

In a succinct (less than three sides) case report from the Mental Institute of Frankfurt am Main in 1907, Alois Alzheimer described the clinical course and pathological findings of a woman who presented, aged 51 years, with jealousy of her husband. This was followed by progressive memory loss, bewilderment, paranoid ideas (including towards her physician), hallucinations, agitation, disturbances of speech and language, screaming, incontinence, loss of self care and finally apathy. She died after 4.5 years, with bedsores, despite nursing care. Brain sections demonstrated generalized atrophy, neurofibrillary changes (in silver-stained sections) and loss of cortical neurons. The entire cortex showed miliary foci resulting from the deposition of a ‘unique substance’[1].
What is now known as Alzheimer's disease (AD) is the commonest age-related dementia. It is distinct from vascular dementia associated with brain infarction, although it may co-exist with cerebrovascular disease as it did in the index case [1]. Not only does AD horrifyingly deconstruct what it is to be human, it represents a monumental and expanding public health problem as populations age worldwide. The intention of this editors' view is modest: to attempt to set the scene for what pharmacology might have to contribute to addressing this overwhelming need, and why the clinical pharmacology in this area of need is so challenging.
Immediate cause(s) of AD and current treatments
Alzheimer's case report identified correctly the main pathological features of AD. These are amyloid (literally ‘starch-like’– so named for their appearance but now known to be proteinaceous rather than carbohydrate in composition) plaques, neurofibrillary tangles and loss of neurons, especially cholinergic neurons in the basal forebrain (see [2] for a text book account). Amyloid plaques are believed to be the main cause of neuronal death in AD. They are deposits of misfolded protein aggregates and are not as ‘unique’ as Alzheimer supposed since they are also a feature of several other neurodegenerative diseases including Parkinson's disease, Creutzfeldt-Jakob disease, Huntington's disease and amyotrophic lateral sclerosis (motor neuron disease). Amyloid deposits in liver, heart, gastrointestinal and other organs outside the central nervous system occur in the various forms of systemic primary or secondary amyloidosis (often associated with myeloma or with prolonged over-stimulation of the immune system – the horses used to manufacture anti-tetanus toxin tended to die of secondary amyloidosis, and historically paraplegics with chronic unhealed bedsores persisting for many years were at risk of secondary amyloidosis), but such systemic organ deposits are not a feature of neurodegenerative disease of the central nervous system (CNS). (Peripheral neuropathies, including autonomic peripheral neuropathies, are another matter – amyloid deposition in association with systemic amyloidosis is one well-recognized cause). Protein misfolding is promoted by various mutations and early stages may be reversed by molecular chaperones (proteins that assist the non-covalent folding or unfolding and the assembly or disassembly of other macromolecular structures). Misfolded protein, if not repaired or cleared, can form oligomers which, if not dealt with by cellular disposal mechanisms, can form insoluble aggregates within or without neuronal cell bodies that cause neuronal damage through mechanisms that remain incompletely understood [3].
The amyloid plaques of AD consist of aggregates of beta amyloid (Aβ) fragments consisting of 36–43 amino acid residues derived from amyloid precursor protein (APP) which is a normal component of cell membranes of healthy neurons. The gene for APP is located on chromosome 21 and people with trisomy 21 (Down's syndrome) almost always develop AD by the age of 40 years [4, 5]. The main physiological pathway for APP (Figure 1) is via cleavage by α-secretase which releases soluble APP (sAPP) which has several trophic and growth-promoting functions. However, cleavage at different sites by β- or γ-secretase gives rise to various forms of Aβ, especially Aβ40 which is only weakly amyloidogenic. However, various mutations in the gene coding for APP both increase the proportion of APP that is processed by this pathway and also increase the formation of Aβ42 which is more strongly amyloidogenic than Aβ40. Clearance of Aβ is impaired by certain mutations of the apoE4 gene which are epidemiologically associated with AD [6] and mice genetically engineered to over express APP develop neurodegenerative disease with similarities to AD [7–9].
Figure 1.
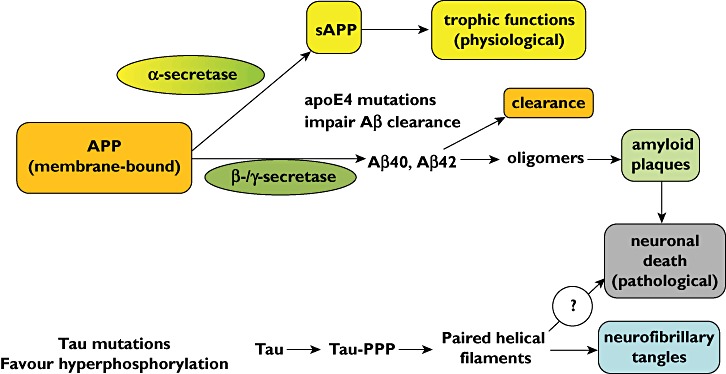
Formation of amyloid plaques in Alzheimer's disease. (APP amyloid precursor protein; sAPP soluble amyloid precursor protein)
Tau proteins [10] stabilize microtubules and are abundant in CNS neurons. When hyperphosphorylated, tau proteins no longer stabilize microtubules properly and are implicated in the other major pathological feature of AD discovered by Alzheimer, neurofibrillary tangles. Hyperphosphylation results in dissociation of tau from microtubules, misfolding and aggregation of paired helical fragments of tau that increase Aβ toxicity. Figure 1 summarizes the pathways implicated in the pathogenesis of AD.
Cholinergic neurons are especially affected in AD. Choline acetyl transferase, acetylcholine, acetylcholinesterase and choline transport are all selectively reduced in the cortex and hippocampus of AD patients compared with the same brain structures from patients with depressive illness or schizophrenia. The reason for this selectivity is as yet unknown [2], but is the rationale for the main group of drugs licensed currently for AD, namely the centrally acting cholinesterase inhibitors donepezil, rivastigmine and galantamine. Regretably, these drugs have only small beneficial effects on cognitive function, reversible on discontinuing the drug [11], as does memantine, a glutamate (NMDA-type) receptor antagonist synthesized by the Eli Lilly company in 1968. Memantine has additional weak actions on various amine receptors and is the only drug other than a cholinesterase inhibitor currently licensed for AD (http://en.wikipedia.org/wiki/Memantine accessed 14th Feb 2012).
Barriers to developing new drugs for AD
Can drug treatment favourably modify the progression of AD? Several rational strategies based on the pathophysiological mechanisms summarized in Figure 1 have been proposed. Several ‘old’ drugs have biochemical activity on processes implicated in the pathogenesis of AD and have been investigated, along with new drugs (including biologicals) specifically targeted at seemingly logical targets, mainly with disappointing results. These include β-secretase inhibition (the anti-diabetic drug rosiglitazone was ineffective), γ-secretase inhibition or modulation, inhibition of Aβ oligomerization, promotion of Aβ clearance, inhibition of tau phosphorylation (the mood stabilizer lithium has some activity against tau phosphorylation in animal models of AD and has been reported to reduce P-tau in the cerebrospinal fluid of patients with amnesic mild cognitive impairment [12]) and inhibition of tau fragment dimerization. None has so far proven its worth in the clinic, although some trials are ongoing.
In this present issue we publish a review of new pharmacological strategies for treatment of AD with a focus on disease modifying drugs [13]. We hope that this will prove inspirational. However, drug development in this field is beset by fearsome difficulties:
limitations of animal models
an unpredictable natural history of the human disease unfolding over years (decades when one considers preclinical progression)
preclinical biomarkers that are potentially crucial yet are incompletely validated
the invasive nature of cerebrospinal fluid (CSF) sampling
the potential for drugs to cause CNS damage (including vasogenic oedema)
ethical challenges in investigating healthy humans (with nothing to gain but potentially severe adverse effects)
ethical challenges in investigating vulnerable AD-patients (albeit not necessarily mentally incompetent)
ethical dilemmas in measuring potential biomarkers for AD
Despite these formidable challenges, in the face of overwhelming human need we hope that the pharmaceutical industry remains engaged in this humanitarian imperative [14], despite the need for new approaches explained by Salomone and his colleagues [13].
REFERENCES
- 1.Alzheimer A. Über eine eigenartige Erkrankung der Himrinde. Allgemeine Zeitschrift Für Psychiatrie Und Physisch-Gerichtliche Medizin 1907. Vol. 64. New York: Hafner Press; 1977. pp. 146–48. translated into English by Hochberg CN and Hochberg FH. In: Neurological Classics in Modern Translation eds Rottenberg DA and Hochberg FH. [Google Scholar]
- 2.Rang HP, Dale MM, Ritter JM, Flower RJ, Henderson G. Rang and Dale's Pharmacology. 7th. Edinburgh: Elsevier Churchill Livingstone; 2012. [Google Scholar]
- 3.Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis. 2007;4:349–65. doi: 10.1159/000105156. [DOI] [PubMed] [Google Scholar]
- 4.Nistor M. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol Aging. 2007;28:1493–506. doi: 10.1016/j.neurobiolaging.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging. 2005;26:383–89. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 7.Games D. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 8.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsiao K. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 10.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGleenon BM, Dynan KB, Passmore AP. Acetylcholinesterase inhibitors in Alzheimer's dosease. Br J Clin Pharmacol. 1999;48:471–80. doi: 10.1046/j.1365-2125.1999.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnesic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;190:351–6. doi: 10.1192/bjp.bp.110.080044. [DOI] [PubMed] [Google Scholar]
- 13.Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological strategies for treatment of Alzheimer's disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012;73:504–17. doi: 10.1111/j.1365-2125.2011.04134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Gerven J, Cohen A. Vanishing clinical psychopharmacology. Br J Clin Pharmacol. 2011;72:1–5. doi: 10.1111/j.1365-2125.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]