

Abstract
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) are established first line treatments for hypercholesterolaemia. In addition to the direct effects of statins in reducing concentrations of atherogenic low density lipoprotein cholesterol (LDL-C), several studies have indicated that the beneficial effects of statins may be due to some of their cholesterol-independent, multiple (pleiotropic) effects which may differ between different members of the class. Pitavastatin is a novel synthetic lipophilic statin that has a number of pharmacodynamic and pharmacokinetic properties distinct from those of other statins, which may underlie its potential pleiotropic benefits in reducing cardiovascular risk factors. This review examines the principal pleiotropic effects of pitavastatin on endothelial function, vascular inflammation, oxidative stress and thrombosis. The article is based on a systematic literature search carried out in December 2010, together with more recent relevant publications where appropriate. The available data from clinical trials and in vitro and animal studies suggest that pitavastatin is not only effective in reducing LDL-C and triglycerides, but also has a range of other effects. These include increasing high density lipoprotein cholesterol, decreasing markers of platelet activation, improving cardiac, renal and endothelial function, and reducing endothelial stress, lipoprotein oxidation and, ultimately, improving the signs and symptoms of atherosclerosis. It is concluded that the diverse pleiotropic actions of pitavastatin may contribute to reducing cardiovascular morbidity and mortality beyond that achieved through LDL-C reduction.
Keywords: dyslipidaemia, hypercholesterolaemia, pitavastatin, pleiotropic effects, statin
Introduction
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) inhibit the biosynthesis of cholesterol. Statins have therefore become established in the treatment of hypercholesterolaemia and attained a central place in cardiovascular medicine because of their proven benefits in both primary and secondary prevention of cardiovascular events (for review see [1]).
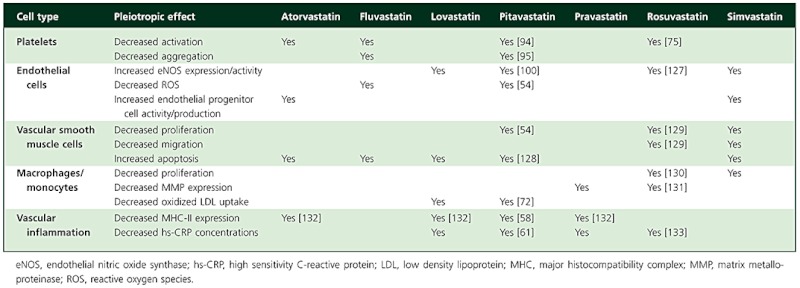
Analyses of major trials, coupled with animal experimentation and studies in vitro, have suggested that some of the benefits of statins may be due to multiple, pleiotropic effects independent of direct reductions in low density lipoprotein cholesterol (LDL-C). Such effects can include improvements in endothelial function, decreases in smooth muscle proliferation and vascular inflammation and antiplatelet actions (Table 1), as well as effects on other lipids and lipoproteins [2–4]. Pleiotropic effects may play an important part in reducing cardiovascular mortality and morbidity [3], and differences in the spectrum of such effects between members of the class could be relevant when formulating management plans for individual patients. Furthermore, different enzymes are involved in the metabolism of different statins, leading to unique drug–drug interaction profiles. Such differences are particularly important for patients at high risk of developing atherosclerosis, who are frequently polymedicated.
Table 1.
Pleiotropic effects of statins. Adapted from Sadowitz et al. [2]
 |
In an era in which clinicians are increasingly aware of the need to tailor therapeutic approaches to individual patients [5], insight into the potential benefits of statins on factors other than LDL-C reduction is particularly important. Recent research, for example, has highlighted the potential of novel statin derivatives to improve the anti-inflammatory [6] and antithrombotic [7] effects of available statins.
Pitavastatin is a novel synthetic lipophilic statin shown to reduce serum concentrations of LDL-C and triglycerides, and increase high density lipoprotein cholesterol (HDL-C), in dyslipidaemic patients [8, 9]. At doses of 2–4 mg daily, pitavastatin has been shown to have a similar effect on LDL-C as atorvastatin 10–20 mg [10], with greater effects on HDL-C [11, 12].
This paper reviews the main pleiotropic effects of pitavastatin on endothelial function, vascular inflammation, oxidative stress and thrombosis. Together with the direct effects of pitavastatin on serum LDL-C concentrations, pleiotropic effects not directly related to cholesterol synthesis may help to explain the unique pharmacological profile of this statin.
Literature search
Publications included in this review were based mainly on a search of the PubMed and EMBASE databases carried out in December 2010. The strategy used was to search for English language publications on pitavastatin in ‘all fields’ on PubMed or in the ‘title/abstract’ field in EMBASE. This global search yielded a total of 756 articles (PubMed 329, EMBASE 427). After exclusion of foreign language references and duplicate entries, a total of 431 non-duplicate individual references were screened for relevance to the pleiotropic effects of pitavastatin. Screening removed all conference abstracts, review articles or correspondence (editorials and letters) and articles not relevant to the pleiotropic effects of pitavastatin, yielding a total of 134 relevant articles for further assessment. Relevant papers published after December 2010 were also included as appropriate. References were then grouped under category headings (endothelial function, inflammation and immunomodulation, oxidative stress and lipoprotein oxidation, effects of pitavastatin on lipids other than LDL-C, thrombosis and fibrinolysis, cardiovascular and organ-protective effects).
Statin pharmacokinetics and metabolism
Statins may be broadly divided into two categories, according to their solubility (Figure 1). Pravastatin and rosuvastatin possess polar side groups attached to hydrophobic ring structures, rendering them hydrophilic, while pitavastatin, atorvastatin, fluvastatin, lovastatin and simvastatin do not, and are consequently classed as lipophilic [2]. Furthermore, the differing chemical structures of individual statins substantially alter the way these molecules are taken up by the liver and catabolized or excreted. Lovastatin and simvastatin, for example, circulate as an inactive, lactone prodrug [13], while atorvastatin and rosuvastatin are biologically active as calcium salts [14, 15]. Rates of biliary excretion, involving a number of transporters, such as breast cancer resistance protein and multidrug resistance-associated protein 2 [16], and hepatic uptake by various transporting polypeptides, are likely to influence the observed pharmacokinetic differences between statins [17].
Figure 1.
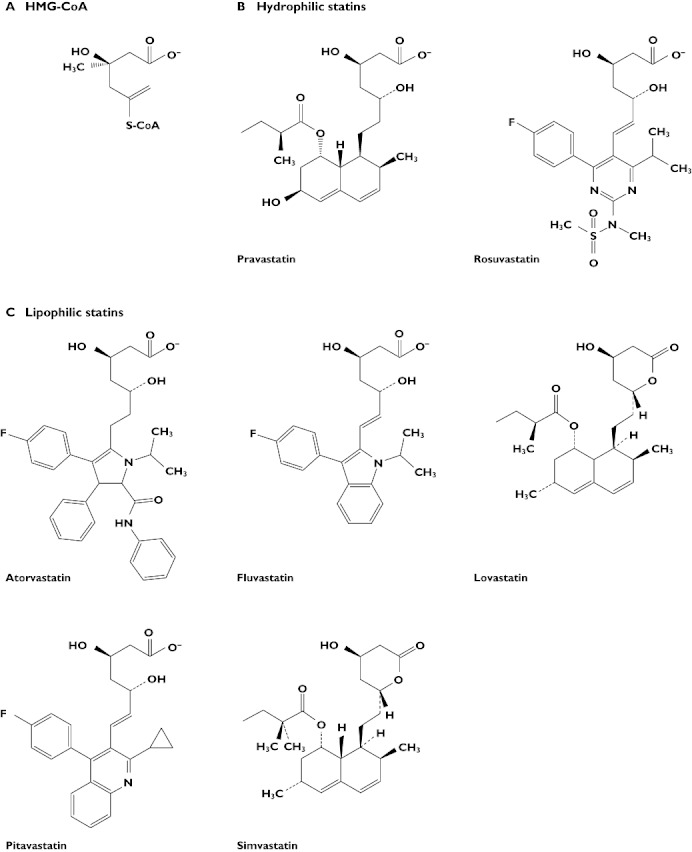
Molecular structures of (A) HMG-CoA and (B) hydrophilic and (C) lipophilic currently available statins. Adapted from Shitara et al. [126]
Hepatic uptake of pitavastatin is mediated by organic anion transporting protein 2 (OATP2 or OATP1B1) [18]. The potential impact of OATPs on statin pharmacokinetics is illustrated by the findings that variants of OATP1B1 alleles are associated with decreased hepatic uptake of pitavastatin [19] and that OAT1B1 388A>G polymorphisms are associated with alterations in pitavastatin disposition and pharmacokinetics [20]. Indeed, a number of factors can influence hepatic uptake of statins, including substrate–transporter affinity, itself highly dependent on the chemical structure of individual statins, the degree of dose dependency between substrate and transporter, the presence of genetic variation in the transporter and competition among transporters. The effect of genetic variation of transporters on the cellular accumulation of statins was demonstrated recently in a comparison of six statins and two transporters [21]. In oocytes, genetic variation in OATP1B1 (OATP1B1*15) reduced cellular uptake of pravastatin, pitavastatin, rosuvastatin and atorvastatin, but not of fluvastatin or simvastatin, whereas variation in the Na+/taurocholate cotransporting polypeptide (NTCP*2) reduced transport of atorvastatin and rosuvastatin but not of pitavastatin.
Pitavastatin has a number of pharmacodynamic and pharmacokinetic properties that are distinct from those of other statins, and may contribute to (or modulate) its lipid lowering and pleiotropic properties. In contrast to other lipophilic statins, pitavastatin undergoes limited metabolism by, and consequently has a limited risk of interaction with drugs metabolized by, cytochrome P450 (CYP) enzymes, particularly CYP3A4 [22, 23].
Statin safety and tolerability
Although the benefits of statins have been shown to outweigh substantially their potential negative effects [24], co-administration of drugs that can interact with the CYP enzymes responsible for metabolizing statins can markedly increase the risk of developing myotoxicity due to reduced statin clearance [25, 26]. Cerivastatin was withdrawn after it emerged that patients exhibited an unacceptably high incidence of rhabdomyolysis, a condition in which severe muscle degradation may lead to kidney function impairment [27].
Almost all statins undergo extensive hepatic metabolism by the CYP isoenzyme systems; lovastatin, simvastatin and atorvastatin are all catabolized primarily by CYP3A4 [17]. The exception is pravastatin, the major metabolites of which are produced by chemical degradation in the stomach [28]. Co-administration of other drugs that interact with CYP3A4, such as the calcium channel antagonist mibefradil or lipid lowering drugs such as gemfibrozil, can substantially increase the risk of patients experiencing severe adverse events [17].
Although statins such as pitavastatin, fluvastatin and rosuvastatin, which are not extensively metabolized by CYP3A4, may be co-administered with such drugs, these statins have their own unique drug–drug interaction profiles. Fluvastatin and rosuvastatin are primarily metabolized by CYP2C9, which interacts with drugs such as diclofenac and phenytoin [17], while pitavastatin is only marginally metabolized by CYP isoenzymes [26].
Although all statins, to a greater or lesser degree, have the potential to cause adverse events if administered in sufficiently high doses, or in combination with other medications that alter their pharmacokinetics [29], multiple long-term safety studies have shown that these drugs are well tolerated by the majority of patients. For example, comparisons between pitavastatin and atorvastatin [11, 12, 30], pravastatin [31], rosuvastatin [31] and simvastatin [32] have shown that these statins have similarly low treatment emergent adverse event profiles when administered in isolation. Furthermore, a 2 year prospective study of over 20 000 patients treated with pitavastatin revealed no unexpected negative side effects of treatment [33], confirming the general long-term safety and tolerability of statins, as observed for atorvastatin [34], simvastatin [34] and rosuvastatin [35] administered for extended periods in previous studies.
Mechanisms of atherosclerosis: the significance of pleiotropic effects of statins
Atherosclerosis is the result of a complex interaction between dyslipidaemia, lipoprotein oxidation, blood flow, endothelial dysfunction, vascular inflammation and platelet activation and aggregation. Although the vascular endothelium is the major regulator of vascular homeostasis and endothelial dysfunction is recognized as an early marker of atherosclerosis, the molecular mechanisms underlying these interactions have only recently begun to emerge. Nitric oxide (NO), a potent vasodilator, is central to the regulation and maintenance of vascular function and is known to inhibit atherogenic LDL oxidation [36]. In addition, hyperlipidaemia is associated with endothelial disturbance, migration of inflammatory cells to sites of endothelial injury and a prolonged inflammatory response [37]. Endothelial damage also promotes platelet aggregation and activation, leading to thrombosis [36].
The pleiotropic effects of statins may act at multiple points in the complex cascade of events leading to atherosclerosis. Decreases in mortality and morbidity seen in the major trials of statins may therefore reflect multi-faceted actions on the atherosclerotic process. Figure 2 illustrates the cellular mechanisms by which statin-induced reductions in serum mevalonate, the direct reaction product of HMG-CoA reductase and HMG-CoA, may influence vascular function. Reductions in protein prenylation (the addition of an isoprene chain such as farnesol, 15 carbon atoms, or geranylgeraniol, 20 carbon atoms) have diverse, multiple effects on atherosclerotic processes including vasodilation, oxidative stress, inflammation and thrombosis. The evidence for the effects of statins, particularly pitavastatin, on reducing the pathophysiological processes that lead to atherosclerosis is described below.
Figure 2.
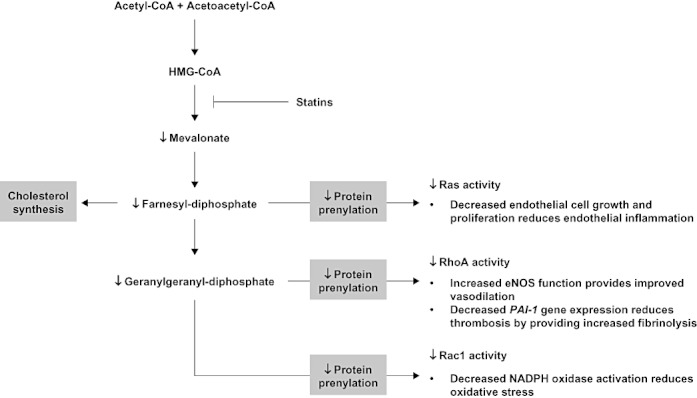
Isoprene synthesis, separate from cholesterol synthesis, modulates cellular function by activating GTPases and G-proteins, such as Ras, Rac1 and RhoA, via addition of an isoprene chain (farnesol or geranylgeraniol). Statin-induced decreases in HMG-CoA reductase activity reduce downstream protein prenylation and have the potential to improve vascular function through multiple pathways. GTP, guanosine triphosphate; PAI-1, plasminogen activator inhibitor-1; Ras, rat sarcoma subfamily; Rac1, Ras-related C3 botulinum toxin substrate 1; RhoA, Ras homologue gene family, A
Endothelial function
Many statin-induced improvements in endothelial function are mediated by transcriptional control of multiple genes, including integrin β4 and thrombomodulin, which affect cell–cell and cell–extracellular matrix interactions and reduce the procoagulant effect of thrombin [38, 39]. Statins have further been shown to promote endothelial progenitor cell proliferation and to modulate the induction of endothelial NO synthase (eNOS), thereby increasing NO production [2].
Animal models have shown that pitavastatin decreases the progression of atherosclerosis, augmenting capillary formation and blood flow recovery and increasing eNOS expression via the Akt (also known as protein kinase B, a serine/threonine-specific protein kinase family) pathway after induction of hind limb ischaemia [40, 41]. Statin therapy also reduces vasoconstriction by inhibiting NADPH oxidase activity and eNOS-dependent production of uncoupled superoxide anion (O2−•) in a rat model of insulin resistance [42]. Furthermore, an in vitro study in human umbilical vein endothelial cells, has shown that pitavastatin increases eNOS production [43, 44] and increases the migration and proliferation of endothelial cells [45].
The cellular mechanisms underlying improvements in endothelial function, and how these interact with the mevalonate pathway downstream of HMG-CoA reductase, have recently begun to emerge. Angiogenesis in response to pitavastatin therapy in a murine hind limb ischaemia model was shown to be mediated by Notch-1, a protein regulating the interactions between adjacent cells [46]. This study further demonstrated that angiogenesis was not dependent on vascular endothelial growth factor, suggesting that growth of new blood vessels was not responsible for the observed recovery of blood flow. Pitavastatin treatment further induced endothelial ephrinB2, a selective marker of neovascularization sites on endothelial and smooth muscle cells, downstream of Notch-1, increasing the density of both capillaries and arterioles in the ischaemic limbs of control mice, while animals without Notch-1 were unaffected [46]. Furthermore, in moderately hypercholesterolaemic rabbits, pitavastatin was found to suppress atherosclerosis via inhibition of macrophage accumulation and foam cell formation [47].
The effects of statins on endothelial cells are associated with significant reductions in coronary artery disease (CAD), cerebrovascular disease and peripheral artery disease [3], and improvements in markers of endothelial function are observed during clinical use of pitavastatin. Fasting and postprandial forearm blood flow increased significantly (P < 0.05) during post ischaemic reactive hyperaemia in patients with CAD following 6 months of treatment with pitavastatin, but not in controls (Figure 3) [48]. Vasodilatation of the brachial artery was also increased after short term (2 weeks) treatment with pitavastatin in patients with primary hypercholesterolaemia. This increase was significantly greater in patients treated with pitavastatin (n = 37) than in those treated with atorvastatin (n = 34) after only 2 weeks of treatment (P < 0.05) and remained higher, although not significantly, in patients treated with pitavastatin for 3 months [30]. Furthermore, improvements in endothelium-dependent flow-mediated vasodilatation have been shown following pitavastatin treatment in people who smoke (Figure 4), an effect likely to reflect protection of endothelial cells against oxidative stress [49].
Figure 3.
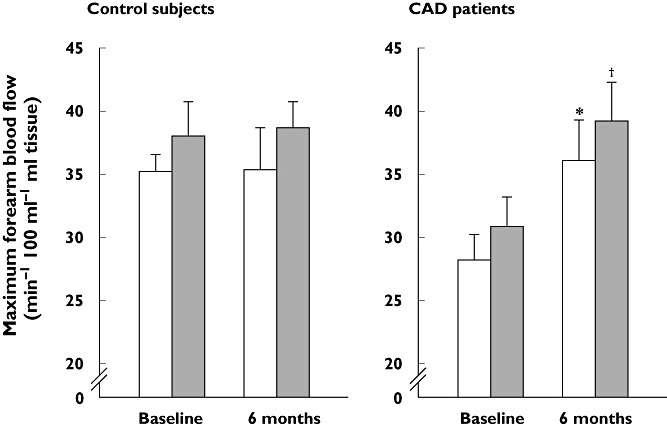
Effects of pitavastatin on forearm blood flow during reactive hyperaemia in patients with coronary artery disease and controls after 6 months' treatment. Blood flow was measured using strain-gauge plethysmography directly before and 2 h after, patients consumed a modified standard test meal (Japan Diabetes Society) after an overnight fast. *P < 0.05 vs. baseline preprandial data, †P < 0.05 vs. baseline postprandial data. Reproduced with permission from Arao et al. [48]. CAD, coronary artery disease. Before meal (□); After meal ( )
)
Figure 4.
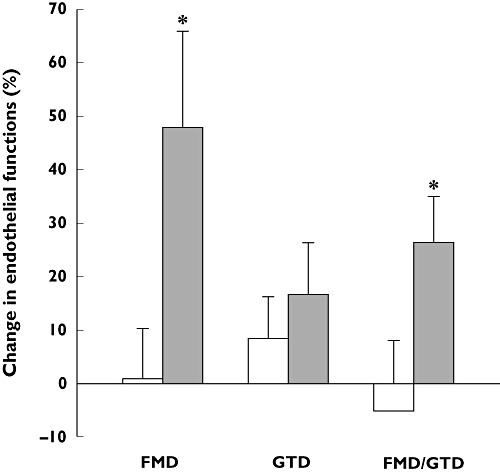
Increase in endothelial-dependent vasodilation following 1 month of treatment with 2 mg pitavastatin daily in chronic smokers. Endothelial function was assessed noninvasively, using high-resolution ultrasound, by measuring endothelium-dependent and -independent dilation of the brachial artery by reactive hyperaemia and glycerol trinitrate, respectively. *P < 0.05 vs. patients not treated with pitavastatin. Reproduced with permission from Yoshida et al. [49]. FMD, endothelium-dependent flow-mediated dilatation; GTD, endotheliumindependent glycerol trinitrate-induced vasodilatation. Controls (□); Pitavastatin ( )
)
Inflammation and immunomodulation
Vascular inflammation is known to play a central role in the development of atherosclerosis [50, 51]. Statins exert a variety of anti-inflammatory effects, including a decrease in T-cell recruitment and activation, reduction in expression of inflammatory cytokines and chemokines, attenuation of expression of cluster of differentiation 40 (CD40, a member of the tumour necrosis factor [TNF] family of proteins) in T-cells and lymphocytes and inhibition of smooth muscle cell proliferation [2].
By inhibiting mevalonate synthesis, statins can further modulate vascular inflammatory responses by reducing leucocyte migration via a mechanism involving the monocyte chemoattractant protein-1 (MCP-1) [52]. Resistance to plaque formation has been demonstrated in mice deficient in MCP-1, or its receptor (C-C motif chemokine receptor 2 or CCR2) [52]. Similarly, pitavastatin has been shown to have effects on inflammation and immunomodulation that may be mediated, in part, by dose-dependent inhibition of monocyte proliferation through down-regulation of the chemokine receptors CCR2 and CCR5 [53], inhibition of lysophosphatidic acid-induced aortic smooth muscle cell proliferation and expression of MCP-1 [54]. Such effects are mediated by suppression of Rac-1 (rat sarcoma [Ras]-related C3 botulinum toxin substrate 1)-induced generation of reactive oxygen species [54], a finding of interest as statins have been shown to down-regulate chemokine receptors in patients with CAD [55]. In retinal ganglion cells, pitavastatin suppresses N-methyl-d-aspartic acid-induced leucocyte recruitment and up-regulation of endothelial adhesion molecules [56]. In a rat model of cerebral aneurysm, pitavastatin decreased expression of nuclear factor (NF)-κB and other pro-inflammatory mediators, effects that were associated with reduced progression of aneurysms [57]. Other anti-inflammatory and anti-stress responses may be modulated by corticotropin-releasing factor type 1 receptors (CRF-1) via mechanisms including CRF signalling, endogenous CRF receptor agonists and urocortin and its associated polypeptides [58].
In terms of its immunomodulatory effects, pitavastatin has been shown to inhibit the production of interferon-α and TNF-α by human plasmacytoid dendritic cells, which are key molecular and cellular pathogenic components in autoimmune diseases. Specifically, statins have suppressive effects on the multiple signal transduction pathways that mediate interferon responses [59].
In addition to the in vitro demonstration of the anti-inflammatory effects of pitavastatin, there is now evidence, as for other statins, of anti-inflammatory effects in humans. Elevated concentrations of high sensitivity C-reactive protein (hs-CRP), a member of the pentraxin family and an inflammatory marker, are associated with high cardiovascular risk [60], and decreased concentrations of hs-CRP have been found in pitavastatin-treated patients with diabetes [61]. Plasma hs-CRP concentrations decreased significantly from a median value of 0.49 mg l−1 at baseline (interquartile range, 0.26–0.87) to 0.37 mg l−1 (0.23–0.79) at 6 months (P < 0.05), an effect that was independent of changes in serum lipids [61]. Furthermore, plasma concentrations of another pentraxin (PTX-3), also a marker of vascular inflammation and atherosclerosis, were reduced after pitavastatin treatment in patients with hypercholesterolaemia [62]. Decreases in PTX-3 concentrations during 6 months of treatment with pitavastatin correlated with decreases in plaque severity score in the carotid artery. This was particularly the case in patients who had high PTX-3 concentrations at baseline, indicating an effect of pitavastatin on asymptomatic atherosclerosis in these patients.
Oxidative stress and lipoprotein oxidation
Oxidative stress
Oxidative stress plays an important role in plaque formation and may be a strong predictor of atherosclerosis, via mechanisms involving oxidized lipoproteins that can trigger inflammation and disrupt normal vascular function [63]. Recent data suggest that leucocyte activation by triglyceride-rich apolipoprotein B (ApoB)-containing lipoproteins can lead to oxidative stress, the production of cytokines and, ultimately, acute endothelial dysfunction. ApoB-containing lipoproteins, however, may also bind to erythrocytes, which may potentially have an anti-atherogenic effect by removing lipoproteins from sites of vascular inflammation [64].
Pitavastatin has been shown to reduce oxidative stress in hypercholesterolaemic rabbits [65] and to reduce elevated levels of reactive oxygen species and NADPH oxidase activity in genetically obese rats [66]. In addition, a reduction in angiotensin II-induced atrial remodelling can be attributed to decreased atrial production of O2−• in pitavastatin-treated mice [67]. Pitavastatin also increases the plasma concentrations of urocortin, which is produced in endothelial cells and inhibits the formation of reactive oxygen species (Figure 5) [68]. This pleiotropic effect, which has not been reported for other statins, is of interest because urocortin is a potent vasodilator that may lower blood pressure and increase K+ATP channel gene expression. Urocortin may also have inotropic and cardio-protective effects in ischaemia–reperfusion injury and beneficial cardiovascular effects in heart failure in humans [69].
Figure 5.
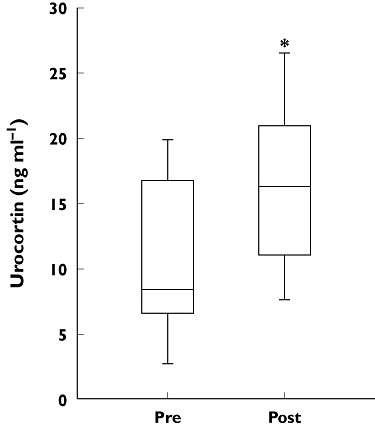
Treatment with pitavastatin (2 mg day−1) for 4 weeks increased serum urocortin concentrations, measured using an enzyme-linked immunosorbent assay, in 15 healthy male volunteers. *P < 0.01 vs. before pitavastatin treatment. Reproduced with permission from Honjo et al. [68]
Lipoprotein oxidation
Statins have been shown to reduce the pro-atherogenic effects of oxidized LDL [2]. Specifically, statin-induced activation of the paraoxonase 1 (PON1) gene (mediated by p44/42 mitogen-activated protein kinase signalling) results in inhibition of LDL oxidation [70, 71], and pitavastatin inhibits the uptake of oxidized LDL by macrophages by down-regulating expression of the type B scavenger receptor CD36 [72]. Pitavastatin reduced lectin-like oxidized LDL receptor 1 (LOX-1) in patients with hypercholesterolaemia, but this was not correlated with a reduction in LDL-C [73]. In another study, decreased measures of oxidative stress and lower cardio-ankle vascular index were seen in patients with type 2 diabetes treated with pitavastatin (Figure 6) [74]. These results are clinically important, as oxidative stress is a key pathogenic factor in diabetes. Patients with diabetes often have dysfunctional HDL, and anti-inflammatory, anti-oxidant and cholesterol efflux mechanisms are impaired [75, 76].
Figure 6.
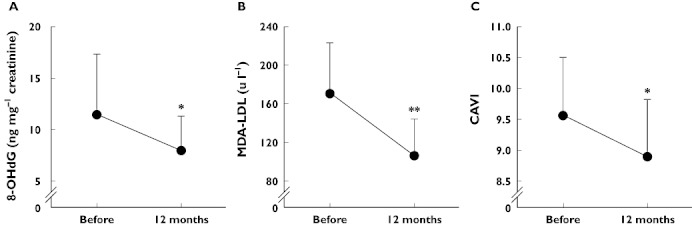
Changes in (A) urinary 8-hydroxy-2′-deoxyguanosine (a marker of oxidative stress), (B) malondialdehyde-LDL (a form of oxidized LDL) and (C) cardio-ankle vascular index in 45 patients with type 2 diabetes mellitus treated with pitavastatin 2 mg day−1 for 12 months (mean ± SD). Urinary 8-hydroxy-2′-deoxyguanosine and malondialdehyde-LDL were measured using enzyme-linked immunosorbent assays. The results for the former were adjusted for serum creatinine concentrations. The cardio-ankle vascular index reflects the stiffness of the aorta and femoral and tibial arteries, independent of blood pressure, and was measured using blood pressure cuffs attached to the biceps and ankles with patients in a supine position. *P < 0.05; **P < 0.01 vs. before treatment, paired t-test. Adapted from Miyashita et al. [74]. 8-OHdG, 8-hydroxy-2′-deoxyguanosine; CAVI, cardio-ankle vascular index; MDA-LDL, malondialdehyde-low-density lipoprotein
Effects of statins on lipids other than LDL-C
The effects of statins on lipids beyond those exerted via LDL-C reduction may play a substantial role in mediating their cardiovascular benefits. The release of pro-inflammatory free cholesterol of erythrocyte membrane (CEM) from red blood cells in atherosclerotic plaques during intraplaque haemorrhage or neoangiogenesis has also been identified as a risk factor for CAD, and a marker of plaque vulnerability, which is independent of serum LDL-C concentrations. A study of 212 patients with acute coronary syndromes undergoing assessment for angina pectoris showed a substantial reduction in CEM in patients receiving statin therapy, which was not associated with changes in serum concentrations of LDL-C, HDL-C or triglycerides [77]. A retrospective database analysis of 105 patients with type III dysbetalipoproteinaemia, a remnant removal disease, has also shown statin-induced regression of lipid deposits in the skin (palmar crease xanthoma) [78].
HDL-C
The anti-inflammatory effects of pitavastatin result in increased expression of macrophage scavenger receptor class B type 1, which plays an important role in bidirectional cholesterol interchange between cells and HDL-C [79]. Pitavastatin also induces endothelial sphingosine-1-phosphate type 1 receptors, which are believed to mediate vascular responses to HDL-C [80].
Differences between statins in their effects on HDL-C may be clinically relevant because of the many anti-atherogenic properties of this lipoprotein fraction. Greater increases in HDL-C with pitavastatin than with atorvastatin were seen in Japanese patients with impaired glucose tolerance and elevated LDL-C [11]. Other statins have less consistent effects on HDL-C [81], which may be related to different mechanisms of action. It has been proposed that statins increase HDL-C by inhibiting cholesteryl ester transfer protein and stimulating ApoAI synthesis [81], potentially via a mechanism mediated by RhoA suppression [82, 83]. There is also evidence that pitavastatin may increase HDL-C by increasing expression of ApoAI and ATP-binding cassette transporter A1, key components of reverse cholesterol transport [84–86].
Studies of the effects of statins on dysfunctional HDL species would be of interest because, currently, only niacin [76] and vitamin E in patients with diabetes with the haptoglobulin 2–2 genotype [87] are known to have beneficial effects on HDL dysfunction. Although statins do affect many of the protein components transported by HDL, such as PON1 which is increased by statin treatment [88] and reduced in patients with congestive heart failure [89], there is, to our knowledge, no study that addresses the effect of pitavastatin treatment on serum PON1 concentrations in humans directly, rather than in cultured cells.
Apolipoproteins
Decreased secretion of ApoB100 from hepatoma cells has been reported in the presence of pitavastatin [90], and pitavastatin was more potent than simvastatin or atorvastatin in inducing ApoAI secretion [82]. There was also a correlation between decreases in ApoB with pitavastatin and a reduction in small, dense LDL-C [91]. Statin therapy has further been shown to reduce postprandial lipaemia, an independent risk factor for CAD, by inhibiting the production of ApoB-containing lipoproteins, thereby increasing the clearance of triglyceride-rich lipoproteins produced in the liver and intestines [92].
Thrombosis and fibrinolysis
Thrombosis associated with unstable plaques represents a serious risk for individuals with CAD, and statins are now well established as part of the pharmacological approach to reducing the risk of cardiovascular events. Their pleiotropic effects include decreasing platelet activation and aggregation [3] and increasing the expression of multiple genes involved in coagulation and fibrinolysis [39, 93].
Pitavastatin has been shown to decrease markers of platelet activation (platelet-derived microparticles), cell adhesion molecules and chemokines (MCP-1) through a mechanism that appears to involve increasing adiponectin concentrations [94, 95]. Pitavastatin also induces endothelial expression of thrombomodulin via inhibition of Rho family G proteins, resulting in an antithrombotic effect (Figure 2) [96]. In addition, pitavastatin reduces the expression of tissue factor (platelet tissue factor or coagulation factor III) and plasminogen activator inhibitor-1, and increases expression of tissue-type plasminogen activator, thereby promoting a balance in favour of fibrinolysis and reducing the risk of thrombus formation at unstable atherosclerotic plaques [97].
Cardiovascular and organ-protective effects
Heart
In an animal model of hypertensive heart failure, pitavastatin inhibits load-induced cardiac hypertrophy and fibrosis through inhibition of RhoA-ERK-serum response factor signalling [98, 99]. Pitavastatin reduces remodelling and improves ventricular function in rat hearts through increased eNOS production associated with PI3K signalling and a decrease in oxidative stress [100]. In the absence of eNOS in a mouse model, pitavastatin also reduced cardiac remodelling induced by angiotensin II and renal insufficiency through inhibition of the transforming growth factor-β-Smad signalling pathway by suppression of oxidative stress [67]. Similar results were obtained by Takuwa et al. [101], who showed that pitavastatin reduces cardiac remodelling, by inhibition of sphingosine kinase 1, and reduces oxidative stress in juvenile mice. Furthermore, in a 12 month randomized, controlled trial in 30 patients with hypercholesterolaemia and preserved left ventricular ejection fraction, treatment with pitavastatin (1 or 2 mg day−1, n = 15) for 1 year significantly reduced carotid arterial stiffness and significantly improved regional left ventricular systolic and diastolic function (P < 0.05) compared with patients given placebo (Figure 7) [102].
Figure 7.
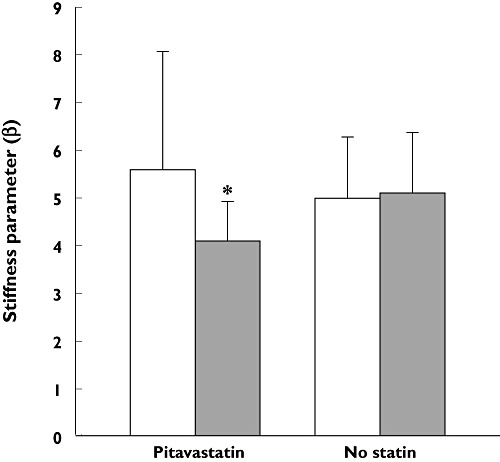
Changes in carotid arterial stiffness in 30 patients with hypercholesterolaemia randomized to receive either pitavastatin (1–2 mg day−1, n = 15) or diet and exercise therapy for 12 months. Carotid ultrasonography was used to reveal end-systolic and end-diastolic diameters of the common carotid artery, Ds and Dd, respectively, and arterial stiffness, β, was calculated using the formula ln(SBP/DBP)/[(Ds – Dd)/Dd], where SBP and DBP are the systolic and diastolic blood pressures, respectively. *P < 0.05 vs. no statin group after treatment. Data from Mizuguchi et al. [102]. Baseline (□); 12 months ( )
)
Brain
Pitavastatin reduces ischaemia-induced neuronal damage in animal models [103, 104]. In the middle cerebral artery occlusion model in the rat, both pitavastatin and simvastatin significantly reduced infarction volume relative to that observed in control animals (P < 0.05 and P < 0.01, respectively), while the reduction attributable to atorvastatin did not reach significance [105]. More recently, a decrease in ischaemia-induced neuronal cell death after treatment with pitavastatin was found to be associated with down-regulation of expression of NF-κB in gerbils [106], and it has been suggested from a study in rats that pitavastatin may strengthen the integrity of the blood–brain barrier through up-regulation of claudin 5 (a functional protein at endothelial tight junctions) [107].
Statins have been shown to augment cerebral blood flow and to confer significant protection against stroke in animal models (for review see [108]). This protective effect in animal models has been replicated in published case studies: two elderly patients (72 and 77 years of age) showed clinically significant increases in cerebral blood flow following 6 months of treatment with pitavastatin 2 mg day−1[109].
Kidney
Pitavastatin improves renal function in diabetic mice, increasing the bioavailability of nitric oxide via a mechanism which suppresses eNOS uncoupling, the process by which eNOS activity is inhibited by an oxidative stress-induced deficiency in the NOS cofactor tetrahydrobiopterin (BH4) [110]. In clinical studies, 6 months of treatment with pitavastatin 1 mg day−1 significantly (P < 0.01) reduced urinary liver-type fatty acid-binding protein (a marker of renal injury), concentrations of 8-hydroxy-2′-deoxyguanosine (a marker of oxidative stress), and proteinuria in 30 non-diabetic patients with chronic kidney disease, independently of any lipid-lowering effects [111]. The beneficial effect on proteinuria was enhanced when co-administered with ezetimibe (Figure 8) [112]. In a 2 year surveillance study, estimated glomerular filtration rate increased significantly (P < 0.001) from baseline (< 60 ml min−1 1.73 m−2) in 958 patients with hypercholesterolaemia and chronic kidney disease [113].
Figure 8.
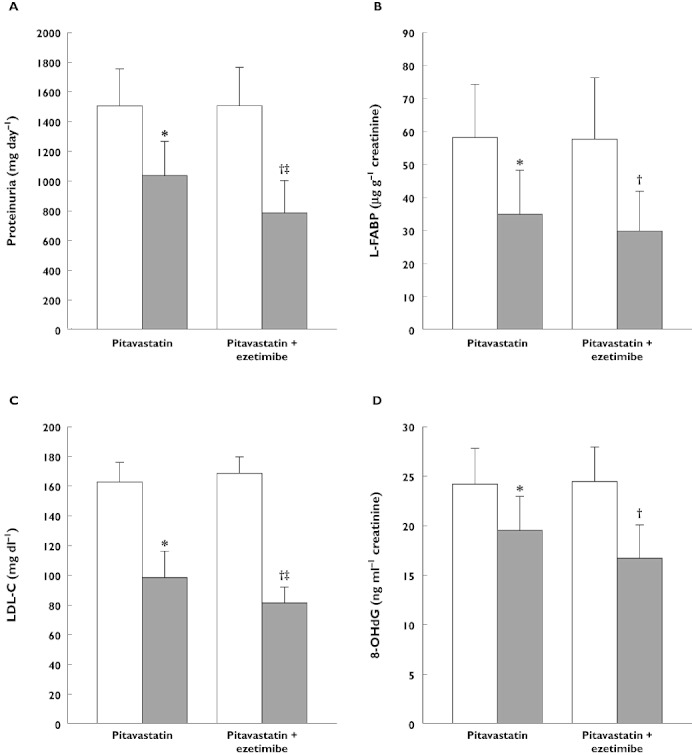
Improvements in markers of (A and B) renal function, (C) serum cholesterol and (D) oxidative stress in patients with chronic kidney disease and dyslipidaemia after treatment with 2 mg pitavastatin daily alone, or in combination with 10 mg ezetimibe, for 6 months. 8-hydroxy-2′-deoxyguanosine and L-fatty acid binding protein were measured enzymatically using enzyme-linked immunosorbent assays, proteinuria was measured using a pyrogallol red method and LDL-C concentrations were calculated using Friedewald's formula. *P < 0.001 vs. before pitavastatin treatment, †P < 0.001 vs. before pitavastatin + ezetimibe treatment, ‡P < 0.05 vs. after pitavastatin treatment alone. Data from Nakamura et al. [112]. Before (□); After ( ). 8-OHdG,8-hydroxy-2′-deoxyguanosine; LDL-C, low density lipoprotein cholesterol; L-FABP, L-fatty acid binding protein
). 8-OHdG,8-hydroxy-2′-deoxyguanosine; LDL-C, low density lipoprotein cholesterol; L-FABP, L-fatty acid binding protein
Clinical evidence for pleiotropic effects
The potential importance of the pleiotropic effects of pitavastatin is highlighted by the results of clinical trials in patients with atherosclerotic disease.
Stabilization of vulnerable carotid plaques was seen within 1 month of treatment with pitavastatin 4 mg day−1 in 33 patients with acute coronary syndrome in a placebo-controlled trial (Figure 9) [114]. In the JAPAN-ACS (Japan assessment of pitavastatin and atorvastatin in acute coronary syndrome) study, a significant (P < 0.001) reduction in coronary plaque volume was seen in 111 patients given pitavastatin 4 mg day−1 for 8–12 months. The decrease in plaque volume with pitavastatin was similar to that seen in 115 patients treated with atorvastatin 20 mg day−1 (−16.9 ± 13.9% vs.−18.1 ± 14.2%) [115]. Stabilization of coronary plaques, determined by colour angioscopy, occurred following treatment with pitavastatin 2 mg day−1 for 52 weeks in patients with CAD. This stabilization was not correlated with changes in LDL-C, suggesting a direct contribution of pleiotropic effects unrelated to cholesterol reduction (Figure 10) [116]. Such angioscopic regression has also been reported with atorvastatin in a previous report but, in contrast to the pitavastatin study, a correlation was observed between changes in the mean angioscopic (yellow) score for plaque stabilization and the change in LDL-C concentrations [117]. Furthermore, in another study, reductions in angioscopic score were correlated with subsequent changes in atheroma volume, measured by intravascular ultrasound, considered to reflect an improvement in plaque vulnerability [118].
Figure 9.
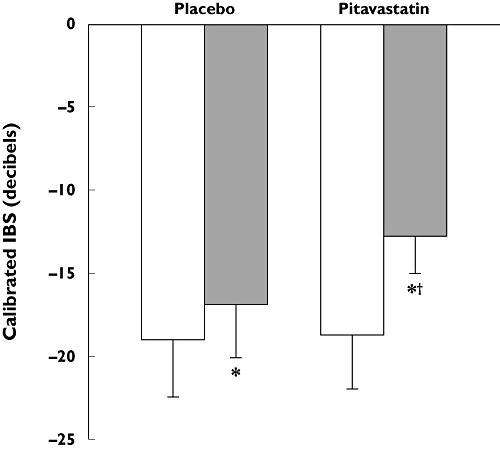
Stabilization of carotid plaques, measured as the change in integrated backscatter (IBS) on carotid ultrasound, following treatment with pitavastatin 4 mg day−1 for 1 month in patients with acute coronary syndromes (mean ± SD). *P < 0.05 vs. baseline within treatment group; †P < 0.05 vs. placebo at 1 month. Data from Nakamura et al. [114]. Baseline (□); 1 month ( )
)
Figure 10.
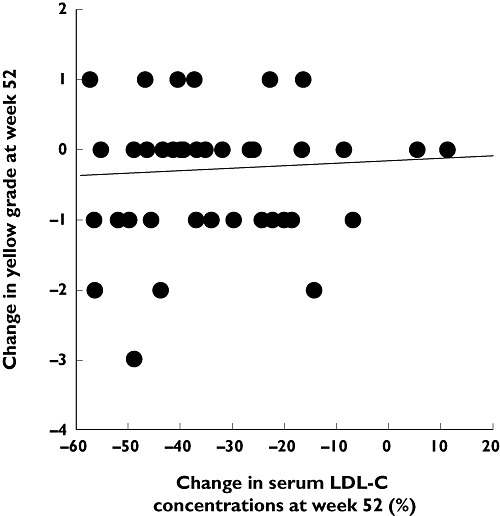
Lack of correlation between stabilization of coronary plaques (expressed as reduction in yellow plaque content) and change in LDL-C in patients with coronary artery disease treated with pitavastatin 2 mg day−1 for 52 weeks. Intravascular ultrasound and coronary angiography were used to grade yellow atherosclerotic plaques and to assess their regression during treatment. Reproduced with permission from Kodama et al. [116]. LDL-C, low density lipoprotein cholesterol
How changes in these secondary markers of cardiovascular disease correlate with hard clinical outcomes, however, remains unclear. Few studies investigate such correlations, and those that do are unable to disentangle clinical improvements due to pleiotropic effects from those attributable to improvements in serum lipid profiles [119]. Furthermore, patient mortality appears to correlate most significantly with concentrations of serum lipids, such as increased LDL-C [120] or apolipoproteins, such as ApoAI and ApoB [121]. One recent retrospective analysis of 743 patients receiving percutaneous coronary intervention, or angioplasty, revealed a significant reduction in major adverse coronary events, including myocardial infarction and sudden cardiac death, in patients treated with adjunctive statin therapy compared with no statin [122]. Further analysis showed that the incidence of coronary events correlated with reductions in LDL-C and increases in HDL-C. Statin therapy also appears to be significantly more effective in reducing the risk of experiencing a major coronary event in patients who display the atherogenic triad (elevated LDL-C and triglycerides, and low HDL-C) than in those presenting with hypercholesterolaemia alone (relative risk [95% CI]; 0.47 [0.32–0.70]vs. 0.91 [0.59–1.41], respectively, P = 0.03) [123].
Nevertheless, long-term follow-up studies are clearly necessary to elucidate precisely how the pleiotropic effects exerted by statins might influence mortality and morbidity in patients at high risk of developing atherosclerosis. Recently, the JUPITER trial has indicated that the incidence of cardiovascular events in apparently healthy subjects with elevated concentrations of the inflammatory marker hs-CRP can be reduced with statin therapy [124]. This suggests not only that inflammation plays an important role in mediating the incidence of cardiovascular events, but also that this effect may be independent of changes in serum lipids, as decreases in hs-CRP did not correlate with reductions in LDL-C. The current consensus appears to be that the anti-atherosclerotic effects of statins are likely to be attributable both to their lipid-lowering action (inhibition of HMG-CoA reductase) and to their wide range of additional pleiotropic effects on pro-atherogenic processes, such as correction of endothelial dysfunction, reduction of LDL oxidation and oxidative stress, and decreased vascular inflammation [125].
Conclusions
Many patients do not achieve their lipid targets on current treatment regimens, and discontinuing or modifying statin therapy based on lipid targets alone potentially ignores the potential valuable cardio- and vasculo-protective pleiotropic properties of these compounds and the benefits they may provide to patients in addition to their ability to modulate serum lipid profiles. Statins provide potent lowering of LDL-C and triglycerides, and elevation of HDL-C, together with diverse pleiotropic effects on pro-atherogenic processes, which are likely to contribute to reductions in cardiovascular mortality and morbidity. Differences in pleiotropic effect profiles between statins, such as the up-regulation of urocortin seen with pitavastatin, are likely to become increasingly important in choosing the most appropriate treatment for individual patients.
Statins also differ in the extent to which they interact with drugs metabolized by cytochrome P450. This is important because combination therapy with statins and other agents is likely to become increasingly common, both to achieve lipid targets and to treat comorbidities. Pitavastatin, which has a low risk of interactions with drugs metabolized by cytochrome P450, therefore offers a useful addition in the management of dyslipidaemia and cardiovascular disease.
Acknowledgments
The author takes full responsibility for the content of the manuscript and thanks Dr David Campbell and Dr Nick Leach (Oxford Pharmagenesis™ Ltd) for medical writing support, editorial assistance and collation and incorporation of comments from the author.
Competing Interests
Professor Jean Davignon, OC, GOQ, MD, MSc, DHC, FRCP(C), FACP, FACN, FAHA, FRSC is or has been a consultant or scientific advisor for Abbott/Solvay, Anthera, Acasti Pharma, Amgen, AstraZeneca, Genzyme, Kowa, McCain, Merck, Pfizer, Pharmena (Cortria) and Roche. He has received research support from AstraZeneca, Merck and Pfizer Canada and has participated in clinical trials for Pfizer, Sanofi-Aventis, Merck, Cortria and Genzyme. The preparation of this manuscript was funded by Kowa Research Europe Ltd.
REFERENCES
- 1.Dembowski E, Davidson MH. A review of lipid management in primary and secondary prevention. J Cardiopulm Rehabil Prev. 2009;29:2–12. doi: 10.1097/HCR.0b013e318192754e. [DOI] [PubMed] [Google Scholar]
- 2.Sadowitz B, Maier KG, Gahtan V. Basic science review: statin therapy – Part I: the pleiotropic effects of statins in cardiovascular disease. Vasc Endovascular Surg. 2010;44:241–51. doi: 10.1177/1538574410362922. [DOI] [PubMed] [Google Scholar]
- 3.Sadowitz B, Seymour K, Costanza MJ, Gahtan V. Basic science review section: statin therapy – Part II: clinical considerations for cardiovascular disease. Vasc Endovascular Surg. 2010;44:421–33. doi: 10.1177/1538574410363833. [DOI] [PubMed] [Google Scholar]
- 4.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–9. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hoqail IA. Personalized medicine in psoriasis: concept and applications. Curr Vasc Pharmacol. 2010;8:432–6. doi: 10.2174/157016110791112223. [DOI] [PubMed] [Google Scholar]
- 6.Ongini E, Impagnatiello F, Bonazzi A, Guzzetta M, Govoni M, Monopoli A, Del Soldato P, Ignarro LJ. Nitric oxide (NO)-releasing statin derivatives, a class of drugs showing enhanced antiproliferative and antiinflammatory properties. Proc Natl Acad Sci U S A. 2004;101:8497–502. doi: 10.1073/pnas.0401996101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Momi S, Impagnatiello F, Guzzetta M, Caracchini R, Guglielmini G, Olivieri R, Monopoli A, Gresele P. NCX 6560, a nitric oxide-releasing derivative of atorvastatin, inhibits cholesterol biosynthesis and shows anti-inflammatory and anti-thrombotic properties. Eur J Pharmacol. 2007;570:115–24. doi: 10.1016/j.ejphar.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi T, Yokote K, Saito Y, Iguchi A. Pitavastatin: efficacy and safety in intensive lipid lowering. Expert Opin Pharmacother. 2007;8:2315–27. doi: 10.1517/14656566.8.14.2315. [DOI] [PubMed] [Google Scholar]
- 9.Saito Y. Critical appraisal of the role of pitavastatin in treating dyslipidemias and achieving lipid goals. Vasc Health Risk Manag. 2009;5:921–36. doi: 10.2147/vhrm.s5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Budinski D, Arneson V, Hounslow N, Gratsiansky N. Pitavastatin compared with atorvastatin in primary hypercholesterolemia or combined dyslipidemia. Clin Lipidol. 2009;4:291–302. [Google Scholar]
- 11.Sasaki J, Ikeda Y, Kuribayashi T, Kajiwara K, Biro S, Yamamoto K, Ageta M, Kobori S, Saikawa T, Otonari T, Kono S. A 52-week, randomized, open-label, parallel-group comparison of the tolerability and effects of pitavastatin and atorvastatin on high-density lipoprotein cholesterol levels and glucose metabolism in Japanese patients with elevated levels of low-density lipoprotein cholesterol and glucose intolerance. Clin Ther. 2008;30:1089–101. doi: 10.1016/j.clinthera.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Yokote K, Bujo H, Hanaoka H, Shinomiya M, Mikami K, Miyashita Y, Nishikawa T, Kodama T, Tada N, Saito Y. Multicenter collaborative randomized parallel group comparative study of pitavastatin and atorvastatin in Japanese hypercholesterolemic patients: collaborative study on hypercholesterolemia drug intervention and their benefits for atherosclerosis prevention (CHIBA study) Atherosclerosis. 2008;201:345–52. doi: 10.1016/j.atherosclerosis.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Pentikainen PJ, Saraheimo M, Schwartz JI, Amin RD, Schwartz MS, Brunner-Ferber F, Rogers JD. Comparative pharmacokinetics of lovastatin, simvastatin and pravastatin in humans. J Clin Pharmacol. 1992;32:136–40. doi: 10.1002/j.1552-4604.1992.tb03818.x. [DOI] [PubMed] [Google Scholar]
- 14.Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42:1141–60. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- 15.White CM. A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin. J Clin Pharmacol. 2002;42:963–70. [PubMed] [Google Scholar]
- 16.Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol. 2005;68:800–7. doi: 10.1124/mol.105.014019. [DOI] [PubMed] [Google Scholar]
- 17.Corsini A, Bellosta S. Drug-drug interaction with statins. Expert Rev Clin Pharmacol. 2008;1:105–13. doi: 10.1586/17512433.1.1.105. [DOI] [PubMed] [Google Scholar]
- 18.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311:139–46. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 19.Chung JY, Cho JY, Yu KS, Kim JR, Oh DS, Jung HR, Lim KS, Moon KH, Shin SG, Jang IJ. Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2005;78:342–50. doi: 10.1016/j.clpt.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Wen J, Xiong Y. OATP1B1 388A > G polymorphism and pharmacokinetics of pitavastatin in Chinese healthy volunteers. J Clin Pharm Ther. 2010;35:99–104. doi: 10.1111/j.1365-2710.2009.01071.x. [DOI] [PubMed] [Google Scholar]
- 21.Choi MK, Shin HJ, Choi YL, Deng JW, Shin JG, Song IS. Differential effect of genetic variants of Na(+)-taurocholate co-transporting polypeptide (NTCP) and organic anion-transporting polypeptide 1B1 (OATP1B1) on the uptake of HMG-CoA reductase inhibitors. Xenobiotica. 2011;41:24–34. doi: 10.3109/00498254.2010.523736. [DOI] [PubMed] [Google Scholar]
- 22.Fujino H, Yamade Y, Kojima J, Hirano M, Matsumoto H, Yoneda M. Studies on the metabolic fate of NK-105 a new inhibitor of HMG-CoA reductase: in vitro metabolism and plasma protein binding in animals and humans. Xenobiotica. 1999;14:415–24. [Google Scholar]
- 23.Sakaeda T, Fujino H, Komoto C, Kakumoto M, Jin JS, Iwaki K, Nishiguchi K, Nakamura T, Okamura N, Okumura K. Effects of acid and lactone forms of eight HMG-CoA reductase inhibitors on CYP-mediated metabolism and MDR1-mediated transport. Pharm Res. 2006;23:506–12. doi: 10.1007/s11095-005-9371-5. [DOI] [PubMed] [Google Scholar]
- 24.Pasternak RC, Smith SC, Jr, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol. 2002;40:567–72. doi: 10.1016/s0735-1097(02)02030-2. [DOI] [PubMed] [Google Scholar]
- 25.Cziraky MJ, Willey VJ, McKenney JM, Kamat SA, Fisher MD, Guyton JR, Jacobson TA, Davidson MH. Statin safety: an assessment using an administrative claims database. Am J Cardiol. 2006;97:61C–68C. doi: 10.1016/j.amjcard.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Corsini A, Ceska R. Drug-drug interactions with statins: will pitavastatin overcome the statins' Achilles' heel? [Review] Curr Med Res Opin. 2011;27:1551–62. doi: 10.1185/03007995.2011.589433. [DOI] [PubMed] [Google Scholar]
- 27.Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346:539–40. doi: 10.1056/NEJM200202143460721. [DOI] [PubMed] [Google Scholar]
- 28.Hatanaka T. Clinical pharmacokinetics of pravastatin: mechanisms of pharmacokinetic events. Clin Pharmacokinet. 2000;39:397–412. doi: 10.2165/00003088-200039060-00002. [DOI] [PubMed] [Google Scholar]
- 29.Mastaglia FL. Iatrogenic myopathies. Curr Opin Neurol. 2010;23:445–9. doi: 10.1097/WCO.0b013e32833c2054. [DOI] [PubMed] [Google Scholar]
- 30.Sakabe K, Fukuda N, Fukuda Y, Wakayama K, Nada T, Morishita S, Shinohara H, Tamura Y. Comparisons of short- and intermediate-term effects of pitavastatin versus atorvastatin on lipid profiles, fibrinolytic parameter, and endothelial function. Int J Cardiol. 2008;125:136–8. doi: 10.1016/j.ijcard.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 31.Saku K, Zhang B, Noda K. Randomized head-to-head comparison of pitavastatin, atorvastatin, and rosuvastatin for safety and efficacy (quantity and quality of LDL): the PATROL trial. Circ J. 2011;75:1493–505. doi: 10.1253/circj.cj-10-1281. [DOI] [PubMed] [Google Scholar]
- 32.Ose L, Budinski D, Hounslow N, Arneson V. Comparison of pitavastatin with simvastatin in primary hypercholesterolaemia or combined dyslipidaemia. Curr Med Res Opin. 2009;25:2755–64. doi: 10.1185/03007990903290886. [DOI] [PubMed] [Google Scholar]
- 33.Yokote K, Shimano H, Urashima M, Teramoto T. Efficacy and safety of pitavastatin in Japanese patients with hypercholesterolemia: LIVES study and subanalysis. Expert Rev Cardiovasc Ther. 2011;9:555–62. doi: 10.1586/erc.11.47. [DOI] [PubMed] [Google Scholar]
- 34.Pedersen TR, Cater NB, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, Larsen ML, Lindahl C, Szarek M. Comparison of atorvastatin 80 mg/day versus simvastatin 20 to 40 mg/day on frequency of cardiovascular events late (five years) after acute myocardial infarction (from the Incremental Decrease in End Points through Aggressive Lipid Lowering [IDEAL] trial) Am J Cardiol. 2010;106:354–9. doi: 10.1016/j.amjcard.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 35.Stein EA, Amerena J, Ballantyne CM, Brice E, Farnier M, Guthrie RM, Harats D, Ma PT, Le Maulf F, Melezinkova H, Gold A, Sager P. Long-term efficacy and safety of rosuvastatin 40 mg in patients with severe hypercholesterolemia. Am J Cardiol. 2007;100:1387–96. doi: 10.1016/j.amjcard.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 36.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23 Suppl 1):III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 37.Bai X, Wang X, Xu Q. Endothelial damage and stem cell repair in atherosclerosis. Vascul Pharmacol. 2010;52:224–9. doi: 10.1016/j.vph.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 38.Morikawa S, Takabe W, Mataki C, Wada Y, Izumi A, Saito Y, Hamakubo T, Kodama T. Global analysis of RNA expression profile in human vascular cells treated with statins. J Atheroscler Thromb. 2004;11:62–72. doi: 10.5551/jat.11.62. [DOI] [PubMed] [Google Scholar]
- 39.Morikawa S, Takabe W, Mataki C, Kanke T, Itoh T, Wada Y, Izumi A, Saito Y, Hamakubo T, Kodama T. The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC, human umbilical vein endothelial cells. J Atheroscler Thromb. 2002;9:178–83. doi: 10.5551/jat.9.178. [DOI] [PubMed] [Google Scholar]
- 40.Sata M, Nishimatsu H, Osuga J, Tanaka K, Ishizaka N, Ishibashi S, Hirata Y, Nagai R. Statins augment collateral growth in response to ischemia but they do not promote cancer and atherosclerosis. Hypertension. 2004;43:1214–20. doi: 10.1161/01.HYP.0000126186.29571.41. [DOI] [PubMed] [Google Scholar]
- 41.Kubo M, Egashira K, Inoue T, Koga J, Oda S, Chen L, Nakano K, Matoba T, Kawashima Y, Hara K, Tsujimoto H, Sueishi K, Tominaga R, Sunagawa K. Therapeutic neovascularization by nanotechnology-mediated cell-selective delivery of pitavastatin into the vascular endothelium. Arterioscler Thromb Vasc Biol. 2009;29:796–801. doi: 10.1161/ATVBAHA.108.182584. [DOI] [PubMed] [Google Scholar]
- 42.Shinozaki K, Nishio Y, Ayajiki K, Yoshida Y, Masada M, Kashiwagi A, Okamura T. Pitavastatin restores vascular dysfunction in insulin-resistant state by inhibiting NAD(P)H oxidase activity and uncoupled endothelial nitric oxide synthase-dependent superoxide production. J Cardiovasc Pharmacol. 2007;49:122–30. doi: 10.1097/FJC.0b013e31802f5895. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Tokoro T, Matsui K, Higa S, Kitajima I. Pitavastatin at low dose activates endothelial nitric oxide synthase through PI3K-AKT pathway in endothelial cells. Life Sci. 2005;76:2257–68. doi: 10.1016/j.lfs.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Xu Z, Kitajima I, Wang Z. Effects of different statins on endothelial nitric oxide synthase and AKT phosphorylation in endothelial cells. Int J Cardiol. 2008;127:33–9. doi: 10.1016/j.ijcard.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 45.Katsumoto M, Shingu T, Kuwashima R, Nakata A, Nomura S, Chayama K. Biphasic effect of HMG-CoA reductase inhibitor, pitavastatin, on vascular endothelial cells and angiogenesis. Circ J. 2005;69:1547–55. doi: 10.1253/circj.69.1547. [DOI] [PubMed] [Google Scholar]
- 46.Kikuchi R, Takeshita K, Uchida Y, Kondo M, Cheng XW, Nakayama T, Yamamoto K, Matsushita T, Liao JK, Murohara T. Pitavastatin-induced angiogenesis and arteriogenesis is mediated by Notch1 in a murine hindlimb ischemia model without induction of VEGF. Lab Invest. 2011;91:691–703. doi: 10.1038/labinvest.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitahara M, Kanaki T, Ishii I, Saito Y. Atherosclerosis induced by chronic inhibition of the synthesis of nitric oxide in moderately hypercholesterolaemic rabbits is suppressed by pitavastatin. Br J Pharmacol. 2010;159:1418–28. doi: 10.1111/j.1476-5381.2009.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arao K, Yasu T, Umemoto T, Jinbo S, Ikeda N, Ueda S, Kawakami M, Momomura S. Effects of pitavastatin on fasting and postprandial endothelial function and blood rheology in patients with stable coronary artery disease. Circ J. 2009;73:1523–30. doi: 10.1253/circj.cj-08-0917. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida O, Kondo T, Kureishi-Bando Y, Sugiura T, Maeda K, Okumura K, Murohara T. Pitavastatin, an HMG-CoA reductase inhibitor, ameliorates endothelial function in chronic smokers. Circ J. 2010;74:195–202. doi: 10.1253/circj.cj-09-0345. [DOI] [PubMed] [Google Scholar]
- 50.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–38. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Romano M, Diomede L, Sironi M, Massimiliano L, Sottocorno M, Polentarutti N, Guglielmotti A, Albani D, Bruno A, Fruscella P, Salmona M, Vecchi A, Pinza M, Mantovani A. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab Invest. 2000;80:1095–100. doi: 10.1038/labinvest.3780115. [DOI] [PubMed] [Google Scholar]
- 53.Fujino M, Miura S, Matsuo Y, Tanigawa H, Kawamura A, Saku K. Pitavastatin-induced downregulation of CCR2 and CCR5 in monocytes is associated with the arrest of cell-cycle in S phase. Atherosclerosis. 2006;187:301–8. doi: 10.1016/j.atherosclerosis.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Kaneyuki U, Ueda S, Yamagishi S, Kato S, Fujimura T, Shibata R, Hayashida A, Yoshimura J, Kojiro M, Oshima K, Okuda S. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vascul Pharmacol. 2007;46:286–92. doi: 10.1016/j.vph.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 55.Waehre T, Damas JK, Gullestad L, Holm AM, Pedersen TR, Arnesen KE, Torsvik H, Froland SS, Semb AG, Aukrust P. Hydroxymethylglutaryl coenzyme a reductase inhibitors down-regulate chemokines and chemokine receptors in patients with coronary artery disease. J Am Coll Cardiol. 2003;41:1460–7. doi: 10.1016/s0735-1097(03)00263-8. [DOI] [PubMed] [Google Scholar]
- 56.Nakazawa T, Takahashi H, Nishijima K, Shimura M, Fuse N, Tamai M, Hafezi-Moghadam A, Nishida K. Pitavastatin prevents NMDA-induced retinal ganglion cell death by suppressing leukocyte recruitment. J Neurochem. 2007;100:1018–31. doi: 10.1111/j.1471-4159.2006.04274.x. [DOI] [PubMed] [Google Scholar]
- 57.Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses formation and progression of cerebral aneurysms through inhibition of the nuclear factor kappaB pathway. Neurosurgery. 2009;64:357–65. doi: 10.1227/01.NEU.0000336764.92606.1D. discussion 365–6. [DOI] [PubMed] [Google Scholar]
- 58.Inada Y, Ikeda K, Tojo K, Sakamoto M, Takada Y, Tajima N. Possible involvement of corticotropin-releasing factor receptor signaling on vascular inflammation. Peptides. 2009;30:365–72. doi: 10.1016/j.peptides.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 59.Amuro H, Ito T, Miyamoto R, Sugimoto H, Torii Y, Son Y, Nakamichi N, Yamazaki C, Hoshino K, Kaisho T, Ozaki Y, Inaba M, Amakawa R, Fukuhara S. Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, function as inhibitors of cellular and molecular components involved in type I interferon production. Arthritis Rheum. 2010;62:2073–85. doi: 10.1002/art.27478. [DOI] [PubMed] [Google Scholar]
- 60.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–43. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 61.Motomura T, Okamoto M, Kitamura T, Yamamoto H, Otsuki M, Asanuma N, Takagi M, Kurebayashi S, Hashimoto K, Sumitani S, Saito H, Kouhara H, Nshii K, Nakao M, Koga M, Sato B, Morimoto Y, Kasayama S. Effects of pitavastatin on serum lipids and high sensitivity C-reactive protein in type 2 diabetic patients. J Atheroscler Thromb. 2009;16:546–52. doi: 10.5551/jat.992. [DOI] [PubMed] [Google Scholar]
- 62.Ohbayashi H, Miyazawa C, Miyamoto K, Sagara M, Yamashita T, Onda R. Pitavastatin improves plasma pentraxin 3 and arterial stiffness in atherosclerotic patients with hypercholesterolemia. J Atheroscler Thromb. 2009;16:490–500. doi: 10.5551/jat.no613. [DOI] [PubMed] [Google Scholar]
- 63.Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411–8. doi: 10.1253/circj.cj-08-1102. [DOI] [PubMed] [Google Scholar]
- 64.Bovenberg SA, Alipour A, Elte JW, Rietveld AP, Janssen JW, van de Geijn GJ, Njo TN, van Mechelen R, Hervas SM, Cabezas MC. Cell-mediated lipoprotein transport: a novel anti-atherogenic concept. Atheroscler Suppl. 2010;11:25–9. doi: 10.1016/j.atherosclerosissup.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Umeji K, Umemoto S, Itoh S, Tanaka M, Kawahara S, Fukai T, Matsuzaki M. Comparative effects of pitavastatin and probucol on oxidative stress, Cu/Zn superoxide dismutase, PPAR-gamma, and aortic stiffness in hypercholesterolemia. Am J Physiol Heart Circ Physiol. 2006;291:H2522–32. doi: 10.1152/ajpheart.01198.2005. [DOI] [PubMed] [Google Scholar]
- 66.Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007;148:160–5. doi: 10.1210/en.2006-1132. [DOI] [PubMed] [Google Scholar]
- 67.Yagi S, Akaike M, Aihara K, Ishikawa K, Iwase T, Ikeda Y, Soeki T, Yoshida S, Sumitomo-Ueda Y, Matsumoto T, Sata M. Endothelial nitric oxide synthase-independent protective action of statin against angiotensin II-induced atrial remodeling via reduced oxidant injury. Hypertension. 2010;55:918–23. doi: 10.1161/HYPERTENSIONAHA.109.146076. [DOI] [PubMed] [Google Scholar]
- 68.Honjo T, Inoue N, Shiraki R, Kobayashi S, Otsui K, Takahashi M, Hirata K, Kawashima S, Yokozaki H, Yokoyama M. Endothelial urocortin has potent antioxidative properties and is upregulated by inflammatory cytokines and pitavastatin. J Vasc Res. 2006;43:131–8. doi: 10.1159/000090132. [DOI] [PubMed] [Google Scholar]
- 69.Zaiou M, Benachour H, Marteau JB, Visvikis-Siest S, Siest G. Genomics and the prospects of existing and emerging therapeutics for cardiovascular diseases. Curr Pharm Des. 2009;15:3193–206. doi: 10.2174/138161209789058011. [DOI] [PubMed] [Google Scholar]
- 70.Arii K, Suehiro T, Ota K, Ikeda Y, Kumon Y, Osaki F, Inoue M, Inada S, Ogami N, Takata H, Hashimoto K, Terada Y. Pitavastatin induces PON1 expression through p44/42 mitogen-activated protein kinase signaling cascade in Huh7 cells. Atherosclerosis. 2009;202:439–45. doi: 10.1016/j.atherosclerosis.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 71.Ota K, Suehiro T, Arii K, Ikeda Y, Kumon Y, Osaki F, Hashimoto K. Effect of pitavastatin on transactivation of human serum paraoxonase 1 gene. Metabolism. 2005;54:142–50. doi: 10.1016/j.metabol.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 72.Han J, Zhou X, Yokoyama T, Hajjar DP, Gotto AM, Jr, Nicholson AC. Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation. 2004;109:790–6. doi: 10.1161/01.CIR.0000112576.40815.13. [DOI] [PubMed] [Google Scholar]
- 73.Matsumoto T, Fujita M, Sawamura T, Kakino A, Sato Y, Fujita Y, Matsuda H, Nakanishi M, Uchida K, Nakae I, Kanda H, Yoshida A, Miwa K, Hayashi H, Mitsunami K, Horie M. Pitavastatin reduces lectin-like oxidized low-density lipoprotein receptor-1 ligands in hypercholesterolemic humans. Lipids. 2010;45:329–35. doi: 10.1007/s11745-010-3402-7. [DOI] [PubMed] [Google Scholar]
- 74.Miyashita Y, Endo K, Saiki A, Ban N, Yamaguchi T, Kawana H, Nagayama D, Ohira M, Oyama T, Shirai K. Effects of pitavastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, on cardio-ankle vascular index in type 2 diabetic patients. J Atheroscler Thromb. 2009;16:539–45. doi: 10.5551/jat.281. [DOI] [PubMed] [Google Scholar]
- 75.Cohen RA, Tong X. Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease. J Cardiovasc Pharmacol. 2010;55:308–16. doi: 10.1097/fjc.0b013e3181d89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, von Eckardstein A, Drexler H, Landmesser U. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation. 2010;121:110–22. doi: 10.1161/CIRCULATIONAHA.108.836346. [DOI] [PubMed] [Google Scholar]
- 77.Tziakas DN, Chalikias GK, Stakos D, Tentes IK, Thomaidi A, Chatzikyriakou S, Mitrousi K, Kortsaris AX, Kaski JC, Boudoulas H, Konstantinides S. Statin use is associated with a significant reduction in cholesterol content of erythrocyte membranes. A novel pleiotropic effect? Cardiovasc Drugs Ther. 2009;23:471–80. doi: 10.1007/s10557-009-6202-7. [DOI] [PubMed] [Google Scholar]
- 78.Blom DJ, Byrnes P, Jones S, Marais AD. Dysbetalipoproteinaemia – clinical and pathophysiological features. S Afr Med J. 2002;92:892–7. [PubMed] [Google Scholar]
- 79.Han J, Parsons M, Zhou X, Nicholson AC, Gotto AM, Jr, Hajjar DP. Functional interplay between the macrophage scavenger receptor class B type I and pitavastatin (NK-104) Circulation. 2004;110:3472–9. doi: 10.1161/01.CIR.0000148368.79202.F1. [DOI] [PubMed] [Google Scholar]
- 80.Igarashi J, Miyoshi M, Hashimoto T, Kubota Y, Kosaka H. Statins induce S1P1 receptors and enhance endothelial nitric oxide production in response to high-density lipoproteins. Br J Pharmacol. 2007;150:470–9. doi: 10.1038/sj.bjp.0707114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sviridov D, Nestel P, Watts G. Statins and metabolism of high density lipoprotein. Cardiovasc Hematol Agents Med Chem. 2007;5:215–21. doi: 10.2174/187152507781058672. [DOI] [PubMed] [Google Scholar]
- 82.Maejima T, Yamazaki H, Aoki T, Tamaki T, Sato F, Kitahara M, Saito Y. Effect of pitavastatin on apolipoprotein A-I production in HepG2 cell. Biochem Biophys Res Commun. 2004;324:835–9. doi: 10.1016/j.bbrc.2004.09.122. [DOI] [PubMed] [Google Scholar]
- 83.Martin G, Duez H, Blanquart C, Berezowski V, Poulain P, Fruchart JC, Najib-Fruchart J, Glineur C, Staels B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–32. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fukutomi T, Takeda Y, Suzuki S, Ito T, Joh T, Itoh M. High density lipoprotein cholesterol and apolipoprotein A-I are persistently elevated during long-term treatment with pitavastatin, a new HMG-CoA reductase inhibitor. Int J Cardiol. 2010;141:320–2. doi: 10.1016/j.ijcard.2008.11.130. [DOI] [PubMed] [Google Scholar]
- 85.Zanotti I, Poti F, Favari E, Steffensen KR, Gustafsson JA, Bernini F. Pitavastatin effect on ATP binding cassette A1-mediated lipid efflux from macrophages: evidence for liver X receptor (LXR)-dependent and LXR-independent mechanisms of activation by cAMP. J Pharmacol Exp Ther. 2006;317:395–401. doi: 10.1124/jpet.105.093930. [DOI] [PubMed] [Google Scholar]
- 86.Maejima T, Sugano T, Yamazaki H, Yoshinaka Y, Doi T, Tanabe S, Nishimaki-Mogami T. Pitavastatin increases ABCA1 expression by dual mechanisms: SREBP2-driven transcriptional activation and PPARalpha-dependent protein stabilization but without activating LXR in rat hepatoma McARH7777 cells. J Pharmacol Sci. 2011;116:107–15. doi: 10.1254/jphs.10241fp. [DOI] [PubMed] [Google Scholar]
- 87.Milman U, Blum S, Shapira C, Aronson D, Miller-Lotan R, Anbinder Y, Alshiek J, Bennett L, Kostenko M, Landau M, Keidar S, Levy Y, Khemlin A, Radan A, Levy AP. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28:341–7. doi: 10.1161/ATVBAHA.107.153965. [DOI] [PubMed] [Google Scholar]
- 88.Deakin S, Leviev I, Guernier S, James RW. Simvastatin modulates expression of the PON1 gene and increases serum paraoxonase: a role for sterol regulatory element-binding protein-2. Arterioscler Thromb Vasc Biol. 2003;23:2083–9. doi: 10.1161/01.ATV.0000096207.01487.36. [DOI] [PubMed] [Google Scholar]
- 89.Tang WH, Wu Y, Mann S, Pepoy M, Shrestha K, Borowski AG, Hazen SL. Diminished antioxidant activity of high-density lipoprotein-associated proteins in systolic heart failure. Circ Heart Fail. 2011;4:59–64. doi: 10.1161/CIRCHEARTFAILURE.110.958348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ooyen C, Zecca A, Bersino AM, Catapano AL. NK-104, a potent 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, decreases apolipoprotein B-100 secretion from Hep G2 cells. Atherosclerosis. 1999;145:87–95. doi: 10.1016/s0021-9150(99)00018-0. [DOI] [PubMed] [Google Scholar]
- 91.Tokuno A, Hirano T, Hayashi T, Mori Y, Yamamoto T, Nagashima M, Shiraishi Y, Ito Y, Adachi M. The effects of statin and fibrate on lowering small dense LDL- cholesterol in hyperlipidemic patients with type 2 diabetes. J Atheroscler Thromb. 2007;14:128–32. doi: 10.5551/jat.14.128. [DOI] [PubMed] [Google Scholar]
- 92.Tentolouris N, Eleftheriadou I, Katsilambros N. The effects of medications used for the management of dyslipidemia on postprandial lipemia. Curr Med Chem. 2009;16:203–17. doi: 10.2174/092986709787002763. [DOI] [PubMed] [Google Scholar]
- 93.Boerma M, Burton GR, Wang J, Fink LM, McGehee RE, Jr, Hauer-Jensen M. Comparative expression profiling in primary and immortalized endothelial cells: changes in gene expression in response to hydroxy methylglutaryl-coenzyme A reductase inhibition. Blood Coagul Fibrinolysis. 2006;17:173–80. doi: 10.1097/01.mbc.0000220237.99843.a1. [DOI] [PubMed] [Google Scholar]
- 94.Nomura S, Shouzu A, Omoto S, Inami N, Shimazu T, Satoh D, Kajiura T, Yamada K, Urase F, Maeda Y, Iwasaka T. Effects of pitavastatin on monocyte chemoattractant protein-1 in hyperlipidemic patients. Blood Coagul Fibrinolysis. 2009;20:440–7. doi: 10.1097/MBC.0b013e32832e0618. [DOI] [PubMed] [Google Scholar]
- 95.Nomura S, Inami N, Shouzu A, Omoto S, Kimura Y, Takahashi N, Tanaka A, Urase F, Maeda Y, Ohtani H, Iwasaka T. The effects of pitavastatin, eicosapentaenoic acid and combined therapy on platelet-derived microparticles and adiponectin in hyperlipidemic, diabetic patients. Platelets. 2009;20:16–22. doi: 10.1080/09537100802409921. [DOI] [PubMed] [Google Scholar]
- 96.Masamura K, Oida K, Kanehara H, Suzuki J, Horie S, Ishii H, Miyamori I. Pitavastatin-induced thrombomodulin expression by endothelial cells acts via inhibition of small G proteins of the Rho family. Arterioscler Thromb Vasc Biol. 2003;23:512–7. doi: 10.1161/01.ATV.0000060461.64771.F0. [DOI] [PubMed] [Google Scholar]
- 97.Markle RA, Han J, Summers BD, Yokoyama T, Hajjar KA, Hajjar DP, Gotto AM, Jr, Nicholson AC. Pitavastatin alters the expression of thrombotic and fibrinolytic proteins in human vascular cells. J Cell Biochem. 2003;90:23–32. doi: 10.1002/jcb.10602. [DOI] [PubMed] [Google Scholar]
- 98.Saka M, Obata K, Ichihara S, Cheng XW, Kimata H, Nishizawa T, Noda A, Izawa H, Nagata K, Murohara T, Yokota M. Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J Cardiovasc Pharmacol. 2006;47:770–9. doi: 10.1097/01.fjc.0000211791.22411.0d. [DOI] [PubMed] [Google Scholar]
- 99.Saka M, Obata K, Ichihara S, Cheng XW, Kimata H, Noda A, Izawa H, Nagata K, Yokota M. Attenuation of ventricular hypertrophy and fibrosis in rats by pitavastatin: potential role of the RhoA-extracellular signal-regulated kinase-serum response factor signalling pathway. Clin Exp Pharmacol Physiol. 2006;33:1164–71. doi: 10.1111/j.1440-1681.2006.04508.x. [DOI] [PubMed] [Google Scholar]
- 100.Kobayashi N, Takeshima H, Fukushima H, Koguchi W, Mamada Y, Hirata H, Machida Y, Shinoda M, Suzuki N, Yokotsuka F, Tabei K, Matsuoka H. Cardioprotective effects of pitavastatin on cardiac performance and remodeling in failing rat hearts. Am J Hypertens. 2009;22:176–82. doi: 10.1038/ajh.2008.333. [DOI] [PubMed] [Google Scholar]
- 101.Takuwa N, Ohkura S, Takashima S, Ohtani K, Okamoto Y, Tanaka T, Hirano K, Usui S, Wang F, Du W, Yoshioka K, Banno Y, Sasaki M, Ichi I, Okamura M, Sugimoto N, Mizugishi K, Nakanuma Y, Ishii I, Takamura M, Kaneko S, Kojo S, Satouchi K, Mitumori K, Chun J, Takuwa Y. S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovasc Res. 2010;85:484–93. doi: 10.1093/cvr/cvp312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mizuguchi Y, Oishi Y, Miyoshi H, Iuchi A, Nagase N, Oki T. Impact of statin therapy on left ventricular function and carotid arterial stiffness in patients with hypercholesterolemia. Circ J. 2008;72:538–44. doi: 10.1253/circj.72.538. [DOI] [PubMed] [Google Scholar]
- 103.Kumagai R, Oki C, Muramatsu Y, Kurosaki R, Kato H, Araki T. Pitavastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, reduces hippocampal damage after transient cerebral ischemia in gerbils. J Neural Transm. 2004;111:1103–20. doi: 10.1007/s00702-004-0150-y. [DOI] [PubMed] [Google Scholar]
- 104.Kurosaki R, Muramatsu Y, Kato H, Araki T. Protective effect of pitavastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, on ischemia-induced neuronal damage. Neurol Res. 2004;26:684–91. doi: 10.1179/016164104225014102. [DOI] [PubMed] [Google Scholar]
- 105.Hayashi T, Hamakawa K, Nagotani S, Jin G, Li F, Deguchi K, Sehara Y, Zhang H, Nagano I, Shoji M, Abe K. HMG CoA reductase inhibitors reduce ischemic brain injury of Wistar rats through decreasing oxidative stress on neurons. Brain Res. 2005;1037:52–8. doi: 10.1016/j.brainres.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 106.Tounai H, Hayakawa N, Kato H, Araki T. Immunohistochemical study on distribution of NF-kappaB and p53 in gerbil hippocampus after transient cerebral ischemia: effect of pitavastatin. Metab Brain Dis. 2007;22:89–104. doi: 10.1007/s11011-006-9040-3. [DOI] [PubMed] [Google Scholar]
- 107.Morofuji Y, Nakagawa S, So G, Hiu T, Horai S, Hayashi K, Tanaka K, Suyama K, Deli MA, Nagata I, Niwa M. Pitavastatin strengthens the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol. 2010;30:727–35. doi: 10.1007/s10571-010-9497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Endres M. Statins and stroke. J Cereb Blood Flow Metab. 2005;25:1093–110. doi: 10.1038/sj.jcbfm.9600116. [DOI] [PubMed] [Google Scholar]
- 109.Horimoto Y, Matsubara M, Mizutani H, Hibino H, Tajima T, Fukagawa K, Kabasawa H. Effects of pitavastatin on cerebral blood flow. Clin Ther. 2009;31:575–9. doi: 10.1016/j.clinthera.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 110.Matsumoto M, Tanimoto M, Gohda T, Aoki T, Murakoshi M, Yamada K, Yamazaki T, Kaneko S, Horikoshi S, Tomino Y. Effect of pitavastatin on type 2 diabetes mellitus nephropathy in KK-Ay/Ta mice. Metabolism. 2008;57:691–7. doi: 10.1016/j.metabol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 111.Nakamura T, Sugaya T, Kawagoe Y, Suzuki T, Inoue T, Node K. Effect of pitavastatin on urinary liver-type fatty-acid-binding protein in patients with nondiabetic mild chronic kidney disease. Am J Nephrol. 2006;26:82–6. doi: 10.1159/000091956. [DOI] [PubMed] [Google Scholar]
- 112.Nakamura T, Sato E, Fujiwara N, Kawagoe Y, Ueda Y, Suzuki T, Ueda S, Fukami K, Okuda S, Yamagishi S. Co-administration of ezetimibe enhances proteinuria-lowering effects of pitavastatin in chronic kidney disease patients partly via a cholesterol-independent manner. Pharmacol Res. 2010;61:58–61. doi: 10.1016/j.phrs.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 113.Kimura K, Shimano H, Yokote K, Urashima M, Teramoto T. Effects of pitavastatin (LIVALO tablet) on the estimated glomerular filtration rate (eGFR) in hypercholesterolemic patients with chronic kidney disease. Sub-analysis of the LIVALO Effectiveness and Safety (LIVES) Study. J Atheroscler Thromb. 2010;17:601–9. doi: 10.5551/jat.3764. [DOI] [PubMed] [Google Scholar]
- 114.Nakamura T, Obata JE, Kitta Y, Takano H, Kobayashi T, Fujioka D, Saito Y, Kodama Y, Kawabata K, Mende A, Yano T, Hirano M, Sano K, Nakamura K, Kugiyama K. Rapid stabilization of vulnerable carotid plaque within 1 month of pitavastatin treatment in patients with acute coronary syndrome. J Cardiovasc Pharmacol. 2008;51:365–71. doi: 10.1097/FJC.0b013e318165dcad. [DOI] [PubMed] [Google Scholar]
- 115.Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, Daida H, Matsuzaki M. Effect of intensive statin therapy on regression of coronary atherosclerosis in patients with acute coronary syndrome: a multicenter randomized trial evaluated by volumetric intravascular ultrasound using pitavastatin versus atorvastatin (JAPAN-ACS [Japan assessment of pitavastatin and atorvastatin in acute coronary syndrome] study) J Am Coll Cardiol. 2009;54:293–302. doi: 10.1016/j.jacc.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 116.Kodama K, Komatsu S, Ueda Y, Takayama T, Yajima J, Nanto S, Matsuoka H, Saito S, Hirayama A. Stabilization and regression of coronary plaques treated with pitavastatin proven by angioscopy and intravascular ultrasound – the TOGETHAR trial. Circ J. 2010;74:1922–8. doi: 10.1253/circj.cj-10-0038. [DOI] [PubMed] [Google Scholar]
- 117.Takano M, Mizuno K, Yokoyama S, Seimiya K, Ishibashi F, Okamatsu K, Uemura R. Changes in coronary plaque color and morphology by lipid-lowering therapy with atorvastatin: serial evaluation by coronary angioscopy. J Am Coll Cardiol. 2003;42:680–6. doi: 10.1016/s0735-1097(03)00770-8. [DOI] [PubMed] [Google Scholar]
- 118.Hirayama A, Saito S, Ueda Y, Takayama T, Honye J, Komatsu S, Yamaguchi O, Li Y, Yajima J, Nanto S, Takazawa K, Kodama K. Qualitative and quantitative changes in coronary plaque associated with atorvastatin therapy. Circ J. 2009;73:718–25. doi: 10.1253/circj.cj-08-0755. [DOI] [PubMed] [Google Scholar]
- 119.Pasterkamp G, van Lammeren GW. Pleiotropic effects of statins in atherosclerotic disease. Expert Rev Cardiovasc Ther. 2010;8:1235–7. doi: 10.1586/erc.10.107. [DOI] [PubMed] [Google Scholar]
- 120.Pekkanen J, Linn S, Heiss G, Suchindran CM, Leon A, Rifkind BM, Tyroler HA. Ten-year mortality from cardiovascular disease in relation to cholesterol level among men with and without preexisting cardiovascular disease. N Engl J Med. 1990;322:1700–7. doi: 10.1056/NEJM199006143222403. [DOI] [PubMed] [Google Scholar]
- 121.Florvall G, Basu S, Larsson A. Apolipoprotein A1 is a stronger prognostic marker than are HDL and LDL cholesterol for cardiovascular disease and mortality in elderly men. J Gerontol A Biol Sci Med Sci. 2006;61:1262–6. doi: 10.1093/gerona/61.12.1262. [DOI] [PubMed] [Google Scholar]
- 122.Maruyama T, Takada M, Nishibori Y, Fujita K, Miki K, Masuda S, Horimatsu T, Hasuike T. Comparison of preventive effect on cardiovascular events with different statins. Circ J. 2011;75:1951–9. doi: 10.1253/circj.cj-10-1163. [DOI] [PubMed] [Google Scholar]
- 123.Ballantyne CM, Olsson AG, Cook TJ, Mercuri MF, Pedersen TR, Kjekshus J. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation. 2001;104:3046–51. doi: 10.1161/hc5001.100624. [DOI] [PubMed] [Google Scholar]
- 124.Ridker PM. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: rationale and design of the JUPITER trial. Circulation. 2003;108:2292–7. doi: 10.1161/01.CIR.0000100688.17280.E6. [DOI] [PubMed] [Google Scholar]
- 125.Zhou Q, Liao JK. Pleiotropic effects of statins. Basic research and clinical perspectives. Circ J. 2010;74:818–26. doi: 10.1253/circj.cj-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112:71–105. doi: 10.1016/j.pharmthera.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 127.Di Napoli P, Taccardi AA, Grilli A, De Lutiis MA, Barsotti A, Felaco M, De Caterina R. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc Res. 2005;66:462–71. doi: 10.1016/j.cardiores.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 128.Tsujimoto A, Takemura G, Mikami A, Aoyama T, Ohno T, Maruyama R, Nakagawa M, Minatoguchi S, Fujiwara H. A therapeutic dose of the lipophilic statin pitavastatin enhances oxidant-induced apoptosis in human vascular smooth muscle cells. J Cardiovasc Pharmacol. 2006;48:160–5. doi: 10.1097/01.fjc.0000245989.89771.1b. [DOI] [PubMed] [Google Scholar]
- 129.Kiyan J, Kusch A, Tkachuk S, Kramer J, Haller H, Dietz R, Smith G, Dumler I. Rosuvastatin regulates vascular smooth muscle cell phenotypic modulation in vascular remodeling: role for the urokinase receptor. Atherosclerosis. 2007;195:254–61. doi: 10.1016/j.atherosclerosis.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 130.Verreth W, De Keyzer D, Davey PC, Geeraert B, Mertens A, Herregods MC, Smith G, Desjardins F, Balligand JL, Holvoet P. Rosuvastatin restores superoxide dismutase expression and inhibits accumulation of oxidized LDL in the aortic arch of obese dyslipidemic mice. Br J Pharmacol. 2007;151:347–55. doi: 10.1038/sj.bjp.0707231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guo H, Shi Y, Liu L, Sun A, Xu F, Chi J. Rosuvastatin inhibits MMP-2 expression and limits the progression of atherosclerosis in LDLR-deficient mice. Arch Med Res. 2009;40:345–51. doi: 10.1016/j.arcmed.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 132.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 133.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–82. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]