

Abstract

A facile synthetic route utilizing readily available reagents affords a series of regioisomerically pure xanthene dye derivatives. Advantages include relatively mild conditions and good to excellent yields. Nonpolar, highly crystalline intermediates are isolable by standard chromatographic techniques. The intermediates are in the requisite xanthene oxidation state, thus avoiding the need for relatively inefficient oxidation chemistry and/or harsh conditions. During the course of this work, a new boron-mediated 1,2-aryl migration reaction was discovered.
Introduction
Fluorescein and fluorone are structurally related xanthene dyes (Figure 1). Fluorone is formally decarboxylated fluorescein. Fluorone derivatives have found numerous applications. They have been used in the detection of a variety of metal ions,1 sugars,2 phosphorylated molecules,3 HIV-1 nucleocapsid protein,4 reactive oxygen species,5 in screening assays for mitochondrial permeability,6 acetylcholinesterase inhibition,7 and telomerase inhibition.8 We have reported the use of fluorones as sialic acid9 and homocysteine probes.10
Figure 1.

Structures of some common xanthene dyes.
The initial fluorone synthesis was reported by Mohlau and Koch.11 Typical syntheses include the following sequence: (i) formation of the leuco base via the condensation of resorcinol with aldehydes under thermal and acid-catalyzed conditions and (ii) the formation of dye via the oxidation of the leuco base. It has been reported that the oxidation step is often low-yielding. Additionally, there can be purification problems due to the formation of byproducts and the relatively polar nature of fluorone derivatives.12
Recently, a novel and innovative microwave protocol resulted in improved yields in related rosamine derivative syntheses.12d The microwave-assisted condensation and oxidation gave relatively higher yields (typically ranging from 27 to 73%) than traditional thermal condition (typical ranges from 8 to 35%).
Fluorone dye preparations have also been reported earlier by Neckers et al. as part of his extensive and pioneering work on xanthenes, involving heating intermediates in a sealed tube at 200 °C.13 Benzophenones have also been used as condensation partners in high-temperature, acid-catalyzed syntheses of xanthenes.14 A novel oxidation reaction of substituted triphenylmethanes has been reported to occur via Br2/CCl4.15
Herein, we report a simple new protocol for attaining a series of fluorone derivatives. It is based on forming tertiary carbinol leuco bases via Grignard reactions. Treatment of carbinol leuco bases with BBr3 affords the desired dyes. The carbinol intermediate is much less polar than the dye products. It can thus be purified via standard column chromatography. These intermediates are often highly crystalline (vide infra). The carbinol carbon is already in the correct oxidation state of the desired dyes. Therefore, potentially troublesome oxidations are avoided. The dye products can be purified by simple filtration methods without chromatography. The use of low-temperature, basic conditions may serve as an attractive alternative to more common, relatively harsh acid-catalyzed and oxidative methods. In addition, during the course of this work we discovered a novel 1,2-aryl migration mediated by boron that furnishes potentially useful dendrimer cores as well as dye architectures.
Results and Discussion
In our initial investigations, we use free radical bromination of appropriate triaryls as a means to obtain dye precursors with the requisite oxidation state. Tetramethyl ether 2 is obtained in nearly quantitative yield by stirring 116 via known procedures.17 Compound 2 (500 mg in 20 mL of CCl4) is treated with NBS (1.05 equiv) in the presence of a catalytic amount of (PhCO2)2 in CCl4 (Scheme 1).
Scheme 1.

Upon washing the crude brominated product with a saturated NH4Cl solution, carbinol 4 is obtained in a 13% yield. Reverse condensation product 5 and brominated congener 6 are also obtained in 6% and 15% yields, respectively. The mechanism of resorcinarene reverse condensation has previously been studied in great detail.12c,18 Compounds 5 and 6 were synthesized independently.19 The structures of 1, 2, 4, 5, and 6 are verified by single-crystal X-ray structural analysis (Supporting Information).
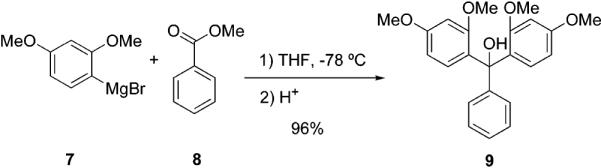
Since solvolysis of bromide 3 occurs readily during workup,15 we decided to synthesize the carbinol precursor directly. It is conveniently accessed by the reaction of readily available Grignard reagents and esters. Compound 9 is obtained in one step in a yield of 96% by reacting 2,4-dimethoxybenzenemagnesium bromide 7 and methyl benzoate 8 (Scheme 2). The structure of 9 is confirmed by X-ray crystallography (Supporting Information). When 9 is treated with BBr3 (6 equiv), monomethyl ether 10 is obtained in 64% yield. Using a greater excess of BBr3 (16 equiv) furnishes fully deprotected 11 in a 65% yield. Compound 11 is conveniently isolated by filtration (Scheme 3).
Scheme 2.
Scheme 3.

It is straightforward to apply the above protocol to the synthesis of a series of regioisomerically pure fluorone dyes. When various methyl benzoates 12a–e are used, the corresponding carbinols 13a–e are obtained in excellent yields (>90%). The reaction of the carbinols 13a–d with 16 equiv of BBr3 furnishes the corresponding fluorone dyes with yields ranging from 70 to 88% while deprotection of 13e affords methyl ether 15 (Table 1). Compound 15 is completely deprotected using 20 equiv of BBr3 to furnish 14e in a 76% yield (Scheme 4). In each case, the fluorone products are obtained without preparative chromatography. The structures of 13b and 13e are confirmed by single-crystal X-ray structure analysis (Supporting Information).
TABLE 1.

| entry | substrate | R1 | R2 | carbinol | carbinol yield (%) | fluorone | fluorone yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 12a | Br | H | 13a | 92 | 14a | 70 |
| 2 | 12b | Ph | H | 13b | 99 | 14b | 87 |
| 3 | 12c | OMe | H | 13c | 83 | 14c | 96 |
| 4 | 12d | H | NO2 | 13d | 91 | 14d | 73 |
| 5 | 12e | NO2 | H | 13e | 95 |
Scheme 4.

To prepare a potential sialic acid sensor 16,9 carbinol 17 was targeted (Scheme 5).
Scheme 5.

The reaction of 7 (2.4 equiv) and 18 (1 equiv), however, affords compound 19 in 13% yield. When 3.6 equiv of 7 is used, the yield of 19 improves to 32%. In each run, 2,4-dimethoxyphenylboronic acid 20 is isolated in trace amounts (Scheme 6). Structures of 18, 19 (Figure 2), and 20 were confirmed by single-crystal X-ray structure analysis (Supporting Information).
Scheme 6.

Figure 2.

X-ray structure for 19.
A proposed mechanism for the formation of compound 19 is shown in Scheme 7. The first equivalent of 7 reacts with boron to form the tetrahedral anionic boronate 21. This is supported by the fact that 18 is quantitatively recovered when 1 equiv of 7 is added to a solution of 18 and stirred overnight before quenching with water. The second and third equivalents of 7 react with the ester to afford the tertiary oxide 22. Upon workup, a 1,2-aryl shift mediated by boron affords 19.
Scheme 7.

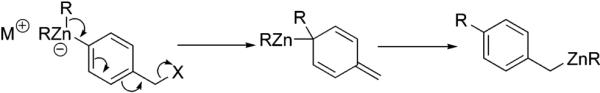
1,2-Alkyl shifts have been known for decades.20 In 1963, Matteson described the prototypical 1,2-alkyl shift involving α-haloalkyl borates.21 Negishi and co-workers established the analogous 1,2-rearrangement of 1-chloroalkyl complexes of Al, Mg, Zn, Cd, Ti, Zr, Hf, V, Cr, Mn, Fe, Co, Ni, and Cu.22 It has also been found that organoboronate complexes bearing α,β-unsaturation and leaving groups at the α- and γ-carbons can undergo a 1,2-alkyl group migration to afford homologated organoboron compounds.23 A recent article described a transformation whereby an organozincate bearing a leaving group at the remote benzylic position would rearrange with concomitant carbon–carbon bond formation to furnish the benzylzincate22j,24 (Scheme 8). In the current case (Scheme 7), the boronic acid is not found in the final product. This is the first observation of a 1,2-aryl group migration mediated by an arylboronate complex involving a leaving group at a remote benzylic position. Compound 19 and congeners may serve as functional dye and/or dendrimer substrates.
Scheme 8.
Conclusion
Carbinol leuco bases are easily prepared and purified precursors for fluorone dyes. A novel reaction that involves a boron-mediated 1,2-aryl shift reaction with a leaving group at a remote benzylic position is described. The methods described herein are now being utilized in our lab toward the synthesis of new naphthofluorescein dye architectures currently unattainable via other methodologies.
Experimental Section
5,5′-Dichloro-2,2′,4,4′-tetramethoxytrityl Alcohol (4)
In a 50-mL round-bottom flask, compound 2 (0.500 g, 1.18 mmol) and NBS (0.315 g, 1.77 mmol) are dissolved in 20 mL of CCl4. A catalytic amount of benzoyl peroxide (~10 mg) is added. The reaction mixture is heated at reflux for 1 h with vigorous stirring. It is cooled to room temperature and quenched with 50 mL of saturated NH4Cl (aq). The mixture is extracted with CH2Cl2 (3 × 15 mL). The combined extracts are dried over MgSO4 and filtered, and the filtrate is evaporated to dryness. The solid residue is purified by flash column chromatography (silica gel; CH2Cl2) to give 72 mg (14%) of 4 along with 18 mg (6%) of 5 and 43 mg (15%) of 6. The structures of 5 and 6 were verified via independent syntheses (vide infra). Data for 4: 1H NMR (DMSO-d6, 250 MHz) δ 7.23–7.16 (m, 5H), 7.05 (s, 2H), 6.76 (s, 2H), 5.52 (s, 1H), 3.88 (s, 6H); 13C NMR (DMSO-d6, 62.9 MHz) δ 156.6, 154.4, 146.0, 129.0, 127.3, 127.3, 127.0, 126.4, 111.3, 98.8, 78.0, 56.2, 55.9. MALDI-TOF m/z 431.517 [M − OH]+, 447.544 [M]+.
5-Chloro-2,4-dimethoxybenzophenone (5)
A 20-mL volume of CH3CN solution containing 2,4-dimethoxybenzophenone (0.500 g, 2.07 mmol) and NCS (0.200 g, 2.17 mmol) is mixed with 100 mg of 60 Å silica gel. The reaction mixture is heated at reflux for 5 h with vigorous stirring and cooled to room temperature. Silica gel is removed via suction filtration, and CH3CN is removed under reduced pressure. The residue is purified by flash column chromatography (silica gel; CH2-Cl2) to afford 0.470 g (82%) of 5. 1H NMR (DMSO-d6, 250 MHz) δ 7.68–7.60 (m, 3H), 7.52–7.70 (m, 2H), 7.39 (s, 1H), 6.89 (s, 1H), 3.97 (s, 3H), 3.69 (s, 3H); 13C NMR (DMSO-d6, 62.9 MHz) δ 193.6, 157.7, 157.6, 137.6, 133.1, 130.1, 129.1, 128.5, 121.0, 112.5, 98.1, 56.6, 56.2. MALDI-TOF m/z 276.1 [M]+, 277.4 [M + H]+, 299.0 [M + Na]+, 314.9 [M + K]+.
1-Bromo-5-chloro-2,4-dimethoxy-benzene (6)
This compound was prepared following the procedures above for compound 5 except that 2,4-dimethoxybromobenzene (1.507 g, 6.9 mmol) is used instead of 2,4-dimethoxybenzophenone. The yield of 6 (1.22 g) is 70%. 1H NMR (DMSO-d6, 250 MHz) δ 7.55 (d, J = 1.2 Hz, 1H), 6.83 (d, J = 1.2 Hz, 1H), 3.89 (s, 3H), 3.88 (s, 3H); 13C NMR (DMSO-d6, 62.9 MHz) δ 155.4, 155.0, 132.1, 112.7, 100.5, 98.6, 56.6, 56.5. MALDI-TOF m/z 247.890 [M]+.
2,2′,4,4′-Tetramethoxytrityl Alcohol (9)
Magnesium turnings (0.543 g, 22.3 mmol) and a few crystals of I2 are placed in a 250-mL three-neck round-bottom flask fitted with a dropping funnel and a condenser. A solution of 2,4-dimethoxybromobenzene (5.0 g, 23.0 mmol) in 20 mL of anhydrous THF is added dropwise to the magnesium. The mixture is stirred for 20–30 min. The resulting Grignard reagent (2,4-dimethoxyphenyl-magnesium bromide) is cooled in a dry ice/acetone bath before a solution of methyl benzoate (1.25 g, 9.38 mmol) in 40 mL of dry THF is added dropwise. The mixture is stirred overnight and then quenched with 100 mL of distilled water and neutralized with 2 N HCl. The unreacted 2,4-dimethoxybromobenzene as well as THF are removed by steam distillation. The resulting mixture is extracted with CH2Cl2 (3 × 30 mL). The combined extracts are dried over MgSO4 and filtered, and the filtrate is evaporated to dryness. The residue is purified by flash chromatography (silica gel; EtOAc–hexane, 20:80) to afford 3.49 g (96%) of 9. Compound 9 was prepared previously via other methodology.25 NMR data is in agreement with the assigned structure: 1H NMR (DMSO-d6, 300 MHz) δ 7.21–7.10 (m, 5H), 6.86 (d, J = 8.6 Hz, 2H), 6.52 (d, J = 2.2 Hz, 2H), 6.42 (dd, J = 8.6, 2.2 Hz, 2H), 5.17 (s, 1H), 3.73 (s, 6H), 3.40 (s, 6H); 13C NMR (DMSO-d6, 75.5 MHz) δ 159.7, 157.9, 147.4, 129.2, 127.4, 126.7, 127.9, 104.1, 99.5, 79.1, 55.4, 55.1.
9-Phenyl-6-methoxy-3-fluorone (10)
A solution of 8 (0.178 g, 0.468 mmol) in 10 mL of dry CH2Cl2 is cooled to −78 °C using a dry ice/acetone bath before BBr3 (0.935 g, 3.74 mmol) is added dropwise. The mixture is allowed to warm to room temperature gradually before being quenched with 20 mL of distilled H2O. The mixture is extracted with CH2Cl2 (3 × 10 mL). The combined extracts are dried over MgSO4 and filtered, and the filtrate is concentrated under reduced pressure. The residue is purified by flash chromatography (silica gel, EtOAc), affording 90 mg (64%) of 10. 1H NMR (DMSO-d6, 250 MHz) δ 7.63–7.61 (m, 3H), 7.47–7.43 (m, 2H), 7.21 (d, J = 2.4 Hz, 1H), 7.08 (d, J = 9.0 Hz, 1H), 7.01 (d, J = 9.7 Hz, 1H), 6.94 (dd, J = 9.0, 2.4 Hz, 1H), 6.42 (dd, J = 9.7, 1.8 Hz, 1H), 6.21 (d, J = 1.8 Hz, 1H), 3.90 (s, 3H); 13C NMR (DMSO-d6, 62.9 MHz) δ 183.8, 164.1, 158.4, 154.0, 149.2, 132.4, 130.8, 129.7, 129.6, 129.5, 129.4, 128.8, 117.0, 113.9, 113.6, 104.7, 100.6, 56.3. MALDI-TOF m/z 303.154 [M + H]+.
9-Phenyl-6-hydroxy-3-fluorone (11)
To a stirred solution of 8 (0.400 g, 1.05 mmol) in 10 mL of dry CH2Cl2 at −78 °C, BBr3 (4.20 g, 16.8 mmol) is added dropwise. The mixture is warmed to room temperature gradually before being quenched with 20 mL of distilled H2O. After being stirred for 20 min, filtration leads to a collection of a red precipitate (11). A sample for analytical purposes is obtained by flash chromatography (silica gel, EtOAc–MeOH 9:1). A quantity of 197 mg (65%) of 11 is collected. Compound 11 is known; however, no characterization data was previously reported via the older synthetic methods.261H NMR (DMSO-d6, 250 MHz) δ 7.63–7.60 (m, 3H), 7.46–7.43 (m, 2H), 7.01 (d, J = 9.2 Hz, 2H), 6.60 (dd, J = 9.2, 2.1 Hz, 2H) 6.60 (d, J = 2.1 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 156.3, 149.8, 132.5, 130.5, 129.5, 129.3, 128.8, 114.6, 103.4.
2,2′,4,4′-Tetramethoxy-4″-bromotrityl Alcohol (13a)
This compound was prepared following the protocol described above for compound 9 except that 4-bromo methyl benzoate 12a (2.01 g, 9.38 mmol) was used as the ester. The yield of 13a (3.97 g) is 92%. 1H NMR (DMSO-d6, 300 MHz) δ 7.25 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.42 (d, J = 2.0 Hz, 2H), 6.34 (dd, J = 8.6, 2.0 Hz, 2H), 5.17 (s, 1H), 3.63 (s, 6H), 3.32 (s, 6H); 13C NMR (DMSO-d6, 75.5 MHz) δ 159.9, 157.8, 147.2, 129.7, 129,4, 129.1, 125.9, 118.9, 104.2, 99.4, 78.5, 55.3, 55.1. MALDI-TOF m/z 457.880 [M]+, 441.521 [M − OH]+, 481.532 [M + K]+.
2,2′,4,4′-Tetramethoxy-4″-phenyltrityl Alcohol (13b)
This compound is prepared following the procedure described above for compound 9 except that 4-phenyl methyl benzoate 12b (1.13 g, 5.33 mmol) is used. The yield of 13b (2.43 g) is 99%. 1H NMR (DMSO-d6, 250 MHz) δ 7.64 (d, J = 7.6 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.43 (t, J = 7.8 Hz, 2H), 7.32 (t, J = 7.1 Hz, 1H), 7.19 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H), 6.55 (d, J = 2.3 Hz, 2H), 6.45 (dd, J = 8.6, 2.3 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 159.7, 157.9, 146.7, 140.0, 137.6, 129.2, 128.9, 128.1, 127.1, 126.6, 126.5, 125.0, 104.1, 99.5, 78.9, 55.4, 55.1. MALDI-TOF m/z 439.654 [M − OH]+, 479.599 [M + Na]+.
2,2′,4,4′-Tetramethoxy-4″-methoxytrityl Alcohol (13c)
This compound is prepared following the procedures described above for compound 9 except that 4-methoxy methyl benzoate 12b (1.55 g, 9.38 mmol) is used. The yield of 13c (3.2 g) is 83%. 1H NMR (CDCl3, 300 MHz) δ 7.16 (d, J = 6.9 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 6.9 Hz, 2H), 6.49 (d, J = 2.4 Hz, 2H), 6.37 (dd, J = 8.6, 2.4 Hz, 2H), 3.79 (s, 9H), 3.54 (s, 6H); 13C NMR (CDCl3, 75.5 MHz) δ 160.3, 158.7, 158.3, 139.7, 130.4, 129.1, 127.6, 112.7, 103.8, 100.2, 80.6, 55.9, 55.5, 55.4. MALDI-TOF m/z 409.490 [M]+.
2,2′,4,4′-Tetramethoxy-3″-nitrotrityl Alcohol (13d)
This compound is prepared following the procedure described above for compound 9 except that 3-nitro methyl benzoate 12d (1.70 g, 9.38 mmol) is used. The yield of 13d (3.86 g) is 95%. 1H NMR (DMSO-d6, 300 MHz) δ 8.00–7.98 (m, 2H), 7.55–7.42 (m, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.54 (d, J = 1.9 Hz, 2H), 6.50 (dd, J = 8.5, 1.9 Hz, 2H), 5.59 (s, 1H), 3.75 (s, 6H), 3.39 (s, 6H); 13C NMR (DMSO-d6, 75.5 MHz) δ 160.2, 157.6, 150.3, 146.7, 134.2, 129.1, 127.8, 125.0, 122.0, 120.7, 104.4, 99.4, 78.1, 55.1. MALDI-TOF m/z 424.481 [M]+, 448.565 [M + Na]+, 464.538 [M + K]+.
2,2′,4,4′-Tetramethoxy-4″-nitrotrityl Alcohol (13e)
This compound is prepared following the procedure described above for compound 9 except that 4-nitro methyl benzoate 12e (1.70 g, 9.38 mmol) is used. The yield of 13e (3.51 g) is 91%. 1H NMR (DMSO-d6, 300 MHz) δ 8.04 (d, J = 8.9 Hz, 2H), 7.35 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 6.54 (d, J = 2.4 Hz, 2H), 6.49 (dd, J = 8.4, 2.4 Hz, 2H), 5.53 (s, 1H), 3.75 (s, 6H), 3.40 (s, 6H); 13C NMR (DMSO-d6, 75.5 MHz) δ 160.2, 157.6, 155.9, 145.4, 129.0, 128.6, 124.9, 121.7, 104.4, 99.4, 78.2, 55.1. MALDI-TOF m/z 424.701 [M]+, 408.624 [M − OH]+.
9-(4-Bromophenyl)-6-hydroxy-3-fluorone (14a)
This compound was prepared following the procedure above for compound 11 except that compound 13a (0.400 g, 0.871 mmol) is used. The yield of 14a (223 mg) is 70%. 1H NMR (DMSO-d6, 250 MHz) δ 7.88 (d, J = 7.5 Hz, 2H), 7.47 (d, J = 7.5 Hz, 2H), 7.29 (d, J = 9.4 Hz, 2H), 6.90 (d, J = 2.1 Hz, 2H), 6.88 (dd J = 9.4, 2.1 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 172.1, 158.0, 132.5, 131.9, 131.7, 124.2, 120.9, 115.6, 102.8. MALDI-TOF m/z 367.228 [M + H]+.
9-(4-Biphenyl)-6-hydroxy-3-fluorone (14b)
This compound is prepared following the procedure above for compound 11 except that compound 22 (0.500 g, 1.09 mmol) is used. The yield of 14b (330 mg) is 87%. 1H NMR (DMSO-d6, 250 MHz) δ 7.91 (d, J = 7.8 Hz, 2H), 7.79 (d, J = 7.9 Hz, 2H), 7.55–7.49 (m, 5H), 7.09 (d, J = 9.0 Hz, 2H), 6.59 (dd, J = 9.0, 2.2 Hz, 2H), 6.54 (d, J = 2.2 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 157.2, 150.4, 141.9, 140.1, 132.6, 131.4, 131.0, 130.0, 128.9, 127.8, 127.7, 122.6, 115.0, 104.3. MALDI-TOF m/z 365.312 [M + H]+, 387.322 [M + Na]+.
9-(4-Hydroxyphenyl)-6-hydroxy-3-fluorone (14c)
This compound is prepared following the procedure above for compound 11 except that compound 13c (1.26 g, 3.07 mmol) is used. The yield of 14c (900 mg) is 96%. 1H NMR (CDCl3, 250 MHz) δ 10.07 (s, 1H), 7.25 (d, J = 8.2 Hz, 2H), 7.14 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 8.2 Hz, 2H), 6.59 (d, J = 9.4 Hz, 2H), 6.52 (s, 2H); 13C NMR (CDCl3, 62.9 MHz) δ 158.6, 156.4, 150.4, 131.1, 130.8, 122.6, 115.5, 114.6, 105.5, 103.3. MALDI-TOF m/z 305.127 [M + H]+, 327.100 [M + Na]+, 341.221 [M + K]+.
9-(3-Nitro-phenyl)-6-hydroxy-3-fluorone (14d)
This compound is prepared following the procedure above for compound 11 except that compound 14d (0.480 g, 1.13 mmol) is used. The yield of 14d (274 mg) is 73%. 1H NMR (DMSO-d6, 300 MHz) δ 8.42 (d, J = 7.0 Hz, 1H), 8.26 (s, 1H), 7.89 (m, 2H), 6.96 (d, J = 9.1 Hz, 2H), 6.55 (dd, J = 9.1, 2.9 Hz, 2H), 6.53 (d, J = 2.9 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 156.3, 138.0, 147.0, 136.0, 134.3, 130.5, 130.3, 124.3, 114.3, 103.5. MALDI-TOF m/z 334.186 [M + H]+, 356.167 [M + Na]+.
9-(4-Nitrophenyl)-6-hydroxy-3-fluorone (14e)
This compound was prepared following the procedure above for compound 11 except that compound 15 (30 mg) is used. The eluent for flash chromatography is EtOAc–MeOH 8:2. The yield of 14e (21 mg) is 76%. 1H NMR (DMSO-d6, 250 MHz) δ 8.44 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H), 6.90 (dd, J = 8.9, 1.1 Hz, 2H), 6.54 (d, J = 8.9 Hz, 2H), 6.52 (d, J = 1.1 Hz, 2H); 13C NMR (DMSO-d6, 62.9 MHz) δ 174.8, 156.3, 148.0, 147.3, 139.7, 131.0, 130.1, 123.4, 122.1, 113.5, 103.6. MALDI-TOF m/z 334.273 [M + H]+, 356.301 [M + Na]+.
9-(4-Nitrophenyl)-6-methoxy-3-fluorone (15)
This compound is prepared following the procedure above for compound 11 except that compound 13e (0.500 g, 1.18 mmol) is used. The yield of 15 (346 mg) is 85%. 1H NMR (DMSO-d6, 300 MHz) δ 8.46(d, J = 8.6 Hz, 2H), 7.25 (d, J = 2.3 Hz, 1H), 7.07 (d, J = 9.4 Hz, 1H), 6.98(d, J = 9.8 Hz, 1H), 6.93 (dd, J = 9.4, 2.3 Hz, 1H), 6.44 (dd, J = 9.8, 1.8 Hz, 1H), 6.26 (d, J = 1.8 Hz, 1H); 13C NMR (DMSO-d6, 75.5 MHz) δ 183.8, 164.3, 158.2, 154.0, 148.2, 139.3, 131.1, 130.5, 129.8, 129.5, 127.1, 124.0, 123.2, 117.2, 113.68, 113.4, 105.0, 100.8, 56.4. MALDI-TOF m/z 348.419 [M + H]+.
Bis-(2,4-Dimethoxyphenyl)-4-(2, 4-dimethoxyphenyl)-phenyl Methane (19)
This compound is prepared following the procedure described above for compound 9 except that 18 (1.64 g, 6.26 mmol) is used. The yield of 19 (1.00 g) is 32%. 1H NMR (CDCl3, 300 MHz) δ 7.37 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 4.1 Hz, 1H), 7.05 (d, J = 8.1 Hz, 2H), 6.77 (d, J = 8.3 Hz, 2H), 6.55 (d, J = 4.1 Hz, 1H), 6.53 (s, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.38 (dd, J = 8.3, 2.3 Hz, 2H), 6.05 (s, 1H), 3.83 (s, 3H), 3.78 (s, 9H), 3.71 (s, 6H); 13C NMR (CDCl3, 75.5 MHz) δ 160.2, 159.3, 158.3, 157.6, 142.7, 135.6, 131.4, 130.7, 129.0, 129.0, 127.8, 104.7, 103.7, 99.1, 98.9, 55.9, 55.7, 55.6, 55.4, 41.8. MALDI-TOF m/z, 500.935 [M]+, 523.990 [M + Na]+.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health for funding this research via Grant R01 EB002044. The purchase of the Nonius Kappa CCD diffractometer was made possible by Grant No. LEQSF-(1999–2000)-ESH-TR-13, administered by the Louisiana Board of Regents.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra, Ortep drawings for 1, 2, 4, 5, 6, 9, 13b, 13e, 18, 19, and 20, and CIF files for 1, 2, 4, 5, 6, 9, 13b, 13e, 18, 19, and 20. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Hirano T, Kikichi K, Urano Y, Higuchi T, Nagono T. Angew. Chem., Int. Ed. 2000;39:1052. doi: 10.1002/(sici)1521-3773(20000317)39:6<1052::aid-anie1052>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]; (b) Liu S, Xie Y, Yong G, Dai Y. J. Agric. Food. Chem. 2000;48:5860. doi: 10.1021/jf0001379. [DOI] [PubMed] [Google Scholar]; (c) Li Z, Tang J, Pan J. Analyst. 2001;126:1154. doi: 10.1039/b101698i. [DOI] [PubMed] [Google Scholar]; (d) Fujita Y, Mori I, Yamaguchi T, Hoshino M, Shigemura Y, Yasuyuki, Shimano M. Anal. Sci. 2001;17:853. doi: 10.2116/analsci.17.853. [DOI] [PubMed] [Google Scholar]; (e) Gee KR, Zhou Z-L, Qian W-J, Kennedy R. J. Am. Chem. Soc. 2002;124:776. doi: 10.1021/ja011774y. [DOI] [PubMed] [Google Scholar]; (f) Li Z, You F, Liu Z, Jian T. Talanta. 2004;63:647. doi: 10.1016/j.talanta.2003.12.007. [DOI] [PubMed] [Google Scholar]
- (2).Turchini J. Acta Histochem. 1957;4:15. [PubMed] [Google Scholar]
- (3).U.S. Patent Appl. Publ. CODEN: USXXCO US 2004038306 A1 20040226. 2004:83.
- (4).Stephen AG, Worthy KM, Towler E, Mikovits JA, Sei S, Roberts P, Yang Q, Akee RK, Klausmeyer P, McCloud TG, Henderson L, Rein A, Covell DG, Currens M, Shoemaker RH, Fisher RJ. Biochem. Biophys. Res. Commun. 2002;296:1228. doi: 10.1016/s0006-291x(02)02063-6. [DOI] [PubMed] [Google Scholar]
- (5).Nagano T, Urano Y. Jpn. Kokai Tokyo Koho CODEN: JKXXAF JP 2000321262 A2 20001124. 2000:14. [Google Scholar]
- (6).Blattner JR, He L, Lemasters JJ. Anal. Biochem. 2001;295:220. doi: 10.1006/abio.2001.5219. [DOI] [PubMed] [Google Scholar]
- (7).Mizutani MY, Itai A. J. Med. Chem. 2004;47:4818. doi: 10.1021/jm030605g. [DOI] [PubMed] [Google Scholar]
- (8).Tolman RL, Gamsey S, Mehta S, Pongracz K. Pct Int. Appl. CODEN: PIXXD2 WO 2002076397 A2 20021003. 2002:41. [Google Scholar]
- (9).Yang Y, Lewis PT, Escobedo JO, St. Luce NN, Trealeaven WD, Cooks RL, Strongin RM. Collect. Czech. Chem. Commun. 2004;69:1282. [Google Scholar]
- (10).Wang W, Escobedo JO, Lawrence CM, Strongin RM. J. Am. Chem. Soc. 2004;126:3400. doi: 10.1021/ja0318838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mohlau R, Koch P. Ber. 1894;27:2887. [Google Scholar]
- (12).(a) Swain CJ, Hedberg K. J. Am. Chem. Soc. 1950;72:3373. [Google Scholar]; (b) Tilak BD. Ind. Chim. Belge. 1967;32:50. [Google Scholar]; (c) He M, Johnson RJ, Escobedo JO, Beck PA, Kim KK, St. Luce NN, Davis CJ, Lewis PT, Fronczek FR, Melancon BJ, Mrse AA, Treleaven WD, Strongin RM. J. Am. Chem. Soc. 2002;124:5000. doi: 10.1021/ja017713h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jiao G-S, Castro JC, Thoresen LH, Burgess K. Org. Lett. 2003;5:3675. doi: 10.1021/ol035327u. [DOI] [PubMed] [Google Scholar]
- (13).Shi J, Zhang X, Neckers DC. J. Org. Chem. 1992;57:4418. [Google Scholar]
- (14).Chang CJ, Jaworski J, Nolan EM, Sheng M, Lippard SJ. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1129. doi: 10.1073/pnas.0308079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dinger MB, Scott MJ. J. Chem. Soc., Perkin Trans. 1. 2000;11:1741. [Google Scholar]
- (16).Jpn. Kokai Tokkyo Koho CODEN: JKXXAF JP 07138200 A2 19950530 Heisei. 1995. p. 5. [Google Scholar]
- (17).Heaney H, Hollinshead JH, Ley SV. Tetrahedron. 1995;51:7741. [Google Scholar]
- (18).(a) Weinelt F, Schneider H-J. J. Org. Chem. 1991;56:5527. [Google Scholar]; (b) Fronczek FR, St. Luce NN, Strongin RM. Acta Crystallogr. 2001;C57:1423. doi: 10.1107/s0108270101015621. [DOI] [PubMed] [Google Scholar]
- (19).Detailed synthetic route is described in the Experimental Section.
- (20).Selected references:Brown HC. Organic Synthesis via Boranes. Wiley-Interscience; New York: p. 1975.. Negishi E. In: Comprehensive Organometallic Chemistry. Wilkinson G, Stone FGA, Abel EW, editors. Vol. 5. Pergamon Press; Oxford: 1982. p. 255.. Pelter A, Smith K, Brown HS. Borane Reagents. Academic Press; London: 1988.
- (21).Matteson DS, Mah RWH. J. Am. Chem. Soc. 1963;85:2599. [Google Scholar]
- (22).(a) Kitatani K, Hiyama T, Nozaki H. Bull. Chem. Soc. Jpn. 1977;50 [Google Scholar]; (b) Negishi E, Akiyoshi K. J. Am. Chem. Soc. 1988;110:646. [Google Scholar]; (c) Negishi E, Akiyoshi K, O'Connor B, Takagi K, Wu G. J. Am. Chem. Soc. 1989;111:3089. [Google Scholar]; (d) Kocienski P, Wadman S, Cooper K. J. Am. Chem. Soc. 1989;111:2363. [Google Scholar]; (e) Miller JA. J. Org. Chem. 1989;54:998. [Google Scholar]; (f) Knochel P, Jeong N, Rozema MJ, Yeh MCP. J. Am. Chem. Soc. 1989;111:6474. [Google Scholar]; (g) Stocks M, Kocienski P, Donald KK. Tetrahedron Lett. 1990;31:1637. [Google Scholar]; (h) AchyuthaRao S, Rosema MJ, Knochel P. J. Org. Chem. 1993;58:2694. [Google Scholar]; (i) Kakiya H, Inoue R, Shinokubo H, Oshima K. Tetrahedron Lett. 1997;38:3275. [Google Scholar]; (j) Harada T, Kaneko T, Fujiwara T, Oku A. J. Org. Chem. 1997;62:8966. [Google Scholar]; (k) Fillion E, Carson RJ, Trepanier VE, Goll JM, Remorova AA. J. Am. Chem. Soc. 2004;126:15354. doi: 10.1021/ja045783t. [DOI] [PubMed] [Google Scholar]
- (23).(a) Harada T, Hara D, Hattori K, Oku A. Tetrahedron Lett. 1988;29:3821. [Google Scholar]; (b) Harada T, Katsuhira T, Osada A, Iwazaki K, Maejima R, Oku A. J. Am. Chem. Soc. 1996;118:11377. [Google Scholar]
- (24).(a) Harada T, Kaneko T, Fujiwara T, Oku A. Tetrahedron. 1998;54:9317. [Google Scholar]; (b) Harada T, Chiba M, Oku A. J. Org. Chem. 1999;64:8210. doi: 10.1021/jo990937m. [DOI] [PubMed] [Google Scholar]
- (25).Yonezawa N, Hino T, Matsuda K, Matsuki T, Narushima O, Kobayashi M, Ikeda T. J. Org. Chem. 2000;65:941. doi: 10.1021/jo990524l. [DOI] [PubMed] [Google Scholar]
- (26).Ramart-Lucas P. Bull. Soc. Chim. 1945;12:477. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.