

Abstract
GABAergic neurons in the thalamic reticular nucleus (TRN) form powerful inhibitory connections with several dorsal thalamic nuclei, thereby controlling attention, sensory processing, and synchronous oscillations in the thalamocortical system. TRN neurons are interconnected by a network of GABAergic synapses, but their properties and their role in shaping TRN neuronal activity are not well understood. Using recording techniques aimed to minimize changes in the intracellular milieu, we show that synaptic GABAA receptor activation triggers postsynaptic depolarizations in mouse TRN neurons. Immunohistochemical data indicate that TRN neurons express very low levels of the Cl− transporter KCC2. In agreement, perforated-patch recordings show that intracellular Cl− levels are high in TRN neurons, resulting in a Cl− reversal potential (ECl) significantly depolarized from rest. Additionally, we find that GABAA receptor-evoked depolarizations are amplified by the activation of postsynaptic T-type Ca2+ channels, leading to dendritic Ca2+ increases and the generation of burst firing in TRN neurons. In turn, GABA-evoked burst firing results in delayed and long-lasting feedforward inhibition in thalamic relay cells. Our results show that GABA-evoked depolarizations can interact with T-type Ca2+ channels to powerfully control spike generation in TRN neurons.
Introduction
Located strategically between neocortex and thalamus, the thalamic reticular nucleus (TRN) is critically involved in a number of diverse computational tasks (Jones, 2007). It controls distinct aspects of sensory processing such as receptive field properties (Hartings et al., 2003) and relay cell excitability (Kim et al., 1997), mediates selective attention (Crick, 1984; McAlonan et al., 2006), and, in addition, generates rhythmic synchronous activity in the thalamocortical system (McCormick and Bal, 1997). Therefore, understanding how intrinsic and afferent synaptic connections control TRN neuronal activity is essential to reveal the various roles of TRN under both physiological and pathological conditions (Beenhakker and Huguenard, 2009).
Neurons in the TRN are exclusively GABAergic and form local networks of electrical (Landisman et al., 2002; Deleuze and Huguenard, 2006) and chemical synapses (Sanchez-Vives et al., 1997; Deleuze and Huguenard, 2006). While electrical synapses are thought to mediate synchronous firing among small clusters of TRN neurons (Long et al., 2004), local GABAergic connections are assumed to have an opposing role. TRN plays a key role in generating certain types of synchronous oscillations in the thalamocortical system, most notably sleep spindles (Beenhakker and Huguenard, 2009). Intra-TRN chemical synapses are thought to be critical for limiting the number of TRN neurons that participate in rhythmic oscillatory activity (Sohal and Huguenard, 2003). Accordingly, when intra-TRN synaptic activity is reduced, thalamic circuits that normally mediate sparse synchronous activity can switch into a hypersynchronous state, leading to the generation of seizure-like activity (Huntsman et al., 1999). A role of intra-TRN GABAergic synapses in desynchronizing activity in the thalamocortical system appears to be consistent with studies showing that activation of GABAergic inputs leads to hyperpolarizing or shunting inhibition in TRN neurons (Ahlsén and Lindström, 1982; Sanchez-Vives et al., 1997; Ulrich and Huguenard, 1997; Shu and McCormick, 2002). However, experimental support has mostly relied on recording techniques that perturb [Cl−]i. Thus, our knowledge of GABAergic signaling in the TRN remains incomplete.
By using noninvasive recording techniques and immunohistochemistry, we find that GABAergic synapses in the TRN are depolarizing, in part mediated by an extremely low abundance of the Cl− transporter KCC2. Furthermore, we show that GABAergic synaptic inputs can activate postsynaptic T-type Ca2+ channels, leading to the generation of bursts of action potentials in TRN neurons and the generation of powerful inhibitory inputs in thalamic relay neurons. Thus, rather than acting solely as a shunt, local GABAergic synaptic connections could form an important substrate for the generation of network activity in the TRN.
Materials and Methods
Slice preparation.
Thalamocortical slices (400 μm) were prepared from both male and female C57BL/6J mice, P13–P35, as described previously (Agmon and Connors, 1991). Animals were anesthetized with isoflurane and decapitated, following procedures in accordance with NIH guidelines and approved by the University of Texas Health Science Center at Houston animal welfare committee. Slices were cut in ice-cold solution containing the following (in mm): 111 choline Cl, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 26 NaHCO3, 10 glucose, and 0.5 CaCl2, saturated with 95% O2–5% CO2, using a vibratome (Leica VT1200S) at slicing speeds of 0.2 mm/s and a blade vibration amplitude of 0.8 mm. Slices were transferred to a holding chamber and incubated at 34°C for 40 min, in saline solution containing the following (in mm): 126 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 10 glucose, 2 CaCl2, and 2 MgCl2, and then kept at room temperature before recordings.
Electrophysiology.
Slices were placed on glass coverslips coated with poly-l-lysine (Sigma-Aldrich) and submerged in a recording chamber (Warner Instruments). All experiments were performed at near-physiological temperatures (32–34°C) using an in-line heater (Warner Instruments) while perfusing the recording chamber with solution at 3–4 ml/min using a Minipulse 3 pump (Gilson). Unless noted, experiments were performed in the presence of NBQX (10 μm), 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (R-CPP) (5 μm), (2S)-3-([(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid (CGP 55845) (5 μm), and hexamethonium (100 μm) to block AMPA, NMDA, GABAB, and nicotinic acetylcholine receptors, respectively. Recordings were obtained under infrared–differential interference contrast visualization using an Olympus BX51WI microscope (Olympus Optical) and a CCD camera (Hamamatsu).
Cell-attached recordings were obtained with pipettes (2–3 MΩ) filled with ACSF, following established procedures (Perkins, 2006). For loose-patch recordings in voltage clamp, the seal resistance was 20–100 MΩ. For tight-seal recordings, the seal resistance was 1–4 GΩ. In voltage clamp, holding potential was continually monitored and adjusted to keep the holding current near 0 pA, to minimize any influence on the membrane potential of the recorded cell (Perkins, 2006). Cell-attached current-clamp recordings were performed in tight-seal configuration after the measured resting membrane potential reached a steady state, which typically occurred 5–10 min following initial seal formation.
Perforated-patch recordings were performed as described previously (Ulrich and Huguenard, 1997), using a voltage-clamp internal solution containing the following (in mm): 132 CsCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, 2 Mg-ATP, 0.4 Na-GTP, and 5 QX-314, adjusted to 295 mOsm and pH 7.3. Gramicidin (dissolved in DMSO) was added to the patch-clamp internal solution to obtain a final concentration of 60 μg/ml. Perforated-patch access resistance was monitored by applying 20 mV hyperpolarizing voltage steps following seal formation. Recordings with final access resistances ∼50–100 MΩ were obtained after 15–30 min. The access resistance was calculated by analyzing the transient responses to voltage-clamp steps, following previously established procedures (Ulrich and Huguenard, 1997). Membrane potentials shown are corrected for the voltage drop across the series resistance.
Whole-cell current-clamp recordings from TRN neurons (see Fig. 5) were obtained with an internal solution containing the following (in mm): 108 K-gluconate, 26 KCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, 2 Mg-ATP, and 0.4 Na-GTP, adjusted to 295 mOsm and pH 7.3. For whole-cell voltage-clamp recordings of IPSCs in neurons of the ventrobasal (VB) thalamus (see Fig. 6), the internal solution contained the following (in mm): 120 CsMeSO3, 10 CsCl, 10 HEPES, 11 EGTA, 1 MgCl2, 1 CaCl2, 2 Mg-ATP, 0.3 Na-GTP, and 1 QX-314, adjusted to 295 mOsm and pH 7.3. Cells were held at 0 mV. Whole-cell voltage-clamp recordings from TRN neurons (see Fig. 4E,F) were obtained with patch pipettes (2–3 MΩ) containing the following (in mm): 93 CsMeSO3, 40 CsCl, 10 HEPES, 0.5 EGTA, 2 MgCl2, 0.16 CaCl2, 2 Mg-ATP, 0.4 Na-GTP, and 2 QX-314, adjusted to 295 mOsm and pH 7.3. Cells were held at −70 mV.
Figure 5.

GABA-evoked Ca2+ increases and burst firing. A, Synaptically evoked spike burst in a TRN neuron recorded in whole-cell current clamp (30 mm Cl−). Action potentials were blocked by either depolarizing the neuron to −60 mV or following application of TTA-P2 (1 μm). B, Summary data showing that synaptically evoked burst firing recorded in whole-cell current clamp is controlled by resting membrane potential and blocked by TTA-P2 (1 μm) (n = 5; p < 0.001, paired Student's t test). C, A representative experiment showing that brief application of muscimol (500 μm; 100 ms; time of application indicated by the bar) triggered Ca2+ increases in the distal dendrite of a TRN neuron recorded whole cell current clamp with a K-based internal solution (30 mm Cl−) supplemented with Fluo-4 (125 μm). Ca2+ increases were blocked by TTA-P2 (1 μm). TTA-P2 had little effect on muscimol-induced postsynaptic membrane depolarization. Recordings were performed in presence of TTX (0.5 μm). D, Summary plot showing that muscimol-induced Ca2+ increases were significantly reduced by bath application of TTA-P2 (1 μm; p < 0.05; n = 5; paired Student's t test). The muscimol-induced postsynaptic membrane depolarization is not significantly reduced by TTA-P2. Error bars indicate SEM.
Figure 6.
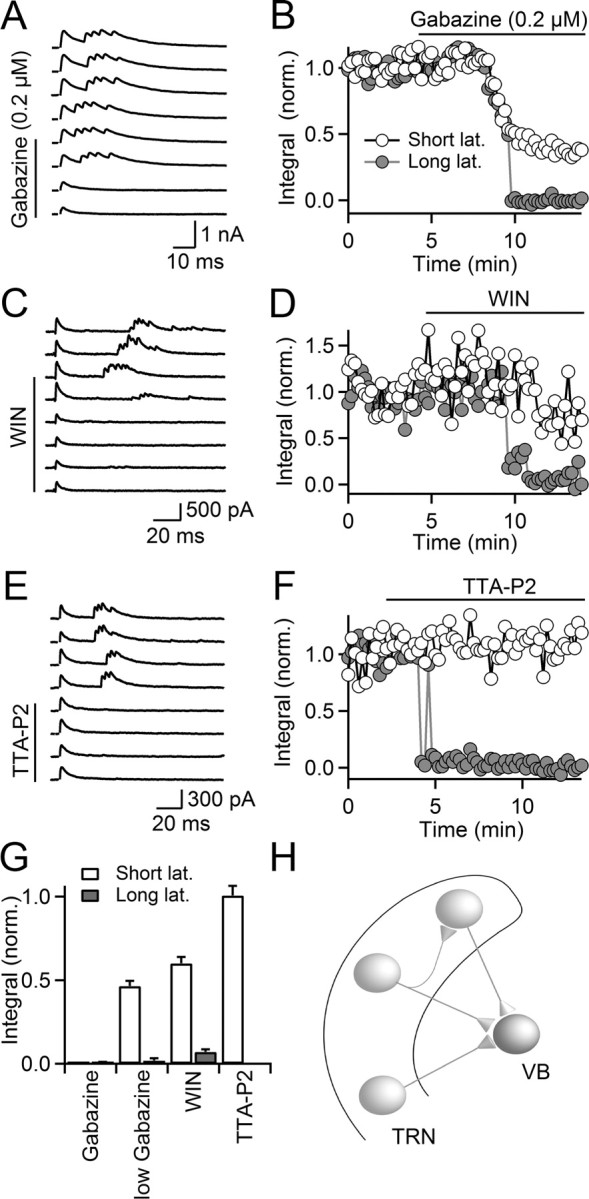
GABA-induced burst firing in TRN mediates long-latency inhibition in VB. VB thalamic neurons were recorded in the presence of blockers of excitatory neurotransmission, using a Cs-based internal solution. Cells were held at 0 mV. IPSCs were evoked with single stimuli, using stimulus electrodes placed in the TRN. A, B, Bath application of subsaturating concentrations of gabazine (0.2 μm) partially reduced short-latency IPSCs but completely eliminated long-latency IPSCs. A, Multiple trials are shown, recorded in control conditions and following wash-in of gabazine (0.2 μm). B, Time course of the IPSC integral (measured as the area underneath the current trace, normalized to baseline) for both short- (open circles) and long-latency responses (filled circles) during wash-in of gabazine (0.2 μm). The neuron is the same as in A. C, D, Bath application of WIN55,212 (WIN) (5 μm) reduced the amplitude of short-latency IPSCs but completely eliminated the long-latency IPSCs. C, Multiple trials are shown, recorded in control conditions and following bath application of WIN (5 μm). D, Time course of IPSC integral of short- (open circles) and long-latency responses (filled circles) during wash-in of WIN. The neuron is the same as in C. E, F, Pharmacological block of T-type Ca2+ channels by application of TTA-P2 (1 μm) eliminated the long-latency IPSC but did not influence short-latency IPSC. E, Multiple trials are shown, recorded in control conditions and following bath application of TTA-P2 (1 μm). F, Time course of IPSC integral for short- (open circles) and long-latency responses (filled circles). The neuron is the same as in E. G, Summary plot quantifying the effect of gabazine (10 μm; n = 5), low concentrations of gabazine (0.2 μm, n = 5), the CB1 receptor agonist WIN (n = 5), and TTA-P2 (n = 5) on short- (open bars) and long-latency IPSCs (gray bars). Error bars indicate SEM. H, Schematic illustrating TRN neuronal circuit mediating short- and long-latency IPSCs in VB neurons. Lat., Latency.
Figure 4.

Activation of postsynaptic T-type Ca2+ channels is required for GABAA-mediated burst firing. A, Top, Synaptically evoked action potentials in a TRN neuron recorded in cell-attached mode (left), blocked by application of the specific T-type Ca2+ channel blocker TTA-P2 (1 μm; right). Bottom, Raster plot showing the timing of spikes in the same neuron evoked by single stimuli applied at t = 0 ms, before and during bath application of TTA-P2 (1 μm). B, Summary data showing the time course of the TTA-P2-induced block in synaptically evoked action potential firing in TRN neurons (n = 5 neurons). C, A representative recording in cell-attached current-clamp mode showing that TTA-P2 (1 μm) blocked burst firing and reduced stimulus-induced membrane depolarization with the remaining component blocked by gabazine (10 μm). Identical results were obtained for four other neurons. D, Time course of the synaptically evoked depolarization (measured as area underneath the recording trace for the first 100 ms, normalized to control) during wash-in of TTA-P2 (1 μm) and gabazine (10 μm), for the same cell shown in C. E, A representative experiment showing that TTA-P2 (1 μm) had no effect on GABAergic PSC amplitude or paired-pulse plasticity. TRN neurons were recorded in whole-cell voltage clamp and intra-TRN GABAergic synapses were activated with paired pulses (20 Hz). Traces are averaged over 20–30 individual trials. F, Summary plot showing that bath application of TTA-P2 (1 μm) did not influence GABAergic PSC amplitude or paired-pulse ratio (PSC2/PSC1) (n = 5; p = 0.71 for amplitude; p = 0.72 for PPR; paired Student's t test). AMP, Amplitude; PPR, paired-pulse ratio.
Extracellular stimuli were evoked every 12 s with patch pipettes filled with ACSF, placed in the TRN. Stimulation intensities ranged from 10 to 30 μA (200 μs stimulus duration). In some experiments, GABAA receptor agonists were locally applied by pressure ejection (100–300 ms duration) via a puffer pipette (6–10 μm tip diameter) controlled by a Picrospritzer II (General Valve) and placed above the slice near the recorded neuron.
Fluorescent imaging of TRN neurons.
TRN neurons were recorded in current clamp with the K-based internal solution (30 mm Cl−) supplemented with Fluo-4 (125 μm) and Alexa 594 (50 μm). Fluo-4 was excited at 480 nm with a monochromator (Polychrome IV; TILL Photonics). The filter set used was 510 nm and 535 LP for emission (Omega Optical). Images were acquired with 25 ms exposures at 20 Hz using a SensiCam CCD camera (PCO Computer Optics).
Data acquisition and analysis.
Data were acquired using pClamp software (Molecular Devices). Recordings were filtered at 2–10 kHz and digitized at 20 kHz with a 16 bit analog-to digital converter (Digidata 1440A; Molecular Devices). Data analysis was performed with custom macros written in Igor Pro (Wavemetrics). Statistical test were performed with the unpaired or paired Student t test. Differences are considered to be significant at p < 0.05. Data are presented as mean ± SEM.
Immunohistochemistry.
Immunohistochemical staining was performed as described previously (Wu et al., 2010). Wild-type C57BL/6 mice at postnatal day 26 were deeply anesthetized and transcardially perfused with ice-cold PBS, pH 7.4, followed by 4% paraformaldehyde in PBS, pH 7.4. The brains were postfixed with the same fixative overnight at 4°C. Brains were sectioned into 50-μm-thick sections with a vibrating microtome (Leica VT1000S) in the coronal plane. Free-floating sections were washed with PBS and permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 min. Sections were then washed with PBS, blocked for 1 h with 3% normal goat serum in PBS at room temperature, and then incubated with a mixture of two primary antibodies: mouse anti-KCC2 (1:200; University of California at Davis/NIH Neuromab Facility) and rabbit anti-GAD67 (1:1000; Invitrogen) in PBS with 2 mg/ml BSA and 1% normal goat serum at 4°C overnight. Sections were then washed with PBS and incubated with the following fluorescent secondary antibodies: goat anti-mouse IgG-Cy3 (Jackson ImmunoResearch Laboratories; 1:500); goat anti-rabbit IgG-Alexa 488 (Invitrogen; 1:500) in PBS at room temperature for 2 h. Following this incubation, sections were counterstained with DAPI (1:10,000; Invitrogen), washed with PBS four times for 10 min each, and mounted with Vectashield (Vector Laboratories). Confocal images were obtained using a Zeiss 710 system.
Results
Activation of GABAergic synapses triggers burst firing in TRN neurons
We examined the properties of intra-TRN synapses by recording from TRN neurons in thalamocortical slices of 2- to 5-week-old mice. To isolate GABAergic synaptic inputs, we blocked excitatory synaptic transmission pharmacologically. We first recorded from TRN cells using loose-patch recordings (Perkins, 2006) to avoid perturbing [Cl−]i. Under these conditions, we found that activation of intra-TRN GABAergic synapses with single stimuli reliably triggered bursts of action potentials in TRN cells (latency, 14.1 ± 1.3 ms; duration of burst, 21.3 ± 1.6 ms; 5.7 ± 0.4 spikes per burst; n = 12). Synaptically evoked burst firing was completely blocked by bath application of the GABAA receptor antagonist gabazine (10 μm) in all cells examined (5.2 ± 0.3 spikes in control; 0 spikes in gabazine; n = 6; p < 0.001; Fig. 1A,B). We have previously shown that endocannabinoids can reduce GABA release from intra-TRN synapses, while having no effect on the postsynaptic membrane properties (Sun et al., 2011). Consistent with an essential role of GABAergic synaptic transmission in triggering burst firing in TRN neurons, synaptically evoked action potentials were eliminated by bath application of the cannabinoid receptor agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN55, 212) (Fig. 1C,D). Together, these results suggest that activation of postsynaptic GABAA receptors by release of GABA from intra-TRN synapses can reliably trigger action potentials in TRN neurons.
Figure 1.
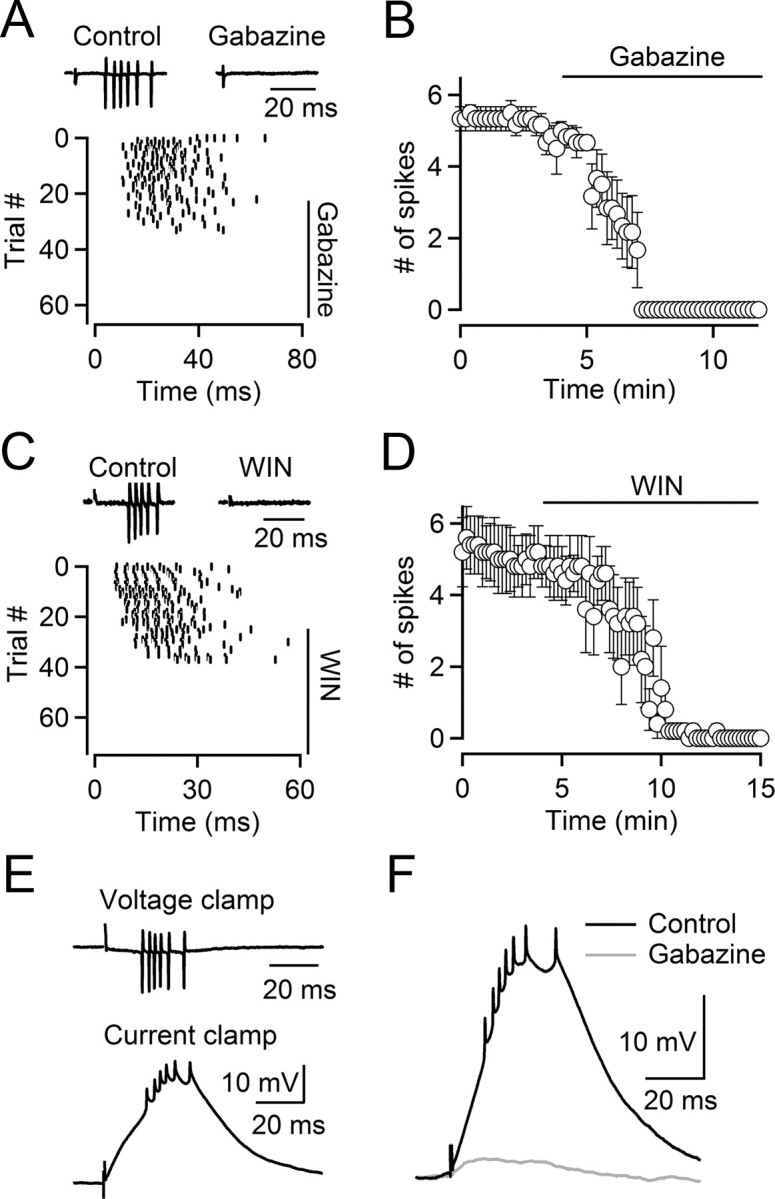
Activation of depolarizing GABAergic inputs triggers bursts in TRN neurons. A, Top, Synaptically evoked action potentials in a TRN neuron recorded in cell-attached mode (left), blocked by the GABAA receptor antagonist gabazine (10 μm; right). Bottom, Raster plot for the same neuron shows timing of action potential firing in multiple trials (collected every 12 s), triggered by single stimuli given at t = 0 ms, before and during wash-in of gabazine (10 μm). B, Summary data showing the time course of the gabazine-induced block in action potential firing (n = 6 neurons). C, D, Burst firing is blocked by bath application of the CB1 receptor agonist WIN55,212 (WIN). C, Top, Synaptically evoked action potentials in a TRN neuron (left), blocked by bath application of WIN (5 μm; right). Bottom, Raster plot indicating the time of synaptically evoked action potentials in the same neuron (stimuli applied at t = 0 ms) over multiple trials (intertrial interval, 12 s), before and during bath application of WIN (5 μm). Spikes were recorded in cell-attached configuration. D, Summary data showing the effect of WIN on stimulus-evoked burst firing in TRN neurons (n = 5 cells). E, Top, Cell-attached voltage-clamp recording of a TRN neuron showing a stimulus-evoked spike burst. Bottom, Cell-attached current-clamp recording of the same cell showing that stimulation elicits a depolarizing postsynaptic potential leading to action potentials. F, A representative experiment showing that both the depolarizing postsynaptic potential and action potentials were blocked by gabazine (10 μm). Identical results were obtained in four other neurons.
Activation of GABAergic synaptic inputs can evoke postsynaptic hyperpolarizations, which for a number of cell types is followed by a rebound depolarization and the generation of bursts of action potentials (Steriade et al., 1993; Aizenman et al., 1998), a mechanism that could in principle explain our findings. To determine the polarity of the GABA-induced postsynaptic membrane potential (PSP) changes in TRN neurons without influencing the intracellular milieu, we used tight-seal cell-attached current-clamp recordings (Perkins, 2006). While the amplitude of the GABA-evoked PSP is likely attenuated in this recording configuration, accurate estimates of both resting membrane potential and polarity of membrane potential changes can be obtained. We found that the average resting membrane potential of TRN neurons was −75.1 ± 1.3 mV (n = 12). Importantly, activation of intra-TRN synapses led to short-latency depolarizations from the resting membrane potential, followed by the generation of action potentials (Fig. 1E), independent of animal age (P13–P35). Both the depolarization and action potentials were blocked by bath application of the GABAA receptor antagonist gabazine (10 μm) (Fig. 1F). Thus, activation of GABAergic synapses evokes postsynaptic depolarizations that lead to bursts of action potentials.
The experiments outlined above were conducted in the presence of CGP 55845 to block GABAB receptors. TRN neurons express GABAB receptors in their dendrites (Ulrich and Huguenard, 1996), so it is possible that their activation attenuates GABAA-induced depolarizations under physiological conditions. However, we found that GABAergic synaptic inputs triggered spikes even in the absence of GABAB receptor blockers (data not shown), consistent with previous work demonstrating that in TRN GABAB receptors only make a minor contribution to the composite GABAergic PSP (Ulrich and Huguenard, 1996).
Activation of TRN somatodendritic GABAA receptors evokes postsynaptic depolarizations
While GABAA receptor-mediated depolarizations have been observed for a number of neuronal types throughout the mature brain, they are often confined to axonal compartments (Jang et al., 2001; Szabadics et al., 2006; Pugh and Jahr, 2011). By contrast, activation of dendritic GABAA receptors typically results in hyperpolarizing or shunting inhibition (Szabadics et al., 2006; Glickfeld et al., 2009), indicating that neurons can maintain different levels of [Cl−]i in distinct subcellular compartments. While the existence of functional intra-TRN GABAergic synaptic connections is well established (Deleuze and Huguenard, 2006; Lam et al., 2006), their ultrastructure and location on the somatodendritic membrane are not well understood (Pinault, 2004). To determine whether a widespread activation of GABAA receptors expressed in TRN neurons mirrors the effect of synaptic activation, we recorded from TRN neurons in loose-patch configuration and applied GABA via brief puffs (300 μm; 100 ms), using a glass pipette placed near the recorded cell. GABAB receptors were blocked pharmacologically. Under these conditions, activation of GABAA receptors led to the generation of action potentials, which could be reversibly blocked by bath application of gabazine (control, 5.1 ± 0.3 spikes; gabazine, 0 spikes; washout, 5.1 ± 1.2 spikes; n = 5; p < 0.001, paired t test; Fig. 2A,C,E). To confirm that action potentials were triggered by a GABAA receptor-mediated depolarization, we recorded from TRN neurons in cell-attached current-clamp recording configuration. In the presence of TTX (500 nm), brief applications of the GABAA receptor agonist muscimol (100 μm; 200 ms duration) induced depolarizing responses in all TRN neurons tested. An example is shown in Figure 2, B and D. Depolarizing responses were reversibly blocked by bath application of gabazine (control, 12.2 ± 1.8 mV; gabazine, 1.1 ± 0.3 mV; washout, 11.7 ± 1.6 mV; n = 5; p < 0.001, paired t test; Fig. 2F). Thus, TRN neurons appear to maintain uniformly high levels of [Cl−]i, suggesting that activation of GABAergic synapses located throughout the somatodendritic region will evoke GABAA receptor-mediated depolarizations. To directly determine the reversal potential of the GABAA receptor-mediated current without perturbing [Cl−]i, we used perforated-patch recordings by including gramicidin in the recording solution (Ulrich and Huguenard, 1997). These experiments were performed in the presence of TTX (500 nm) to avoid contamination of the postsynaptic current by action potential activity. We found that currents evoked by brief applications of the GABAA receptor agonist muscimol (100 μm; 200 ms duration) had a reversal potential of −45.0 mV (range, −46.1 to −39.5 mV; n = 5). An example is shown in Figure 2G. Collectively, our data suggest that TRN neurons maintain a uniformly high [Cl−]i (20–30 mm), causing ECl to be positive to resting membrane potential and GABAA receptor activation to generate depolarizing synaptic responses.
Figure 2.
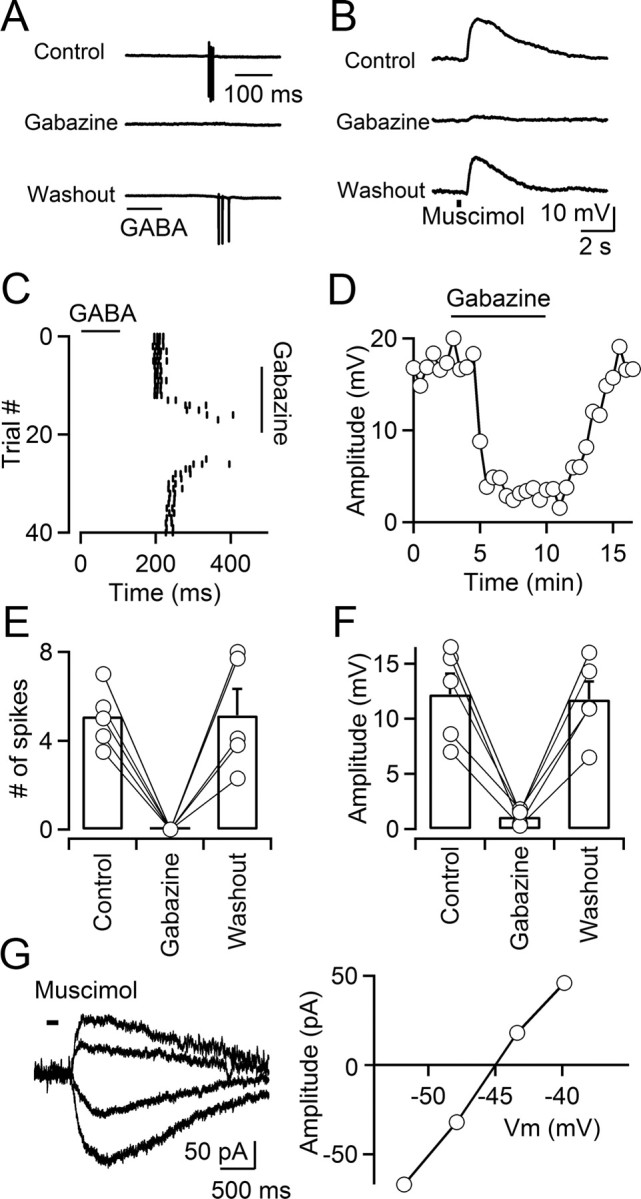
Activation of GABAA receptors depolarizes TRN neurons. A, C, E, GABA induces action potentials in TRN neurons. A, A representative experiment showing that brief puffs of GABA (300 μm; 100 ms; time of application indicated by the bar) elicits action potentials in a TRN neuron in the presence of a GABAB antagonist, recorded in cell-attached voltage clamp. Action potential firing is reversibly blocked by bath application of the GABAA receptor antagonist gabazine (10 μm). C, Raster plot shows timing of GABA-evoked action potential firing over multiple trials during wash-in and washout of gabazine, for the neuron shown in A. E, Summary data showing that GABA-induced action potentials were reversibly blocked by gabazine (n = 5; p < 0.001, paired Student's t test). Error bars indicate SEM. B, D, F, Activation of GABAA receptors elicits postsynaptic depolarizations. B, For a neuron recorded in cell-attached current-clamp mode, brief application of the GABAA receptor agonist muscimol (500 μm; 300 ms; time of application indicated by the bar) evoked postsynaptic depolarizations that were reversibly blocked by gabazine (10 μm). Recordings were performed in the presence of TTX (0.5 μm). D, Plot showing the time course of the muscimol-induced response amplitude during wash-in and washout of gabazine, for the neuron shown in B. F, Summary data showing that muscimol-induced responses were reversibly blocked by gabazine (n = 5; p < 0.001, paired Student's t test). G, Left, Muscimol-induced membrane currents evoked at different command potentials (−63 to −38 mV) measured with gramicidin D perforated-patch recordings. Application of muscimol (100 μm; 200 ms duration) is indicated by the bar. Right, The graph plots the current–voltage relationship of the muscimol-evoked response. Currents were evoked in the presence of TTX (0.5 μm).
Low expression of KCC2 in the TRN
For many types of neurons, [Cl−]i is primarily determined by the two Cl− transporters NKCC1 and KCC2 (Rivera et al., 1999; Blaesse et al., 2009). NKCC1 accumulates Cl− ions via the cotransport of Cl− into cells using the Na+ concentration gradient; KCC2, however, extrudes Cl− from neurons using the K+ concentration gradient. Thus, the high levels of [Cl−]i in TRN neurons could be determined by a low expression of KCC2. To test this hypothesis, we performed immunohistochemical analysis of KCC2 expression in thalamic nuclei of P26 mice. We found that KCC2 was virtually nonexistent in the TRN (Fig. 3A,B), while the neighboring ventrobasal complex [ventral posterolateral nucleus of thalamus (VPL) and ventral posteromedial nucleus of thalamus (VPM)] showed strong expression (Fig. 3A,C), in agreement with the KCC2 expression pattern in rat thalamus (Barthó et al., 2004). Thus, the high levels of [Cl−]i maintained in TRN neurons are at least in part due to a low expression of the chloride transporter KCC2.
Figure 3.

Thalamic nucleus-specific expression of KCC2 A, Expression of GAD67 (A1) and KCC2 (A2) from the same section of mouse thalamus detected by immunohistochemistry. Cell nuclei were labeled by DAPI (A3). An overlay of the three channels is shown in A4. B, Higher magnification view of the immunoreactive signal of GAD67 (B1) and KCC2 (B2) for the area indicated in A1, showing KCC2 is not detectable in TRN neurons (B4). C, Higher magnification view of the immunoreactive signal for GAD67 (C1) and KCC2 (C2) for the area indicated in A1, showing KCC2 is strongly expressed in VPM neurons (C4). B3 and C3 show DAPI staining for TRN and VPM, respectively.
GABA-triggered action potentials in the TRN require activation of T-type Ca2+ channels
If ECl is more positive than spike threshold, GABA-induced depolarizations should be sufficient to trigger the generation of Na+ spikes in the axon of TRN neurons. Alternatively, postsynaptic low-threshold conductances such as T-type Ca2+ channels might be critically involved in amplifying postsynaptic GABAergic responses. In TRN neurons, T-type Ca2+ channels composed of Cav3.2 and Cav3.3. subunits (Talley et al., 1999) are expressed in high densities throughout the dendritic tree (Destexhe et al., 1996; Crandall et al., 2010) and are critically involved in rhythmic burst firing typical for certain stages of sleep (Cueni et al., 2008). Studies on a number of neuronal types have suggested that T-type Ca2+ channels expressed in dendrites might serve to amplify glutamatergic synaptic inputs (Magee et al., 1995; Gillessen and Alzheimer, 1997; Urban et al., 1998; Crandall et al., 2010). However, the lack of a selective antagonist has complicated efforts to isolate the effects of T-type Ca2+ channel activation in a given cell type. Here, we tested the role of T-type Ca2+ channels in GABA-evoked spike generation, by using the selective antagonist 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide(TTA-P2) (Dreyfus et al., 2010). In all TRN cells examined, bath application of TTA-P2 (1 μm) completely eliminated synaptically evoked bursting (4.8 ± 0.4 spikes in control; 0 spikes in TTA-P2; n = 5; p < 0.001, paired t test; Fig. 4A,B), indicating that the activation of T-type Ca2+ channels is an essential requirement for GABA-evoked action potentials in the TRN. For neurons held in cell-attached current clamp, TTA-P2 eliminated fast events reflecting Na+-dependent action potentials, revealing a depolarizing postsynaptic response (Fig. 4C). This remaining component was blocked by bath application of gabazine (10 μm) (Fig. 4C,D), indicating that it likely reflected an isolated GABAA receptor-mediated depolarization. TTA-P2 had no effect on the properties of Na+-dependent action potentials or on the frequency of tonic firing evoked by current steps from a depolarized membrane potential (data not shown), at which T-type Ca2+ channels are inactivated, indicating that TTA-P2 did not influence Na+ channel kinetics, consistent with a previous report (Dreyfus et al., 2010). It is possible that T-type Ca2+ channels are involved in the release of GABA, which in principle could explain the observed block of synaptically evoked spike bursts by TTA-P2. To examine this possibility directly, we recorded GABAergic postsynaptic currents (PSCs) from TRN neurons under whole-cell voltage clamp. As shown in Figure 4, E and F, bath application of TTA-P2 (1 μm) did not influence PSC amplitude (347.0 ± 155.3 pA in control; 342.4 ± 146.1 pA in TTA-P2; n = 5; p = 0.71, paired t test) or paired-pulse ratio (0.79 ± 0.07 in control; 0.80 ± 0.06 in TTA-P2; n = 5; p = 0.72, paired t test), indicating that T-type Ca2+ channels are not involved in regulating GABA release from intra-TRN synapses. Thus, T-type Ca2+ channels expressed in TRN dendrites are required for GABA-induced burst firing in the TRN.
Activation of T-type Ca2+ channels is responsible for Ca2+ influx induced by GABA
To confirm the essential role of T-type Ca2+ channels in amplifying GABAergic responses, we performed whole-cell current-clamp recordings from TRN neurons, using an internal solution with 30 mm Cl− aimed to set ECl to values determined by our perforated-patch recordings outlined above. When neurons were held between −75 and −70 mV, synaptic stimulation triggered bursts of action potentials with properties similar to the results obtained with cell-attached recordings (Fig. 5A,B). The threshold for action potential generation, measured at the time corresponding to the initial peak of the third derivative of the voltage trace, was −35.3 ± 2.3 mV (n = 5). When neurons were depolarized to −60 mV to inactivate Ca2+ channels, GABAergic responses no longer elicited action potentials in most neurons (Fig. 5A,B). For the same neurons held between −75 and −70 mV, pharmacological block of T-type Ca2+ channels with TTA-P2 also eliminated action potential firing (control, 3.8 ± 0.1 spikes; TTA-P2, 0 spikes; p < 0.001, paired t test; Fig. 5A,B). These data confirm that GABA-evoked depolarizations alone are not sufficient to reach the threshold for spike generation. Rather, GABA-induced burst firing is strongly state dependent and relies on the activation of low-threshold Ca2+ channels from a hyperpolarized membrane potential.
To determine the contribution of T-type Ca2+ channels to GABA-evoked Ca2+ influx, we loaded TRN neurons with the Ca2+ indicator Fluo-4 (125 μm) and recorded dendritic Ca2+ transients using a CCD camera. The GABAA receptor agonist muscimol was applied using a puff electrode placed near the recorded neuron, in the presence of TTX to block action potential activity. Brief puffs of muscimol reliably evoked Ca2+ responses in TRN neuron dendrites (Fig. 5C). Bath application of TTA-P2 (1 μm) largely eliminated muscimol-evoked Ca2+ transients (Fig. 5C,D) while having only a modest influence on the muscimol-evoked depolarization. Thus, Ca2+ influx evoked by GABAA receptor activation is primarily mediated by activation of T-type Ca2+ channels.
Feedforward inhibition induced by excitatory GABAergic intra-TRN synapses
Neurons of the TRN provide powerful inhibitory connections to thalamic relay cells (Jones, 2007). We examined the impact of GABA-evoked burst firing in TRN neurons on the dynamics of inhibitory inputs received by relay neurons in the ventrobasal nucleus of the thalamus (VB). We recorded from VB neurons in whole-cell voltage clamp (0 mV holding potential), with excitatory neurotransmission blocked pharmacologically. Single stimuli applied to TRN led to two distinct types of inhibitory responses in VB neurons: short-latency monophasic IPSCs with little latency jitter from trial to trial, and long-latency multiphasic IPSCs with pronounced latency jitter between trials (Fig. 6A,C,E). Both responses were blocked by the GABAA receptor antagonist gabazine (10 μm) (Fig. 6G). As VB does not contain local GABAergic interneurons and receives inhibitory input exclusively from the TRN, both types of responses must have originated in the TRN. Based on the findings described above, we hypothesized that long-latency responses are disynaptic, mediated by stimulus-evoked bursts of action potentials in TRN neurons, which then generate feedforward inhibition in relay neurons. By contrast, short-latency responses are evoked by direct activation of axons formed by TRN neurons targeting VB cells (Fig. 6H). To test this idea, we applied low concentrations of the GABAA receptor antagonist gabazine (0.2 μm), which on average reduced the amplitude of the short-latency response by 53.5 ± 1.7% (n = 5) (Fig. 6A,B,G). By contrast, long-latency responses were completely eliminated, consistent with the idea that attenuated GABAA receptor-mediated depolarizations no longer triggered action potentials in TRN neurons. Similarly, reducing release probability at TRN GABAergic terminals should have similar effects on the short- and long-latency responses. Indeed, we found that reducing transmitter release by bath application of the CB1 receptor agonist WIN55,212 (5 μm) led to a differential reduction of short- and long-latency amplitude (Fig. 6C,D,G), consistent with a block of GABA-induced bursting in the TRN (Fig. 1C,D) and a more moderate reduction in transmitter release at GABAergic synapses in VB (Sun et al., 2011).
As demonstrated above (Fig. 4), GABA-triggered burst firing in TRN neurons is dependent on activation of dendritic T-type Ca2+ channels. If long-latency multiphasic IPSCs in VB neurons are evoked by GABA-triggered bursts of action potentials in TRN neurons, they should be eliminated by the T-type Ca2+ channel blocker TTA-P2. Indeed, bath application of TTA-P2 (1 μm) completely eliminated long-latency responses (Fig. 6E–G), confirming that T-type Ca2+ channels are critical for GABA-evoked TRN burst firing and the generation of disynaptic inhibition in VB neurons, By contrast, short-latency IPSCs were unaffected by TTA-P2 (Fig. 6E–G), confirming that T-type Ca2+ channels are not involved in regulating transmitter release at GABAergic terminals in either TRN (Fig. 4E,F) or VB. Thus, depolarizing intra-TRN GABAergic synapses can generate long-latency multiphasic IPSCs in the VB neurons by recruiting local TRN neurons.
Discussion
Here, we have shown that intra-TRN GABAergic synapses, acting via GABAA receptors, depolarize postsynaptic TRN neurons. We found that TRN neurons maintain high [Cl−]i, resulting in a ECl of approximately −45 mV. High [Cl−]i is generally associated with low KCC2 expression in the plasma membrane (Rivera et al., 1999). In agreement, we found that KCC2 is only weakly expressed in the TRN, whereas relay neurons in the adjacent somatosensory thalamus showed strong expression. Intra-TRN synaptic activity was sufficient to trigger action potentials when TRN neurons were in burst mode, mediated by the activation of T-type Ca2+ channels.
Depolarizing GABAergic synaptic transmission in the TRN
While intra-TRN synapses have been assigned a number of computational tasks such as controlling the degree of thalamic synchrony (Beenhakker and Huguenard, 2009) or mediating attentional processes (Crick, 1984), important questions remain about their anatomical and physiological properties. Previous work examining the impact of intra-TRN GABAergic synapses on their postsynaptic target has led to conflicting results. Bath application of GABAA receptor agonists can depolarize TRN neurons (Spreafico et al., 1988). Similarly, in vivo work in anesthetized cats has shown burst firing in TRN neurons, evoked by depolarizing GABAergic inputs (Bazhenov et al., 1999). In contrast, many studies have concluded that intra-TRN synapses are either shunting or hyperpolarizing from rest (Ahlsén and Lindström, 1982; Sanchez-Vives et al., 1997; Ulrich and Huguenard, 1997; Shu and McCormick, 2002). However, almost all experimental evidence has relied on intracellular recording techniques, which alter the intracellular milieu and introduce unknown measurement errors of the membrane potential (Verheugen et al., 1999).
By using both cell-attached and perforated-patch recording techniques at near-physiological temperatures, we conclude that GABAA receptor-mediated postsynaptic responses are depolarizing from rest. Furthermore, our immunohistochemical data indicate that TRN neurons express extremely low levels of KCC2, in contrast to the strong expression found in relay neurons of the adjacent thalamic nuclei. Similar nucleus-specific expression of KCC2 was previously shown in adult rats (Barthó et al., 2004). These data provide independent evidence for a high [Cl−]i in TRN neurons, suggesting that our electrophysiological data are not the result of a slicing-induced change in Cl− regulation.
Our experimentally determined value for ECl in TRN neurons (−45 mV) is significantly more depolarized compared with the value of −70 mV reported in a previous study (Ulrich and Huguenard, 1997). While using similar techniques, Ulrich and Huguenard (1997) performed their study at room temperature, whereas our experiments were performed at near-physiological temperatures. It is possible that intracellular chloride levels in TRN neurons are strongly temperature dependent, particularly since increases in temperature appear to reduce KCC2 activity, while NKCC1 and -2 activity is increased (Hartmann and Nothwang, 2011). While repeated agonist applications could have influenced ECl as measured under our experimental conditions (Huguenard and Alger, 1986), we want to emphasize that GABAA receptor-evoked depolarizations and spikes were observed following single GABAergic synaptic inputs, which are unlikely to lead to increases in [Cl−]i.
An excitatory role of GABA in TRN neurons is consistent with a growing list of studies suggesting that GABA, by activating ionotropic GABAA receptors, can depolarize distinct neuronal compartments in neurons of the mature or near-mature brain. GABA-induced depolarizations and Ca2+ increases can facilitate transmitter release and increase cell excitability (Price and Trussell, 2006; Stell et al., 2007; Pugh and Jahr, 2011) or facilitate action potential generation under certain conditions (Wagner et al., 1997; Chavas and Marty, 2003; Gulledge and Stuart, 2003; Chiang et al., 2012; Haam et al., 2012). Only a small number of studies have demonstrated that GABA release from interneurons alone can trigger action potentials from resting membrane potential levels (Szabadics et al., 2006; Woodruff et al., 2006), and the evidence is indirect and controversial (Glickfeld et al., 2009). Our study provides direct evidence that single GABAergic synaptic inputs are sufficient to trigger action potentials.
Amplification of excitatory GABAergic synaptic transmission by T-type Ca2+ channels
Our electrophysiological and imaging data show that activation of GABAA receptors leads to the opening of dendritic T-type Ca2+ channels in TRN neurons, thereby amplifying GABA-evoked postsynaptic depolarizations. This extends previous work suggesting that subthreshold glutamatergic EPSPs can be enhanced by the activation of T-type Ca2+ channels (Magee et al., 1995; Gillessen and Alzheimer, 1997; Urban et al., 1998; Crandall et al., 2010). Importantly, we find that the activation of T-type Ca2+ channels is essential for GABA-triggered spike generation. When T-type Ca2+ channels were either blocked pharmacologically or inactivated following depolarization of the postsynaptic membrane, GABAergic synaptic inputs no longer triggered action potentials, indicating that ECl is below the threshold for Na+-dependent action potentials.
The lack of selective antagonists has long made it challenging to assign computational roles to specific types of voltage-gated Ca2+ channels. A case in point is the role of T-type conductances in TRN neurons. Previous work has suggested that these conductances allow for rhythmic bursting typical for non-rapid eye movement sleep (Contreras, 2006; Cueni et al., 2008). However, supporting evidence was often based on pharmacological block of T-type Ca2+ channels using mibefradil, a drug that also interacts with Na+ channels and, in addition, blocks R-type (Cav2.3) Ca2+ channels (Randall and Tsien, 1997), which are expressed in TRN neurons (Zaman et al., 2011). In fact, recent studies in mice lacking Cav2.3 have demonstrated that R-type Ca2+ channels are critical for oscillatory burst discharges (Zaman et al., 2011), further questioning an exclusive role of T-type conductances in mediating rhythmic burst firing. Using the selective antagonist TTA-P2 (Dreyfus et al., 2010), which has only negligible effects on R-type conductances at the concentration used in our study (Choe et al., 2011), we directly demonstrate that the amplification of GABA-triggered postsynaptic responses is mediated by dendritic T-type Ca2+ channels. Interestingly, T-type Ca2+ channels are tightly associated with Ca2+-dependent small-conductance (SK)-type K+ channels in TRN dendrites (Cueni et al., 2008). Therefore, the dendritic amplification of subthreshold glutamatergic and GABAergic synaptic inputs is likely controlled by a dynamic interplay of voltage- and Ca2+-gated conductances.
Functional role of intra-TRN GABAergic synapses
TRN is crucially involved in the generation of rhythmic activity in the thalamocortical system (McCormick and Bal, 1997). Neurons in the TRN send powerful inhibitory inputs to excitatory thalamic relay cells, which in turn form excitatory inputs onto TRN neurons. This reciprocally connected TRN–relay cell circuit mediates synchronous oscillations (e.g., during sleep spindles). Importantly, individual pairs of TRN and relay neurons do not form reciprocal connections (Pinault, 2004) and connectivity between relay neurons is virtually nonexistent. Thus, it is possible that depolarizing GABAergic synapses, along with electrical synapses (Landisman et al., 2002), form a critical substrate for the initiation of synchronous oscillations in the thalamus. This is consistent with modeling (Bazhenov et al., 1999) and experimental (Steriade et al., 1987) studies suggesting that the isolated TRN nucleus can generate rhythmic oscillations.
However, the functional impact of intra-TRN GABAergic synapses is likely to be complex and strongly dependent on the context of their activation (Marty and Llano, 2005). Since ECl is depolarized compared with the resting membrane potential but more hyperpolarized compared with spike threshold, subthreshold GABAergic inputs will likely act as a shunt for glutamatergic inputs that coincide with the opening of the GABAA receptor-mediated chloride conductance, while being excitatory following the closing of the chloride conductance, similar to what was observed for GABAergic inputs in neocortical pyramidal neurons (Gulledge and Stuart, 2003). More sustained activation of GABAergic synapses will tend to clamp the membrane potential near the equilibrium potential for Cl−, shunting excitatory synaptic inputs and in addition, leading to inactivation of T-type Ca2+ channels. Thus, intra-TRN GABAergic synapses are most likely inhibitory during more sustained levels of synaptic and neuronal activity, consistent with a role of preventing pathological forms of hypersynchrony in thalamic circuits (Huntsman et al., 1999).
GABAergic synapses between TRN neurons could also form an important substrate for the rapid interaction between distinct thalamic nuclei. TRN is composed of multiple sectors, each of which primarily serves an individual sensory, motor, or limbic modality (Pinault, 2004). Work in vitro has shown that activity in one thalamic nucleus can lead to inhibition in a functionally distinct thalamic nucleus (Crabtree et al., 1998; Crabtree and Isaac, 2002). As thalamic nuclei do not form functional connections with each other, such cross-modal actions are likely mediated by a synaptic pathway passing through the TRN. Therefore, local circuits of synaptically connected TRN neurons might allow TRN to select appropriate intrathalamic pathways for distinct sensory, motor, and cognitive tasks and could mediate cross-modal modulation of TRN neurons during attentional processes (Yu et al., 2009).
Footnotes
This work was supported in part by NIDA Grant DA029381 and NICHD Grant HD065561 (H.-C.L.), and NINDS Grant NS077989, the American Heart Association, The Whitehall Foundation, and the Epilepsy Foundation (M.B.). We thank Drs. Adam Carter, Barry Connors, and Jay Gibson for comments on a previous version of this manuscript and the Baylor Intellectual and Developmental Disabilities Research Center core facility (NIH Grant HD024064) for confocal microscopy access.
V.N.U. and J.J.R. are employees of Merck & Co., Inc., and potentially own stock and/or stock options in the company.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Ahlsén G, Lindström S. Mutual inhibition between perigeniculate neurones. Brain Res. 1982;236:482–486. doi: 10.1016/0006-8993(82)90731-4. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Manis PB, Linden DJ. Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron. 1998;21:827–835. doi: 10.1016/s0896-6273(00)80598-x. [DOI] [PubMed] [Google Scholar]
- Barthó P, Payne JA, Freund TF, Acsády L. Differential distribution of the KCl cotransporter KCC2 in thalamic relay and reticular nuclei. Eur J Neurosci. 2004;20:965–975. doi: 10.1111/j.1460-9568.2004.03562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Self-sustained rhythmic activity in the thalamic reticular nucleus mediated by depolarizing GABAA receptor potentials. Nat Neurosci. 1999;2:168–174. doi: 10.1038/5729. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PH, Wu PY, Kuo TW, Liu YC, Chan CF, Chien TC, Cheng JK, Huang YY, Chiu CD, Lien CC. GABA is depolarizing in hippocampal dentate granule cells of the adolescent and adult rats. J Neurosci. 2012;32:62–67. doi: 10.1523/JNEUROSCI.3393-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic-Todorovic V, Todorovic SM. TTA-P2 is a potent and selective blocker of T-type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol. 2011;80:900–910. doi: 10.1124/mol.111.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D. The role of T-channels in the generation of thalamocortical rhythms. CNS Neurol Disord Drug Targets. 2006;5:571–585. doi: 10.2174/187152706779025526. [DOI] [PubMed] [Google Scholar]
- Crabtree JW, Isaac JT. New intrathalamic pathways allowing modality-related and cross-modality switching in the dorsal thalamus. J Neurosci. 2002;22:8754–8761. doi: 10.1523/JNEUROSCI.22-19-08754.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW, Collingridge GL, Isaac JT. A new intrathalamic pathway linking modality-related nuclei in the dorsal thalamus. Nat Neurosci. 1998;1:389–394. doi: 10.1038/1603. [DOI] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci. 2010;30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci. 2006;26:8633–8645. doi: 10.1523/JNEUROSCI.2333-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci. 1996;16:169–185. doi: 10.1523/JNEUROSCI.16-01-00169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillessen T, Alzheimer C. Amplification of EPSPs by low Ni2+- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Neurophysiol. 1997;77:1639–1643. doi: 10.1152/jn.1997.77.3.1639. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Excitatory actions of GABA in the cortex. Neuron. 2003;37:299–309. doi: 10.1016/s0896-6273(02)01146-7. [DOI] [PubMed] [Google Scholar]
- Haam J, Popescu IR, Morton LA, Halmos KC, Teruyama R, Ueta Y, Tasker JG. GABA is excitatory in adult vasopressinergic neuroendocrine cells. J Neurosci. 2012;32:572–582. doi: 10.1523/JNEUROSCI.3826-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, Temereanca S, Simons DJ. State-dependent processing of sensory stimuli by thalamic reticular neurons. J Neurosci. 2003;23:5264–5271. doi: 10.1523/JNEUROSCI.23-12-05264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann AM, Nothwang HG. Opposite temperature effect on transport activity of KCC2/KCC4 and N(K)CCs in HEK-293 cells. BMC Res Notes. 2011;4:526. doi: 10.1186/1756-0500-4-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Alger BE. Whole-cell voltage-clamp study of the fading of GABA-activated currents in acutely dissociated hippocampal neurons. J Neurophysiol. 1986;56:1–18. doi: 10.1152/jn.1986.56.1.1. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Jang IS, Jeong HJ, Akaike N. Contribution of the Na–K–Cl cotransporter on GABAA receptor-mediated presynaptic depolarization in excitatory nerve terminals. J Neurosci. 2001;21:5962–5972. doi: 10.1523/JNEUROSCI.21-16-05962.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The thalamus. Ed 2. Cambridge, UK; New York: Cambridge UP; 2007. [Google Scholar]
- Kim U, Sanchez-Vives MV, McCormick DA. Functional dynamics of GABAergic inhibition in the thalamus. Science. 1997;278:130–134. doi: 10.1126/science.278.5335.130. [DOI] [PubMed] [Google Scholar]
- Lam YW, Nelson CS, Sherman SM. Mapping of the functional interconnections between thalamic reticular neurons using photostimulation. J Neurophysiol. 2006;96:2593–2600. doi: 10.1152/jn.00555.2006. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. Electrical synapses in the thalamic reticular nucleus. J Neurosci. 2002;22:1002–1009. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Landisman CE, Connors BW. Small clusters of electrically coupled neurons generate synchronous rhythms in the thalamic reticular nucleus. J Neurosci. 2004;24:341–349. doi: 10.1523/JNEUROSCI.3358-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Christofi G, Miyakawa H, Christie B, Lasser-Ross N, Johnston D. Subthreshold synaptic activation of voltage-gated Ca2+ channels mediates a localized Ca2+ influx into the dendrites of hippocampal pyramidal neurons. J Neurophysiol. 1995;74:1335–1342. doi: 10.1152/jn.1995.74.3.1335. [DOI] [PubMed] [Google Scholar]
- Marty A, Llano I. Excitatory effects of GABA in established brain networks. Trends Neurosci. 2005;28:284–289. doi: 10.1016/j.tins.2005.04.003. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Attentional modulation of thalamic reticular neurons. J Neurosci. 2006;26:4444–4450. doi: 10.1523/JNEUROSCI.5602-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Price GD, Trussell LO. Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of Held. J Neurosci. 2006;26:11432–11436. doi: 10.1523/JNEUROSCI.1660-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Jahr CE. Axonal GABAA receptors increase cerebellar granule cell excitability and synaptic activity. J Neurosci. 2011;31:565–574. doi: 10.1523/JNEUROSCI.4506-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology. 1997;36:879–893. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Bal T, McCormick DA. Inhibitory interactions between perigeniculate GABAergic neurons. J Neurosci. 1997;17:8894–8908. doi: 10.1523/JNEUROSCI.17-22-08894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, McCormick DA. Inhibitory interactions between ferret thalamic reticular neurons. J Neurophysiol. 2002;87:2571–2576. doi: 10.1152/jn.00850.2001. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Huguenard JR. Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci. 2003;23:8978–8988. doi: 10.1523/JNEUROSCI.23-26-08978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico R, de Curtis M, Frassoni C, Avanzini G. Electrophysiological characteristics of morphologically identified reticular thalamic neurons from rat slices. Neuroscience. 1988;27:629–638. doi: 10.1016/0306-4522(88)90294-1. [DOI] [PubMed] [Google Scholar]
- Stell BM, Rostaing P, Triller A, Marty A. Activation of presynaptic GABAA receptors induces glutamate release from parallel fiber synapses. J Neurosci. 2007;27:9022–9031. doi: 10.1523/JNEUROSCI.1954-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G, Deschênes M. The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol. 1987;57:260–273. doi: 10.1152/jn.1987.57.1.260. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Sun YG, Wu CS, Lu HC, Beierlein M. Target-dependent control of synaptic inhibition by endocannabinoids in the thalamus. J Neurosci. 2011;31:9222–9230. doi: 10.1523/JNEUROSCI.0531-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. GABAB receptor-mediated responses in GABAergic projection neurones of rat nucleus reticularis thalami in vitro. J Physiol. 1996;493:845–854. doi: 10.1113/jphysiol.1996.sp021427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NN, Henze DA, Barrionuevo G. Amplification of perforant-path EPSPs in CA3 pyramidal cells by LVA calcium and sodium channels. J Neurophysiol. 1998;80:1558–1561. doi: 10.1152/jn.1998.80.3.1558. [DOI] [PubMed] [Google Scholar]
- Verheugen JA, Fricker D, Miles R. Noninvasive measurements of the membrane potential and GABAergic action in hippocampal interneurons. J Neurosci. 1999;19:2546–2555. doi: 10.1523/JNEUROSCI.19-07-02546.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Castel M, Gainer H, Yarom Y. GABA in the mammalian suprachiasmatic nucleus and its role in diurnal rhythmicity. Nature. 1997;387:598–603. doi: 10.1038/42468. [DOI] [PubMed] [Google Scholar]
- Woodruff AR, Monyer H, Sah P. GABAergic excitation in the basolateral amygdala. J Neurosci. 2006;26:11881–11887. doi: 10.1523/JNEUROSCI.3389-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Zhu J, Wager-Miller J, Wang S, O'Leary D, Monory K, Lutz B, Mackie K, Lu HC. Requirement of cannabinoid CB1 receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci. 2010;32:693–706. doi: 10.1111/j.1460-9568.2010.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XJ, Xu XX, He S, He J. Change detection by thalamic reticular neurons. Nat Neurosci. 2009;12:1165–1170. doi: 10.1038/nn.2373. [DOI] [PubMed] [Google Scholar]
- Zaman T, Lee K, Park C, Paydar A, Choi JH, Cheong E, Lee CJ, Shin HS. Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron. 2011;70:95–108. doi: 10.1016/j.neuron.2011.02.042. [DOI] [PubMed] [Google Scholar]