

Abstract
Reaction of cellular thiols with the 1,2-dithiolan-3-one 1-oxide moiety of leinamycin triggers the generation of DNA-damaging reactive intermediates. Studies with small, synthetic analogues of leinamycin reveal that the macrocyclic portion of the natural product imparts remarkable hydrolytic stability to the 1,2-dithiolan-3-one 1-oxide heterocycle without substantially compromising its thiol-sensing property.
The propensity to undergo intracellular bioactivation is an important property of many DNA-damaging natural products.1-5 Characterization of these processes is relevant to both medicinal chemistry and toxicology as it has the potential to reveal novel and chemically interesting strategies for the intracellular generation of biologically active reactive intermediates.6 The interior of cells is relatively rich in thiols7 and reaction with intracellular thiols represents a common means for unmasking DNA-damaging intermediates from natural products. For example, calicheamicin, dynemicin, neocarzinostatin, acylfulvenes related to the illudins, myrocin C, 2-crotonyloxymethyl-2-cycloalkeneones, lissoclinotoxin A, varacin, thiarubrin C, and some analogues of mitomycin C can be activated by reactions with thiols.8-24.
Leinamycin (1) is a thiol-activated, Streptomyces-derived secondary metabolite that displays potent activity against human cancer cell lines.25-29 Leinamycin contains a unique 1,2-dithiolan-3-one 1-oxide heterocycle that serves as its “thiol sensing” unit. Reaction of thiols with this moiety initiates rearrangement of leinamycin into a DNA-alkylating episulfonium ion 4 and converts the attacking thiol into a persulfide 2 that can mediate generation of reactive oxygen species (Scheme 1).17,30-44 The 1,2-dithiolan-3-one 1-oxide heterocycle is hydrolytically labile; however, in the context of leinamycin this reaction is slow relative to the thiol-mediated activation of the natural product.45 Indeed, it seems likely that resistance to hydrolysis coupled with high thiol-reactivity combine to allow efficient and selective bioactivation of leinamycin inside cells.
Scheme 1.
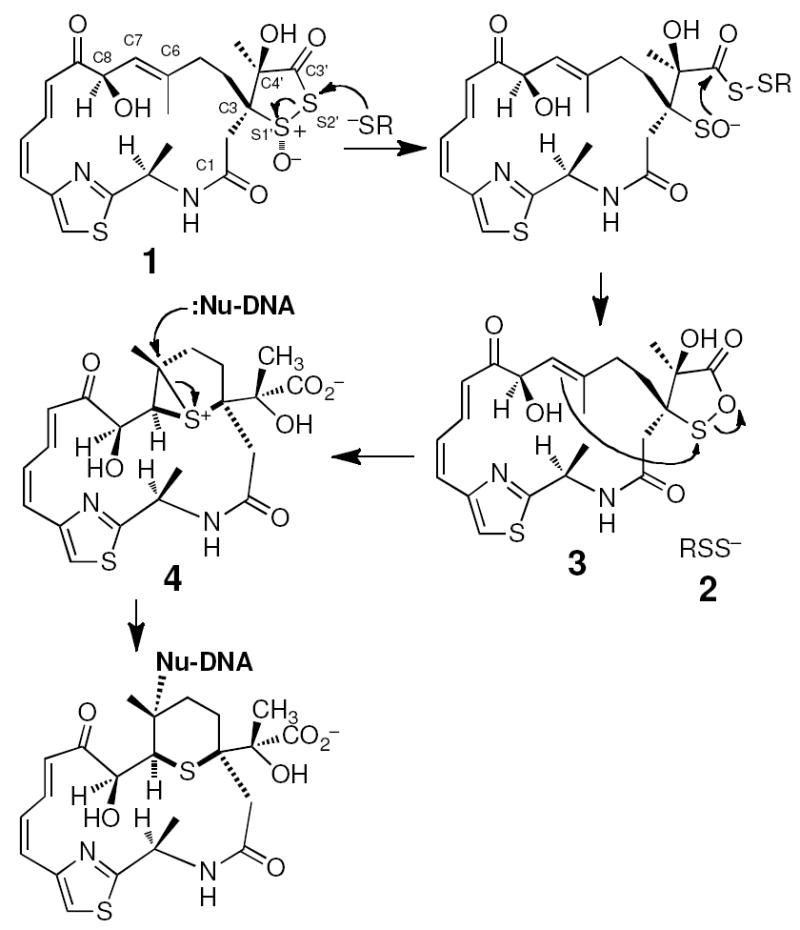
Thiol-activated DNA alkylation by leinamycin.
To better understand the chemical events underlying the bioactivation of leinamycin, we examined the inherent reactivity of the thiol-sensing unit found in the natural product. Accordingly, we synthesized this fragment of leinamycin via a modification of the route described by Pattenden and Shuker (Scheme 2).46 The published route employing NaSH for the epoxide ring-opening was ineffective in our hands, perhaps due to the notoriously impure nature of commercially available NaSH reagents.47 Instead, we used benzyl mercaptan as a protected hydrogen sulfide surrogate. In addition, we employed dimethyldioxirane for the final oxidation step rather than m-CPBA.48 The final oxidation reaction gave two major products and proton NMR analysis suggested that these are a 4:1 mixture of two diastereomers. These compounds were separable by thin layer chromatography and column chromatography on silica gel. The major isomer showed three methyl resonances at 1.74, 1.37, and 1.19 ppm, while resonances for the methyl groups in the minor isomer appeared at 1.58, 1.62, and 1.28 ppm. HMBC experiments were used to unambiguously assign the chemical shifts of the 4-methyl groups as the resonances at 1.37 ppm and 1.58 ppm for the major and minor isomer, respectively, via observation of three-bond coupling between methyl hydrogens and the carbonyl carbon. Literature precedents indicate that the proton NMR resonance of a methyl group in the cis orientation relative to a sulfoxide oxygen in a five-membered ring will be shifted downfield relative to a trans methyl group.49-52 Thus, the NMR results immediately suggested that the major isomer has the methyl group in the 4-position in a trans relationship to the sulfoxide oxygen as shown in 5 (Scheme 2). Consistent with this assignment, an NOE experiment on the major isomer revealed a much stronger through space interaction of the 4-methyl substituent with the 5-methyl group that displays a resonance at 1.19 ppm (trans to the sulfoxide oxygen) than with the 5-methyl group at 1.74 ppm (cis to the sulfoxide oxygen). Analysis of 13C-chemical shifts further supported the structural assignment. An HMQC experiment, along with the HMBC and NOE experiments described above, allowed us to assign the chemical shifts of each methyl group in the major isomer. The methyl group at C4 appears at 19.1 ppm. The methyl group at C5 with a cis relationship to the C4 methyl appears at 20.1 ppm and the methyl group at C5 with a trans relationship to the C4 methyl appears at 17.1. Literature precedents indicate that a methyl group with a cis relationship to the oxygen of an adjacent sulfoxide will appear upfield relative to a methyl group in the trans orientation.49,53 Thus, our data showing that the shielded, upfield methyl at C5 is trans to the C4 methyl, suggested that, in the major isomer, the methyl group at C5 which has a cis relationship to the sulfoxide oxygen also has a cis relationship to the hydroxyl group at C4. This is consistent with structure 5 for the major isomer. Thus, we were able to assign the major isomer as 5, in which the 4-hydroxyl group is cis to the sulfoxide oxygen, and the minor isomer as 6, in which the 4-hydroxyl group is trans to the sulfoxide oxygen.54 In the oxidation leading to 5 and 6, hydrogen bonding may direct dimethyldioxirane to the same face as the hydroxyl substituent leading to favored production of 5 during the synthesis.55 For the remainder of this paper, 5 will be referred to as the cis isomer and 6 as the trans isomer.
Scheme 2.
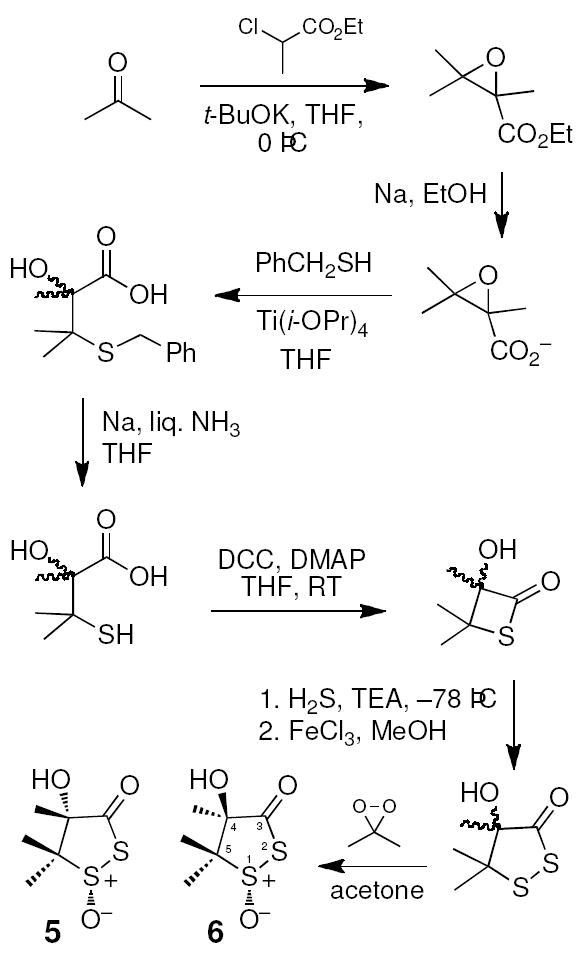
Synthesis of 1,2-dithiolan-3-one 1-oxides related to leinamycin.
With the cis and trans isomers of leinamycin’s thiol-sensing unit in hand, we examined their stability at 24 °C in an aqueous buffered solution containing the compound (5 or 6, 70 μM), MOPS buffer (250 mM, pH 7), and acetonitrile (25% v/v). Disappearance of the compounds was monitored by HPLC over the course of three half-lives and the data fit to a first-order decay process to determine apparent rate constants and half-lives for the decomposition process (Fig. 1).56
Fig. 1.
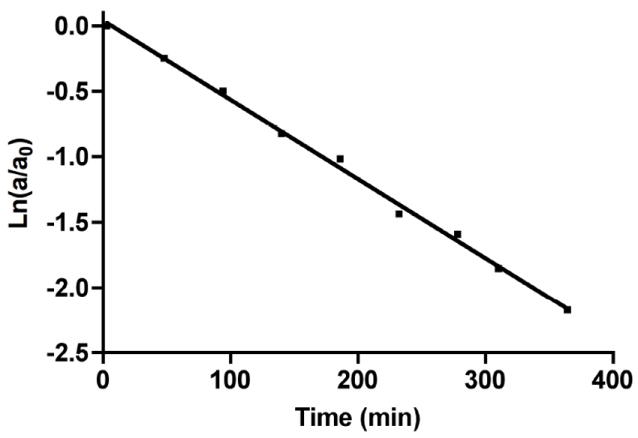
A representative plot showing the decomposition of 5 in aqueous buffered solution (5 (70 μM); MOPS (250 mM, pH 7); MeCN (25% v/v); 24 °C). The disappearance of 5 was monitored by HPLC using a C18 reverse phase column (Varian microsorb-MV 100 Å pore size, 5 μm particle size, 250 mm length, 4.6 mm diam.) eluted with acetonitrile: water (35:65) at a flow rate of 1.0 mL/min.
The apparent rate constants for the decomposition of 5 and 6 under these conditions were measured at kobs = 13.2 ± 0.01 × 10−3 min-1 (t1/2 = 53 min) and kobs = 6.2 ± 0.6 × 10−3 min-1 (t1/2 = 112 min), respectively (Table 1). The decomposition rate of 6 increases with increasing buffer concentration (kobs = 1.33 ± 0.02 × 10−2 min-1 at 100 mM MOPS, kobs = 1.5 ± 0.2 × 10−2 min-1 and 300 mM MOPS, and kobs = 1.9 ± 0.2 × 10−2 min-1 at 500 mM MOPS). While the contribution of buffer is significant, at buffer concentrations used in our studies the buffer-independent reaction predominates. Near the physiological pH range, the rate at which 6 decomposes increases with increasing pH (kobs = 8.7 × 10−3 min-1 at pH 6.5, kobs = 1.5 × 10−2 min-1 at pH 7.0, and kobs = 4.7 × 10−2 min-1 at pH 7.5; all at 300 mM MOPS). This mirrors the effect of pH observed previously on the stability of leinamycin in sodium phosphate buffer containing 10% methanol.57 The pH effects are consistent with literature precedent indicates that hydroxide rather than water is the kinetically relevant species involved in the hydrolysis of thiosulfinates and thioesters.58-60 We find that the decomposition of leinamycin under the same conditions used for the 5 and 6 above occurs with a rate constant of kobs = 0.408 ± 0.001 × 10−3 min-1 (t1/2 = 27 h). This value is generally consistent with a previous measurement of leinamycin’s stability (100 μM 1 in HEPES, 250 mM, pH 7, 24 °C, containing no organic co-solvent) that gave a rate constant of 0.72 × 10−3 min-1 (t1/2 = 16.1 h).45,61 Clearly, the 1,2-dithiolan-3-one 1-oxide heterocycle, when embedded in the context of the natural product, enjoys remarkably increased stability compared to the simple analogues 5 and 6.
Table 1.
Rate constants for 1, 5, and 6.
| Compound | Conditions | kobs (min-1) | t1/2 |
|---|---|---|---|
|
| |||
| 5 no thiol | 5 (70 μM); MOPS (250 mM, pH 7); MeCN (25% v/v); 24 °C | 13.2 ± 0.01 × 10−3 | 53 min |
|
| |||
| 6 no thiol | 6 (70 μM); MOPS (250 mM, pH 7); MeCN (25% v/v); 24 °C | 6.2 ± 0.6 × 10−3 | 112 min |
| 1 no thiol | 1 (70 μM); MOPS (250 mM, pH 7); MeCN (25% v/v); 24 °C | 0.408 ± 0.001 × 10−3 | 27 h |
| 5 + thiol | 5 (70 μM); GSH (700 μM) MOPS (300 mM, pH 7); MeCN (25% v/v); 24 °C | 24.3 ± 0.1 × 10−2 | 2.8 min |
| 6 + thiol | 5 (70 μM); GSH (700 μM) MOPS (300 mM, pH 7); MeCN (25% v/v); 24 °C | 18.3 ± 0.2 × 10−2 | 3.8 min |
| 1 + thiol | 5 (70 μM); GSH (700 μM) MOPS (300 mM, pH 7); MeCN (25% v/v); 24 °C | 6.67 ± 0.01 × 10−2 | 10.4 min |
We next examined the reaction of 5, 6, and leinamycin (70 μM) with the biological thiol glutathione (GSH, 700 μM) in MOPS buffer (300 mM, pH 7) containing acetonitrile (25% v/v). The pseudo-first-order rate constants for the disappearance of 5 and 6 under these conditions were measured at kobs = 24.3 ± 0.1 × 10−2 min-1 (t1/2 = 2.8 min) and kobs = 18.3 ± 0.2 × 10−2 min-1 (t1/2 = 3.8 min), respectively. From these values, one can estimate second-order rate constants of 344 M-1 min-1 for 5 and 261 M-1 min-1 for 6. The apparent rate constant for the reaction of leinamycin with GSH under these conditions was kobs = 6.67 ± 0.01 × 10−2 min-1 (t1/2 = 10.4 min, 95 M-1 min-1). This is reasonably close to the value of 10.4 M-1 s-1 (624 M-1 min-1) reported previously for the reaction of leinamycin with GSH (in HEPES buffer, 50 mM, pH 7, 24 °C, containing no organic co-solvent).45
The natural product leinamycin was isolated as the single stereoisomer shown in Scheme 1, with a trans relationship between the C4’-OH group and the S1’-sulfinyl oxygen.62 Our work with 5 and 6 show that the naturally-occurring trans isomer is approximately two times more stable than the cis isomer in aqueous buffered solution. However, differences in the hydrolytic stability of the cis/trans isomers 5 and 6 are subtle compared to the dramatic effect that leinamycin’s 18-membered macrocycle brings to the stability of the natural product in aqueous solution. Leinamycin is approximately 30 times more stable than 5 and 15 times more stable than 6 against decomposition in aqueous buffer. It is interesting to consider potential mechanisms by which the macrocycle stabilizes leinamycin against decomposition in aqueous solution. Based upon computational analysis of small model systems, Wu and Greer suggested that an nO –> σS1,* interaction between the amide carbonyl in the macrocycle and the S1’-sulfinyl group of leinamycin stabilizes the 1,2-dithiolan-3-one 1-oxide ring system by ~6 kcal/mol.63 Alternatively, or in addition, the macrocycle of the natural product may present a steric impediment to these approach of hydroxide to the 1,2-dithiolan-3-one 1-oxide heterocycle of leinamycin. The solution structure of leinamycin deserves further consideration in this regard. Regardless of mechanism, our results clearly establish that the macrocyclic portion of leinamycin stabilizes the natural product against decomposition in aqueous solution.
It is striking that the macrocycle increases the aqueous stability of leinamycin without compromising the ability of the natural product to react avidly with thiols. For example, leinamycin is only 3.6 times less reactive toward GSH than is compound 5. This may reflect the fact that thiol-mediated activation of leinamycin proceeds via attack of the mercaptan at the sterically exposed S2’ position of the 1,2-dithiolan-3-one 1-oxide ring system,41 while hydrolysis may proceed primarily via attack of hydroxide on S1’ or C3’ – reactions that may be suppressed by the macrocyclic substituent.
Previous work established three mechanisms by which the macrocycle of leinamycin may facilitate efficient DNA alkylation. First, the Z,E-5-(thiazol-4-yl)-penta-2,4-dienone portion of the macrocycle presents a slightly twisted π–surface that confers non-covalent DNA-binding properties to the natural product.32,64,65 Second, the hydroxyl group at C8 of the macrocycle engages the episulfonium ion 5 in a reversible thia-Payne reaction that may stabilize the episulfonium ion against hydrolytic destruction.30 Third, the conformationally rigid macrocycle may accurately position the C6-C7 alkene for efficient reaction with the electrophilic sulfur of 3 in the generation of the alkylating agent 4 (Scheme 1).66 The work presented here establishes an additional role for the macrocyclic portion of leinamycin in facilitating efficient thiol-triggered alkylation of cellular DNA. The macrocyclic portion of leinamycin imparts substantial aqueous stability to the 1,2-dithiolan-3-one 1-oxide without compromising its ability to act as a thiol-sensing unit. Thus, the 18-membered macrocycle of leinamycin enables efficient and selective bioactivation of the natural product in the thiol-rich environment found inside cells.
Acknowledgments
We are grateful to the National Institutes of Health for support of this work (CA83925 and 119131).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Referenes and notes
- 1.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477–2495. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. Carcinogenesis. 2000;21:345–351. doi: 10.1093/carcin/21.3.345. [DOI] [PubMed] [Google Scholar]
- 3.Gates KS. In: Comprehensive Natural Products Chemistry. Kool ET, editor. Vol. 7. Pergamon; New York: 1999. pp. 491–552. [Google Scholar]
- 4.Gates KS. In: Reviews of Reactive Intermediates. Platz MS, Moss RA, Jones MJ, editors. John Wiley and Sons, Inc.; Hoboken: 2007. pp. 333–378. [Google Scholar]
- 5.Gates KS. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cronin MTD. Crit Rev Toxiciol. 2010;40:728–748. doi: 10.3109/10408444.2010.494175. [DOI] [PubMed] [Google Scholar]
- 7.Meister A, Anderson ME. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 8.Chu-Moyer M, Danishefsky SJ. Tetrahedron Lett. 1993;34:3025–3028. [Google Scholar]
- 9.Lee AHF, Chen J, Liu D, Leung TYC, Chan ASC, Li T. J Am Chem Soc. 2002;124:13972–13973. doi: 10.1021/ja020531i. [DOI] [PubMed] [Google Scholar]
- 10.Lee HFL, Chan ASC, Li T. JCS Chem Comm. 2002:2112–2113. doi: 10.1039/b204920c. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee M, Cramer KD, Townsend CA. J Am Chem Soc. 1993;115:3374–3375. [Google Scholar]
- 12.Myers AG, Cohen SB, Tom NJ, Madar DJ, Fraley ME. J Am Chem Soc. 1995;117:7574–7575. [Google Scholar]
- 13.Myers AG, Cohen SB, Kwon BM. J Am Chem Soc. 1994;116:1255–1271. [Google Scholar]
- 14.Myers AG. Tet Lett. 1987;28:4493–4496. [Google Scholar]
- 15.Chatterji T, Gates KS. Bioorg Med Chem Lett. 1998;8:535–538. doi: 10.1016/s0960-894x(98)00066-3. [DOI] [PubMed] [Google Scholar]
- 16.Chatterji T, Gates KS. Bioorganic Med Chem Lett. 2003;13:1349–1352. doi: 10.1016/s0960-894x(03)00103-3. [DOI] [PubMed] [Google Scholar]
- 17.Chatterji T, Keerthi K, Gates KS. Bioorg Med Chem Lett. 2005;15:3921–3924. doi: 10.1016/j.bmcl.2005.05.110. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Koreeda M, Chatterji T, Gates KS. J Org Chem. 1998;63:8644–8645. [Google Scholar]
- 19.McMorris TC, Kelner MJ, Wang W, Moon S, Taetle R. Chem Res Toxicol. 1990;3:574–579. doi: 10.1021/tx00018a013. [DOI] [PubMed] [Google Scholar]
- 20.Paz M, Tomasz M. Org Lett. 2001;3:2789–2792. doi: 10.1021/ol015517+. [DOI] [PubMed] [Google Scholar]
- 21.Wang S, Kohn H. J Org Chem. 1999;42:788–790. doi: 10.1021/jm9806796. [DOI] [PubMed] [Google Scholar]
- 22.Fekry MI, Price N, Zang H, Huang C, Harmata M, Brown P, Daniels JS, Gates KS. Chem Res Toxicol. 2011;24:217–228. doi: 10.1021/tx100282b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamilton DS, Zhang X, Ding Z, Hubatsch I, Mannervik B, Houk KN, Ganem B, Creighton DJ. J Am Chem Soc. 2003;125:15049–15058. doi: 10.1021/ja030396p. [DOI] [PubMed] [Google Scholar]
- 24.Lee SH, Kohn H. J Am Chem Soc. 2004;126:4281–4292. doi: 10.1021/ja030577r. [DOI] [PubMed] [Google Scholar]
- 25.Gates KS. Chem Res Toxicol. 2000;13:953–956. doi: 10.1021/tx000089m. [DOI] [PubMed] [Google Scholar]
- 26.Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi K, Nakano H. J Antibiotics. 1989;42:1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]
- 27.Hara M, Saitoh Y, Nakano H. Biochemistry. 1990;29:5676–5681. doi: 10.1021/bi00476a005. [DOI] [PubMed] [Google Scholar]
- 28.Hara M, Takahashi I, Yoshida M, Kawamoto I, Morimoto M, Nakano H. J Antibiotics. 1989;42:333–335. doi: 10.7164/antibiotics.42.333. [DOI] [PubMed] [Google Scholar]
- 29.Bassett S, Urrabaz R, Sun D. Anti-Cancer Drugs. 2004;15:689–696. doi: 10.1097/01.cad.0000136886.72917.6f. [DOI] [PubMed] [Google Scholar]
- 30.Asai A, Hara M, Kakita S, Kanda Y, Yoshida M, Saito H, Saitoh Y. J Am Chem Soc. 1996;118:6802–6803. [Google Scholar]
- 31.Behroozi SB, Kim W, Gates KS. J Org Chem. 1995;60:3964–3966. [Google Scholar]
- 32.Fekry M, Szekely J, Dutta S, Breydo L, Zang H, Gates KS. J Am Chem Soc. 2011;132:17641–17651. doi: 10.1021/ja2046149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zang H, Gates KS. Chem Res Toxicol. 2003;16:1539–1546. doi: 10.1021/tx0341658. [DOI] [PubMed] [Google Scholar]
- 34.Nooner T, Dutta S, Gates KS. Chem Res Toxicol. 2004;17:942–949. doi: 10.1021/tx049964k. [DOI] [PubMed] [Google Scholar]
- 35.Shipova K, Gates KS. Bioorg Med Chem Lett. 2005;15:2111–2113. doi: 10.1016/j.bmcl.2005.02.058. [DOI] [PubMed] [Google Scholar]
- 36.Dutta S, Chowdhury G, Gates KS. J Am Chem Soc. 2007;129:1852–1853. doi: 10.1021/ja067294u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viswesh V, Gates KS, Sun D. Chem Res Toxicol. 2010;23:99–107. doi: 10.1021/tx900301r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gates KS, Nooner T, Dutta S. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 39.Behroozi SJ, Kim W, Dannaldson J, Gates KS. Biochemistry. 1996;35:1768–1774. doi: 10.1021/bi952257t. [DOI] [PubMed] [Google Scholar]
- 40.Mitra K, Kim W, Daniels JS, Gates KS. J Am Chem Soc. 1997;119:11691–11692. [Google Scholar]
- 41.Breydo L, Gates KS. J Org Chem. 2002;67:9054–9060. doi: 10.1021/jo020568l. [DOI] [PubMed] [Google Scholar]
- 42.Chatterji T, Kizil M, Keerthi K, Chowdhury G, Posposil T, Gates KS. J Am Chem Soc. 2003;125:4996–4997. doi: 10.1021/ja029169y. [DOI] [PubMed] [Google Scholar]
- 43.Keerthi K, Gates KS. Org Biomol Chem. 2007;5:1595–1600. doi: 10.1039/b701179b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zang H, Breydo L, Mitra K, Dannaldson J, Gates KS. Bioorganic Med Chem Lett. 2001;11:1511–1515. doi: 10.1016/s0960-894x(01)00196-2. [DOI] [PubMed] [Google Scholar]
- 45.Breydo L, Zang H, Mitra K, Gates KS. J Am Chem Soc. 2001;123:2060–2061. doi: 10.1021/ja003309r. [DOI] [PubMed] [Google Scholar]
- 46.Pattenden G, Shuker AJ. J Chem Soc Perkin 1. 1992:1215–1221. [Google Scholar]
- 47.Hughes MN, Centelles MN, Moore KP. Free Rad Biol Med. 2009;47:1346–1353. doi: 10.1016/j.freeradbiomed.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 48.Glass RS, Liu Y. Tetrahedron Lett. 1994;35:3887–3888. [Google Scholar]
- 49.Barbarella G, Rossini S, Bongini A, Tugnoli V. Tetrahedron. 1985;41:4691–4701. [Google Scholar]
- 50.Barbarella G, Garbesi A, Fava A. J Am Chem Soc. 1975;97:5883–5889. [Google Scholar]
- 51.Rigau JJ, Bacon CC, Johnson CR. J Org Chem. 1970;35:3655–3657. [Google Scholar]
- 52.Carey FA, Dailey ODJ, Fromuth TE. Phosphorus, Sulfur Silcon Relat Elem. 1981;10:163–168. [Google Scholar]
- 53.LaLonde RT, Wong CF, Tsai AI-M, Wróbel JT, Ruszkoska J, Kabzinska K, Martin TI, MacLean DB. Can J Chem. 1976;54:3860–3868. [Google Scholar]
- 54.Synthesis of 4-hydroxy-4,5,5-trimethyl-1,2-dithiolan-3-one-1-oxide (5 and 6). To a stirred solution of 4-hydroxy-4,5,5-trimethyl-1,2-dithiolan-3-one (50 mg, 0.28 mmol) in acetone (0.5 mL) in an ice bath was added freshly prepared dimethyldioxirane (dropwise using pipette). The disappearance of the starting material was closely monitored by TLC (5:1 hexane:ethyl acetate) and the reaction was complete in about 5 min. Flash column chromatography on silica gel eluted with ethyl acetate/dichloromethane/hexane (1:3:4) gave the diastereomers 5 and 6 as colorless solids (50 mg total, 90%). Compound 5: Rf = 0.36; 1H NMR (300 MHz, CDCl3) δ 1.19 (3H, s, C5-CH3 trans to sulfoxide oxygen), 1.37 (3H, s, C4-CH3), 1.74 (3H, s, C5-CH3 cis to sulfoxide oxygen); 13C-NMR (CDCl3, 62.9 MHz) δ 201.8 (C=O), 84.9 (C4), 66.7 (C5), 20.1 (C5-CH3 trans to sulfoxide oxygen), 19.1 (C4-CH3), 17.1 (C5-CH3 cis to sulfoxide oxygen); HRMS: (EI) calcd for C6H10O3S2 [M + Li]+ 201.0225, found 201.0215. Compound 6: Rf = 0.24 1H NMR (300 MHz, CDCl3) δ 1.27 (3H, s), 1.60 (3H, s), 1.62 (3H, s), 2.9 (1H, s, OH); 13C-NMR (CDCl3, 62.9 MHz) δ 84.7, 65.9, 24.2, 18.8, 16.8; HRMS: (EI) calcd for C6H10O3S2 [M + Na]+ 216.9963, found 216.9962.
- 55.Waldemar A, Smerz AK. J Org Chem. 1996;61:3506–3510. [Google Scholar]
- 56.Espenson JH. Chemical Kinetics and Reaction Mechanisms. 2. McGraw-Hill, Inc.; New York: 1995. [Google Scholar]
- 57.Asai A, Saito H, Saitoh Y. Bioorg Med Chem. 1997;5:723–729. doi: 10.1016/s0968-0896(97)00015-1. [DOI] [PubMed] [Google Scholar]
- 58.Kice JL, Rogers TE. J Am Chem Soc. 1974;96:8009–8015. [Google Scholar]
- 59.Nagy P, Ashby MT. Chem Res Toxicol. 2007;20:1364–1372. doi: 10.1021/tx700168z. [DOI] [PubMed] [Google Scholar]
- 60.Castro E. Chem Rev. 1999;99:3505–3524. doi: 10.1021/cr990001d. [DOI] [PubMed] [Google Scholar]
- 61.The decomposition of leinamycin in water at 37 °C is faster with a half-life of 8 h in sodium phosphate (10 mM, pH 7) containing methanol (10% v/v).57
- 62.Kanda Y, Fukuyama T. J Am Chem Soc. 1993;115:8451–8452. [Google Scholar]
- 63.Wu S, Greer A. J Org Chem. 2000;65:4883–4887. doi: 10.1021/jo000145o. [DOI] [PubMed] [Google Scholar]
- 64.Breydo L, Zang H, Gates KS. Tetrahedron Lett. 2004;45:5711–5716. [Google Scholar]
- 65.Breydo L, Barnes CL, Gates KS. Acta Cryst C. 2002;C58:o447–0449. doi: 10.1107/s0108270102007503. [DOI] [PubMed] [Google Scholar]
- 66.Keerthi K, Gates KS. Manuscript submitted. [Google Scholar]