concentration. Accordingly, the data were fitted to a three parameter logistic equation to estimate IC50 values, which were 0.8 μM NS-398 for wildtype PGHS-1 and 2.1 μM NS-398 for the R108Q variant (Fig. 5). The aspirin concentration used in these experiments, 1.0 mM, is far below aspirin’s Ki value for both enzymes. As a result, the dissociation constants for the NS-398 complexes that compete with aspirin binding (i.e., the Ki values for NS-398) will be only slightly lower than the corresponding IC50 values for NS-398. Thus, NS-398 has a weaker affinity as an aspirin antagonist for R108Q than wildtype PGHS-1.
Fig. 5.
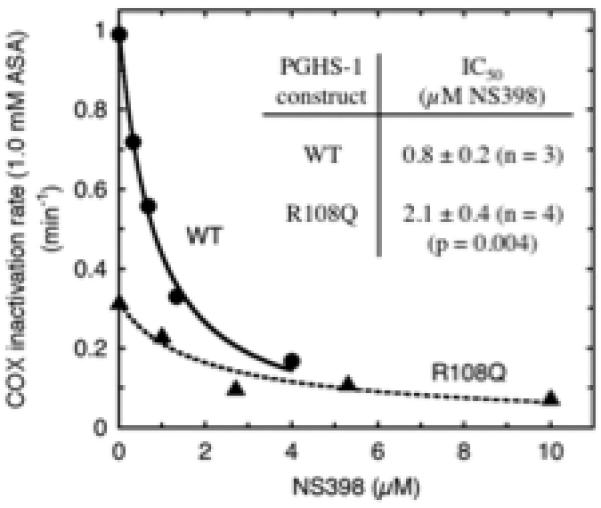
Effect of NS398 on kinetics of COX inactivation by aspirin in PGHS-1 wildtype (WT) and R108Q variant. Each enzyme was preincubated with 1.0 mM aspirin and the indicated levels of NS-398 for varying lengths of time to determine apparent rate constants (k2’) for loss of COX activity. The k2’ vs. [NS-398] data from each experiment were analyzed to estimate IC50 values as described in Methods. The IC50 values shown are averages ± SD from 3-4 experiments.
Effects of NS-398 on COX-1 activity
The observation that NS-398 slowed the rate of acetylation in PGHS-1 (Fig. 5) suggested that NS-398 is a competing ligand at the COX active site. This possibility was tested directly by measuring the substrate dependence of wildtype PGHS-1 COX activity at 0, 8 and 20 μM NS-398. The results are presented as a double reciprocal plot in Fig. 6. The observed pattern of intersecting lines on the 1/V axis indicates that NS-398 increases the apparent Km value while leaving the Vmax value largely unchanged. This is the behavior expected for a simple competitive inhibitor. Further, a plot of the apparent Km value as a function of the NS-398 level (inset to Fig. 6) exhibits the linear relationship expected for a competitive inhibitor. Fitting the inset data to the equation Km’ = Km (1 + I/Ki) gives a Km value of 1.8 μM arachidonate and a Ki value of 6.7 μM NS-398. Results from a second, similar experiment indicated a Km value of 1.7 μM arachidonate and a Ki value of 5.1 μM NS-398 (data not shown). The average Ki value of ~6 μM for NS-398 as arachidonate antagonist is considerably higher than the Ki of ~1 μM NS-398 as aspirin antagonist (see above). This substantial difference between the dissociation constants for the coxib suggests that two distinct modes of NS398 binding are involved in antagonism of PGHS-1 interactions with aspirin and with arachidonate.
Fig. 6.
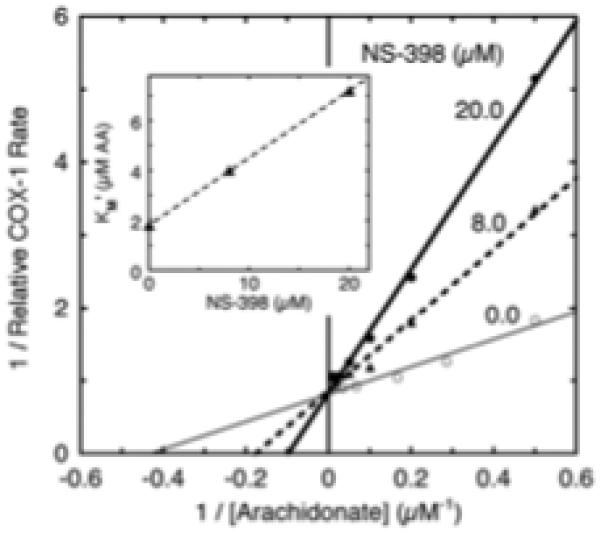
Analysis of COX inhibition type for NS-398 in wildtype PGHS-1. The lines are transformed from the non-linear least squares fits of data at each NS-398 level to the Michaelis-Menten equation. Inset: apparent Km values for arachidonate (AA) calculated from data in the main figure are shown as function of the NS-398 level. A linear fit to the data is shown. Details are described in Methods.
DISCUSSION
Variants and catalytic activity
COX activity has now been examined for a number of PGHS-1 variants using endogenous proteins in human platelets [38] as well as recombinant proteins in detergent extracts [15] and in purified form (present study) (Table 3). As expected, variants in the cleaved signal peptide (R8W and P17L) had little effect on COX activity. Except for V481I, each of the seven variants in the mature PGHS-1 protein was found to have a COX specific activity consistently lower than that of wildtype PGHS-1. This may reflect an intrinsically slower COX catalysis in these variants. However, there may be other contributions to the lower COX specific activity, such as batch-to-batch differences during expression, solubilization or purification of the proteins. In this study, we evaluated the COX/POX ratio as a way to control for such expression/purification variables.
Table 3.
Comparison of COX specific activities in PGHS-1 variants.
| PGHS-1 variant | Heterozygositya | COX activity (relative to wildtype) | ||
|---|---|---|---|---|
| Present studyb | Lee et al. (2007)c | Halushka et al. (2003)d | ||
| Wildtype | 1.00 | 1.00 ± 0.07 | 1.00 ± 0.07 | |
| W8Re | 0.091 | ND | 1.04 ± 0.10 | 0.84 ± 0.18 |
| P17L | 0.180 | ND | 1.13 ± 0.07 | 0.78 ± 0.10 |
| R53H | 0.004 | 0.46 | 0.35 ± 0.05h | ND |
| R78W | NDf | ND | 0.36 ± 0.04h | ND |
| R108Q | (0.04)g | 0.71 | ND | ND |
| K185T | 0.018 | ND | 0.59 ± 0.06h | ND |
| G230S | 0.005 | ND | 0.57 ± 0.04 h | ND |
| L237M | 0.043 | 0.78 | 0.51 ± 0.03h | ND |
| V481I | 0.015 | 0.53 | 1.21 ± 0.10 | ND |
Current dbSNP value [13].
Normalized by Coomassie-stained protein density. Wildtype: 34 nmol O2/min/μg PGHS-1 protein.
Normalized by immunoreactive protein density [15]. Wildtype: 20 μM O2/min/mg total protein
Normalized to 5 × 105 platelets [38]. Wildtype: 970 ng PGF2α/ml/30 s.
Sometimes listed as R8W.
Not determined.
Minor allele frequency [57].
P-value < 0.05 compared to wildtype.
An altered COX/POX ratio was seen in R108Q, L237M and perhaps R53H (Table 1). Alterations in relative velocity of the two PGHS-1 catalytic reactions are of particular interest because of the mechanistic linkage between the POX and COX catalytic cycles [39]. Thuresson et al. [40] conducted a systematic mutagenic analysis of ~20 residues in the PGHS-1 COX site and found that many substitutions in these residues decreased the COX/POX ratio. There were only two active site residues where substitutions consistently increased the COX/POX ratio by more than 10% above the wildtype value: Phe517 and Met521 (human PGHS-1 numbering). Interestingly, the F517A, F517M, F517Y, M521A and M521L mutants had relative COX/POX ratios of 1.3-1.6 [40], in the range we found for the L237M and R108Q variants (Table 1). Both Phe517 and Met521 are some distance from the residues studied here [10], so the significance of this overlap in COX/POX ratios remains to be seen. COX and POX catalysis each involve multiple reaction steps [39], and further studies will be needed to identify the particular step(s) affected in the PGHS-1 variants that have an elevated COX/POX ratio.
Variants and COX Km value
Thuresson et al. [40] used arachidonate Km values to probe the effects of COX active site mutations in PGHS-1. For approximately a third of the ~20 residues targeted in that study, mutations that retained > 10% of wildtype COX activity exhibited increases of over 50% in the arachidonate Km value. Some Km measurements have been made for PGHS-1 mutations outside the COX site, including histidine-tagged PGHS-1 [41], serine substitutions at Cys313, Cys512, and Cys540 [42], and Y504F [20]. In each of these mutants, the Km for arachidonate was found to be the same or less than the wildtype value. Thus, the increased Km observed with R108Q at high and low detergent levels (Table 1) groups this variant among PGHS-1 mutants with specific structural modifications in the COX active site. The 86% increase in Km value in R108Q (under standard conditions) is comparable to the 70-100% increases seen in the Y355L, F518Y, and M522L mutants, but much smaller than the many-fold increases seen in other COX site mutants [40]. It seems reasonable to conclude that the R108Q substitution leads to an altered active site interaction with arachidonate, though not sufficient to cause a major change in product profile, such as observed with site-directed mutations at Val349, Trp387, or Leu534 [40].
Variants and inhibition by aspirin
Some of the PGHS-1 variants tested are clearly less reactive toward aspirin than the wildtype protein (Table 2). A key question is how much such lower aspirin reactivity would affect COX inhibition in platelets exposed to the drug. Aspirin is rather stable in the buffer used in our COX assays, with a t1/2 of ~18 hours (Methods). In blood, however, the drug is rapidly hydrolyzed by the platelet-activating factor acetylhydrolase in red cells [43], giving a t1/2 of only ~15 min [44]. Aspirin’s hydrolysis product is salicylic acid, which is a negligible COX-1 inhibitor [6]. Ingestion of 500-650 mg aspirin results in a brief drug pulse in the plasma, peaking at 30-80 μM after ~30 min [45, 46]. Conceptually, a slower rate of PGHS-1 reaction with aspirin (as in the R108Q variant) should increase the fraction of platelet COX activity surviving such an aspirin pulse. However, the quantitative relationship between differences in aspirin reactivity and in surviving COX is not obvious, particularly if one considers the cumulative effect of repeated aspirin pulses, as would be encountered with a daily aspirin regimen.
To examine this in more detail, we developed a computational model (see Methods) to predict how much of an increase in surviving platelet COX might be expected from the 60% lower aspirin reactivity seen in the R108Q variant. Homodimeric structure was assumed for wildtype and variant PGHS-1. We modeled the lower aspirin reactivity in R108Q by a proportionately smaller rate constant for the acetyl transfer step (k2 in Reaction 2): 0.33 s−1 (20 min−1) for wildtype PGHS-1 and 0.13 s−1 (8 min−1) for R108Q. The model produces a peak blood aspirin level of ~40 μM at 25 min (Fig. 7A), a reasonable simulation of circulating aspirin levels after one or two standard (325 mg) aspirin tablets [45, 46]. Fig. 7A also shows the predicted declines in COX activity for PGHS-1 wildtype and R108Q variant during an aspirin pulse. In each case, surviving COX activity declines quickly during the aspirin pulse, but levels off after 120 min, when most aspirin has been hydrolyzed. A key prediction is that R108Q retains over 18% of its COX activity after the first aspirin pulse, vs. only 1.4% for wildtype PGHS-1. Simulations in which the lower aspirin reactivity of R108Q was modeled by decreasing the value of k1 rather than that of k2 predicted almost exactly the same outcome (data not shown). Thus, a 60% decrease in aspirin reactivity predicts a 12-fold increase in the fraction of COX-1 activity surviving one cycle of aspirin exposure.
Fig. 7.
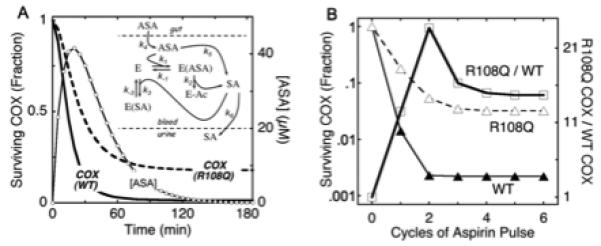
Predicted effects of aspirin reactivity on COX inhibition by pulsatile aspirin exposure. A) Simulation of blood aspirin pulse after single oral dosing and prediction of consequent COX activity loss in wildtype PGHS-1 (WT) and R108Q variant. Inset: mechanistic scheme used for model. B) Prediction of surviving COX activity after repeated cycles of aspirin exposure. Details are described in Methods.
Additional aspirin pulses were simulated in the model to predict the cumulative effects of repeated aspirin exposure, as experienced by circulating platelets in an individual taking aspirin daily. Platelets have an average lifespan of ~7 days, and their replacement by new platelets with new PGHS-1 was mimicked in the model by replacing 15% of all enzyme intermediates between simulated aspirin cycles. The prediction, shown in Fig. 7B, is that the surviving COX activities decline to a “steady-state” level after 3-5 aspirin cycles, with 3.2% of the original COX activity surviving in R108Q and 0.22% in wildtype PGHS-1. The predicted ratio of R108Q to wildtype COX activities transiently peaks after the second cycle and then levels off at ~15 (Fig. 7B). Thus, the 60% decrease in aspirin reactivity in the R108Q variant is predicted to retain some 15-fold more COX activity after repeated aspirin exposure, compared to the PGHS-1 wildtype.
The other PGHS-1 variants showed smaller decreases in aspirin reactivity (Table 2). To predict the response to repeated aspirin dosing in these variants (and in hypothetical variants over a wider range of aspirin reactivity), further computation simulation of COX surviving repeated aspirin was performed, with k2 values set at 0.2-1.25 times the wildtype value. The resulting predictions (Fig. 8) show the expected inverse relationship between the relative k2 value and the relative surviving COX activity. Using the data in Fig. 8 as a standard curve, the aspirin reactivities in Table 2 predict that compared to wildtype, R53H, L237M and V481I would have 1.5-, 1.8-, and 2.1-fold more COX activity surviving repeated aspirin treatment. These predicted increases in surviving COX activity are much smaller than those for the R108Q variant. However, even modest levels of residual thromboxane production in patients taking aspirin can be associated with increased risk of thrombotic events [47]. It will be of interest to test the effects of the PGHS-1 variants, especially R108Q, on aspirin effectiveness against COX-1 in human platelets.
Fig. 8.
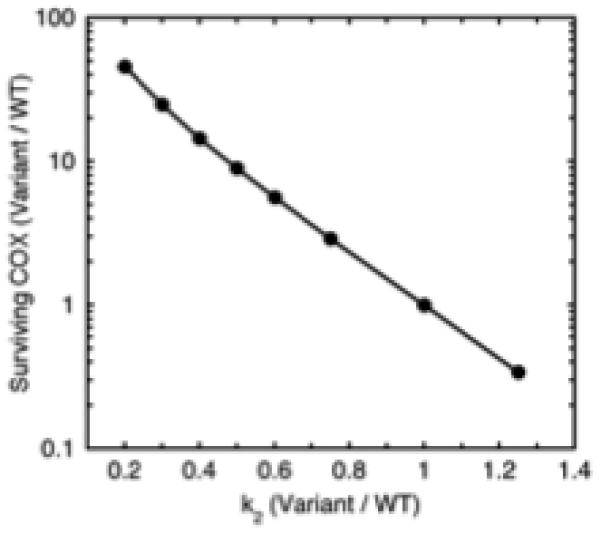
Predicted effect of relative aspirin reactivity of PGHS-1 variant on relative level of COX activity surviving repeated aspirin treatment cycles. Details are described in Methods.
Although the best-studied pathological roles of COX-1 involve the cardiovascular system, there is considerable evidence for participation of COX-1 in other pathologies, such as pain and cancer [48]. Chemopreventive actions are well established for aspirin in the case of colorectal cancer, and possible mechanisms for this beneficial effect of aspirin include inhibition of COX-1 in colonic mucosa and/or platelets [49]. The decreased aspirin reactivity observed here for the R108Q variant of COX-1 suggests that individuals with this variant may have less chemopreventive benefit from aspirin, at least at the lower doses used for cardioprotection.
Other effects of non-synonymous PTGS1 polymorphisms
Halushka et al. [38] reported that COX activity in platelets from individuals heterozygous for P17L (along with a completely linked polymorphism in the promoter) was more fully inhibited by aspirin than COX in platelets from individuals homozygous for the common haplotype. The P17L variant is in the cleaved N-terminal signal peptide, not in the mature PGHS-1 protein, so the increased aspirin effectiveness does not reflect a direct structural effect. Significant gene-NSAID interactions were reported for the P17L polymorphism with respect to risk of colorectal polyps [12]. Whereas there was no appreciable difference in adenoma or hyperplastic polyp risk associated with R8W, P17L, or L237M, a >3-fold increased risk was observed for individuals heterozygous for the rare L15-L16del polymorphism. In 2009 Agundez et al. [50] reviewed allele frequencies and published functional effects data and identified the likely candidates for variable platelet response to aspirin as P17L and L237M among Europeans and R53H and K185T in African populations. A later case-cohort study found no indication of association between P17L, R53H or L237M polymorphisms and risk of coronary heart disease or stroke [51]. However, individuals heterozygous for the L237M PGHS-1 variant recently were found to have a 42% reduction in hemodynamic response to visual stimulation [52]; this effect was interpreted in terms of the decreased COX activity reported earlier for this variant by Lee et al. (2007). We also found a decreased COX specific activity in the L237M variant, though smaller than that reported by Lee et al. (Table 3).
Variants and coxib actions
Several reports have noted that PGHS-1 exhibits lower IC50 values for coxibs as antagonists of time-dependent inactivation by aspirin and other agents than for the same coxibs as inhibitors of COX-1 activity [53-55]. An extreme example is NS-398, which was reported to be a potent antagonist of PGHS-1 inactivation by aspirin but to lack effect on COX catalysis even at low arachidonate levels [56]. This apparent ability of coxibs to interfere with time-dependent COX inhibitors without direct COX inhibition led to the recent proposal of an allosteric mechanism [56]. In this mechanism, coxib binding to the COX site in one monomer of the PGHS-1 dimer alters the COX site conformation in the other monomer to prevent the action of aspirin and similar agents but still allows normal COX catalysis. However, our analysis of NS-398 effects over a wide range of arachidonate levels showed that this coxib is actually a reversible, competitive inhibitor of COX-1 catalysis (Fig. 6). This behavior supports an earlier proposal that coxibs have a competitive relationship with both arachidonate and aspirin [55]. Nevertheless, we did find that the Ki for NS-398 as an aspirin competitor was some seven-fold lower than its Ki as an arachidonate competitor. This difference suggests that NS-398 has two modes for PGHS-1 binding as a COX site competitor, but it remains to be seen whether the competition involves one or both monomers in the PGHS-1 dimer.
Arg108 as a novel structural determinant of COX function
Mutagenic studies have identified several COX active site residues that influence the interactions of PGHS-1 with substrates or inhibitors [39]. The changes in arachidonate Km, aspirin Ki and k2, and NS-398 antagonism vs. aspirin observed with the R108Q variant indicate that the sidechain structure at position 108 affects binding interactions within the COX site. However, the Arg108 sidechain is not in the COX active site, but rather on the surface of the protein (Fig. 9). Our results with the R108Q variant thus identify Arg108 as a novel, indirect determinant of COX site function. The crystallographic data show an interaction between Arg108 and Gln357, which is in a loop comprising residues 353-361 (Fig. 9). Another residue in this loop, Tyr354, is a prominent feature of the COX active site, and its sidechain makes contacts with substrates and inhibitors. A shift in the positions of Gln357 and Tyr354 in R108Q might explain COX site effects from a structural change on the protein surface.
Fig. 9.
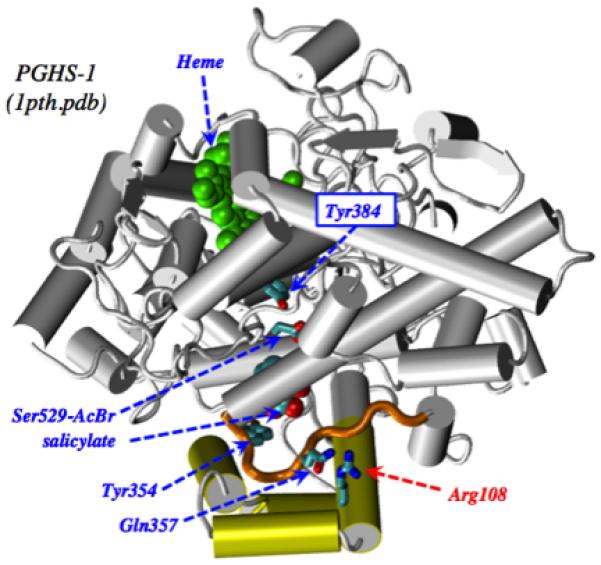
Crystallographic structure for PGHS-1 acylated by treatment with bromo-aspirin [10]. The COX active site contains Tyr384, acylated Ser529, salicylate, and Tyr354. Heme is in the POX site. Arg108 is shown interacting with Gln357 on the 353-361 loop (gold color). The membrane anchor helices are shown in yellow.
Limitations
We have characterized the purified recombinant PGHS-1 wildtype and variants as homodimers, and the computational model is set up for PGHS-1 homodimers. However, the low prevalence of the variants means that almost all individuals carrying a variant allele are heterozygotes, so their PGHS-1 protein should be a 1:2:1 mixture of wildtype homodimers, wildtype/variant heterodimers, and variant homodimers. The properties of both types of homodimers were determined in the present studies. PGHS-1 and -2 dimers show half-of-sites behavior, raising the possibility of dominant negative or dominant positive effects in heterodimers. Thus, further studies will be needed to characterize COX catalysis and inhibition in heterodimers to predict the in vivo functional impact for individual with PGHS-1 variants such as R108Q.
Concluding summary
The present results have shown that the R108Q variant of human PGHS-1 has altered COX kinetic properties and decreased reactivity with aspirin, identifying Arg108 as a novel, external structural determinant of COX site interactions. The slower action of aspirin in R108Q predicts markedly less effective COX inhibition of this variant in platelets or other tissues exposed to the transient pulses of circulating aspirin produced by daily dosing with the drug. Although the prevalence of R108Q is low, aspirin is taken by much of the population in many countries, so that a large number of individuals are likely to carry this PGHS-1 variant. Further studies will be needed to determine if these individuals derive less cardioprotective and chemopreventative benefit from current aspirin dosing regimens, and whether these individuals would benefit from higher aspirin doses. We found at least mildly decreased aspirin reactivity in each of the four PGHS-1 variants studied. However, even small decreases in aspirin reactivity are predicted to substantially increase COX activity surviving exposure to repeated pulses of aspirin and it will be of interest to examine aspirin inhibition kinetics in the other human PGHS-1 variants.
Acknowledgments
FUNDING: Grant (NIH CA 114467) from the US Public Health Service.
ABBREVIATIONS
- PGHS-1
prostaglandin H synthase-1
- COX
cyclooxygenase
- POX
peroxidase
- ASA
acetylsalicylic acid (aspirin)
- SA
salicylic acid
- SNP
single nucleotide polymorphism.
Footnotes
CONFLICTS OF INTEREST: None declared.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 2.Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA. 2007;297:2018–2024. doi: 10.1001/jama.297.18.2018. [DOI] [PubMed] [Google Scholar]
- 3.Patrono C, Rocca B. Drug insight: aspirin resistance--fact or fashion? Nat Clin Pract Cardiovasc Med. 2007;4:42–50. doi: 10.1038/ncpcardio0728. [DOI] [PubMed] [Google Scholar]
- 4.Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–140. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 5.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 6.Rome LH, Lands WE. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci U S A. 1975;72:4863–4865. doi: 10.1073/pnas.72.12.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeWitt DL, el-Harith EA, Kraemer SA, Andrews MJ, Yao EF, Armstrong RL, et al. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J Biol Chem. 1990;265:5192–5198. [PubMed] [Google Scholar]
- 8.Roth GJ, Machuga ET, Ozols J. Isolation and covalent structure of the aspirin-modified, active-site region of prostaglandin synthetase. Biochemistry. 1983;22:4672–4675. doi: 10.1021/bi00289a010. [DOI] [PubMed] [Google Scholar]
- 9.Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest. 1975;56:624–632. doi: 10.1172/JCI108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol. 1995;2:637–643. doi: 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]
- 11.Maree AO, Curtin RJ, Chubb A, Dolan C, Cox D, O’Brien J, et al. Cyclooxygenase-1 haplotype modulates platelet response to aspirin. J Thromb Haemost. 2005;3:2340–2345. doi: 10.1111/j.1538-7836.2005.01555.x. [DOI] [PubMed] [Google Scholar]
- 12.Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, et al. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004;13:889–893. [PubMed] [Google Scholar]
- 13.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, et al. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 15.Lee CR, Bottone FG, Jr., Krahn JM, Li L, Mohrenweiser HW, Cook ME, et al. Identification and functional characterization of polymorphisms in human cyclooxygenase-1 (PTGS1) Pharmacogenet Genomics. 2007;17:145–160. doi: 10.1097/01.fpc.0000236340.87540.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 17.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren Y, Loose-Mitchell DS, Kulmacz RJ. Prostaglandin H synthase-1: evaluation of C-terminus function. Arch Biochem Biophys. 1995;316:751–757. doi: 10.1006/abbi.1995.1100. [DOI] [PubMed] [Google Scholar]
- 19.Ulrich CM, Bigler J, Sibert J, Greene EA, Sparks R, Carlson CS, et al. Cyclooxygenase 1 (COX1) polymorphisms in African-American and Caucasian populations. Hum Mutat. 2002;20:409–410. doi: 10.1002/humu.9080. [DOI] [PubMed] [Google Scholar]
- 20.Rogge CE, Liu W, Kulmacz RJ, Tsai AL. Peroxide-induced radical formation at TYR385 and TYR504 in human PGHS-1. J Inorg Biochem. 2009;103:912–922. doi: 10.1016/j.jinorgbio.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Poole EM, Ulrich CM, Kulmacz RJ. Polymorphic human prostaglandin H synthase-2 proteins and their interactions with cyclooxygenase substrates and inhibitors. Pharmacogenomics J. 2011;11:337–347. doi: 10.1038/tpj.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peterson GL. Determination of total protein. Methods Enzymol. 1983;91:95–119. doi: 10.1016/s0076-6879(83)91014-5. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 25.Kulmacz R, Lands W. Cyclo-oxygenase: measurement, purification and properties. In: Benedetto C, McDonald-Gibson R, Nigam S, Slater T, editors. Prostaglandins and related substances: a practical approach. IRL Press; Washington DC: 1987. pp. 209–227. [Google Scholar]
- 26.Liu W, Cao D, Oh SF, Serhan CN, Kulmacz RJ. Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. Faseb J. 2006;20:1097–1108. doi: 10.1096/fj.05-5273com. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Poole EM, Ulrich CM, Kulmacz RJ. Polymorphic human prostaglandin H synthase-2 proteins and their interactions with cyclooxygenase substrates and inhibitors. Pharmacogenomics J. 2010 doi: 10.1038/tpj.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulmacz RJ, Lands WE. Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti-inflammatory agents. J Biol Chem. 1985;260:12572–12578. [PubMed] [Google Scholar]
- 29.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Gerngross O, Kersasp H. Uber salze der acetylsalicylsaure. Eur J Org Chem. 1914;406:240–260. [Google Scholar]
- 31.Choudhury S, Mitra AK. Kinetics of aspirin hydrolysis and stabilization in the presence of 2-hydroxypropyl-beta-cyclodextrin. Pharm Res. 1993;10:156–159. doi: 10.1023/a:1018953920081. [DOI] [PubMed] [Google Scholar]
- 32.Kulmacz RJ. Concerted loss of cyclooxygenase and peroxidase activities from prostaglandin H synthase upon proteolytic attack. Prostaglandins. 1989;38:277–288. doi: 10.1016/0090-6980(89)90133-0. [DOI] [PubMed] [Google Scholar]
- 33.Salmon JA, Flower RJ. Extraction and thin-layer chromatography of arachidonic acid metabolites. Methods Enzymol. 1982;86:477–493. doi: 10.1016/0076-6879(82)86219-8. [DOI] [PubMed] [Google Scholar]
- 34.Bakar SK, Niazi S. Stability of aspirin in different media. J Pharm Sci. 1983;72:1024–1026. doi: 10.1002/jps.2600720914. [DOI] [PubMed] [Google Scholar]
- 35.Kulmacz RJ, van der Donk WA, Tsai AL. Comparison of the properties of prostaglandin H synthase-1 and -2. Prog Lipid Res. 2003;42:377–404. doi: 10.1016/s0163-7827(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 36.Doyen JR, Yucer N, Lichtenberger LM, Kulmacz RJ. Phospholipid actions on PGHS-1 and -2 cyclooxygenase kinetics. Prostaglandins Other Lipid Mediat. 2008;85:134–143. doi: 10.1016/j.prostaglandins.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capdevila JH, Morrow JD, Belosludtsev YY, Beauchamp DR, DuBois RN, Falck JR. The catalytic outcomes of the constitutive and the mitogen inducible isoforms of prostaglandin H2 synthase are markedly affected by glutathione and glutathione peroxidase(s) Biochemistry. 1995;34:3325–3337. doi: 10.1021/bi00010a023. [DOI] [PubMed] [Google Scholar]
- 38.Halushka MK, Walker LP, Halushka PV. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther. 2003;73:122–130. doi: 10.1067/mcp.2003.1. [DOI] [PubMed] [Google Scholar]
- 39.Tsai AL, Kulmacz RJ. Prostaglandin H synthase: Resolved and unresolved mechanistic issues. Arch Biochem Biophys. 2010;493:103–124. doi: 10.1016/j.abb.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thuresson ED, Lakkides KM, Rieke CJ, Sun Y, Wingerd BA, Micielli R, et al. Prostaglandin endoperoxide H synthase-1: the functions of cyclooxygenase active site residues in the binding, positioning, and oxygenation of arachidonic acid. J Biol Chem. 2001;276:10347–10357. doi: 10.1074/jbc.M009377200. [DOI] [PubMed] [Google Scholar]
- 41.Smith T, Leipprandt J, DeWitt D. Purification and characterization of the human recombinant histidine-tagged prostaglandin endoperoxide H synthases-1 and -2. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy TA, Smith CJ, Marnett LJ. Investigation of the role of cysteines in catalysis by prostaglandin endoperoxide synthase. J Biol Chem. 1994;269:27357–27364. [PubMed] [Google Scholar]
- 43.Zhou G, Marathe GK, Willard B, McIntyre TM. Intracellular Erythrocyte Platelet-activating Factor Acetylhydrolase I Inactivates Aspirin in Blood. J Biol Chem. 2011;286:34820–34829. doi: 10.1074/jbc.M111.267161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rance MJ, Jordan BJ, Nichols JD. A simultaneous determination of acetylsalicylic acid, salicylic acid and salicylamide in plasma by gas liquid chromatography. J Pharm Pharmacol. 1975;27:425–429. doi: 10.1111/j.2042-7158.1975.tb09472.x. [DOI] [PubMed] [Google Scholar]
- 45.Rowland M, Riegelman S, Harris PA, Sholkoff SD, Eyring EJ. Kinetics of acetylsalicylic acid disposition in man. Nature. 1967;215:413–414. doi: 10.1038/215413a0. [DOI] [PubMed] [Google Scholar]
- 46.Davison C. Salicylate metabolism in man. Ann N Y Acad Sci. 1971;179:249–268. doi: 10.1111/j.1749-6632.1971.tb46905.x. [DOI] [PubMed] [Google Scholar]
- 47.Frelinger AL, 3rd, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, et al. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–2596. doi: 10.1161/CIRCULATIONAHA.109.900589. [DOI] [PubMed] [Google Scholar]
- 48.Perrone MG, Scilimati A, Simone L, Vitale P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr Med Chem. 2010;17:3769–3805. doi: 10.2174/092986710793205408. [DOI] [PubMed] [Google Scholar]
- 49.Chan AT, Arber N, Burn J, Kuang Chia J, Elwood P, Hull MA, et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res (Phila) 2011 doi: 10.1158/1940-6207.CAPR-11-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agundez JA, Martinez C, Perez-Sala D, Carballo M, Torres MJ, Garcia-Martin E. Pharmacogenomics in aspirin intolerance. Curr Drug Metab. 2009;10:998–1008. doi: 10.2174/138920009790711814. [DOI] [PubMed] [Google Scholar]
- 51.Lee CR, North KE, Bray MS, Couper DJ, Heiss G, Zeldin DC. Cyclooxygenase polymorphisms and risk of cardiovascular events: the Atherosclerosis Risk in Communities (ARIC) study. Clin Pharmacol Ther. 2008;83:52–60. doi: 10.1038/sj.clpt.6100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahn T, Heinzel S, Plichta MM, Reif A, Lesch KP, Fallgatter AJ. Neurovascular coupling in the human visual cortex is modulated by cyclooxygenase-1 (COX-1) gene variant. Cereb Cortex. 2011;21:1659–1666. doi: 10.1093/cercor/bhq236. [DOI] [PubMed] [Google Scholar]
- 53.Rosenstock M, Danon A, Rimon G. PGHS-2 inhibitors, NS-398 and DuP-697, attenuate the inhibition of PGHS-1 by aspirin and indomethacin without altering its activity. Biochim Biophys Acta. 1999;1440:127–137. doi: 10.1016/s1388-1981(99)00105-5. [DOI] [PubMed] [Google Scholar]
- 54.Rosenstock M, Danon A, Rubin M, Rimon G. Prostaglandin H synthase-2 inhibitors interfere with prostaglandin H synthase-1 inhibition by nonsteroidal anti-inflammatory drugs. Eur J Pharmacol. 2001;412:101–108. doi: 10.1016/s0014-2999(00)00931-6. [DOI] [PubMed] [Google Scholar]
- 55.Ouellet M, Riendeau D, Percival MD. A high level of cyclooxygenase-2 inhibitor selectivity is associated with a reduced interference of platelet cyclooxygenase-1 inactivation by aspirin. Proc Natl Acad Sci U S A. 2001;98:14583–14588. doi: 10.1073/pnas.251543298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rimon G, Sidhu RS, Lauver DA, Lee JY, Sharma NP, Yuan C, et al. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc Natl Acad Sci U S A. 2010;107:28–33. doi: 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi J, Misso NL, Duffy DL, Bradley B, Beard R, Thompson PJ, et al. Cyclooxygenase-1 gene polymorphisms in patients with different asthma phenotypes and atopy. Eur Respir J. 2005;26:249–256. doi: 10.1183/09031936.05.00140104. [DOI] [PubMed] [Google Scholar]