

Abstract
AIMS
In vitro studies indicated CYP3A4 alone was responsible for tolvaptan metabolism. To determine the effect of a CYP3A4 inhibitor (ketoconazole) and a CYP3A4 inducer (rifampicin) on tolvaptan pharmacokinetics (PK) and pharmacodynamics (PD), two clinical trials were performed.
METHODS
For CYP3A4 inhibition, a double-blind, randomized (5:1), placebo-controlled trial was conducted in 24 healthy subjects given either a single 30 mg dose of tolvaptan (n = 19) or matching placebo (n = 5) on day 1 with a 72 h washout followed by a 3 day regimen of 200 mg ketoconazole, once daily with 30 mg tolvaptan or placebo also given on day 5. For CYP3A4 induction, 14 healthy subjects were given a single dose of 240 mg tolvaptan with 48 h washout followed by a 7 day regimen of 600 mg rifampicin, once daily, with 240 mg tolvaptan also given on the seventh day.
RESULTS
When co-administered with ketoconazole, mean Cmax and AUC(0,∞) of tolvaptan were increased 3.48- and 5.40-fold, respectively. Twenty-four hour urine volume increased from 5.9 to 7.7 l. Erythromycin breath testing showed no difference following a single dose of tolvaptan. With rifampicin, tolvaptan mean Cmax and AUC were reduced to 0.13- and 0.17-fold of tolvaptan administered alone. Twenty-four hour urine volume decreased from 12.3 to 8.8 l.
CONCLUSIONS
Tolvaptan is a sensitive CYP3A4 substrate with no inhibitory activity. Due to the saturable nature of tolvaptan's effect on urine excretion rate, changes in the pharmacokinetic profile of tolvaptan do not produce proportional changes in urine output.
Keywords: CYP3A induction, CYP3A inhibition, pharmacodynamics, pharmacokinetics, tolvaptan
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Before these trials were done, the effects of CYP3A4 inhibition and induction on the pharmacokinetics (PK) and pharmacodynamics (PD) of tolvaptan in healthy subjects were unknown. As tolvaptan is a CYP3A4 substrate, knowing the effects of inhibition and induction on CYP3A4-mediated metabolism was important for dosing recommendations.
WHAT THIS STUDY ADDS
This paper describes the changes in tolvaptan PK and PD following inhibition or induction of CYP3A4 and explores the mechanisms behind the disparity seen between tolvaptan PK and effects on urine output. It also discusses the concentrations at which tolvaptan produces its maximal response on urine output and the timing of the onset and offset of this response.
Introduction
Tolvaptan is an orally effective nonpeptide arginine vasopressin (AVP) V2-receptor antagonist, with no intrinsic agonist activity, discovered by Otsuka Pharmaceutical Company [1]. Stimulation of AVP V2-receptors increases cAMP production by adenylcyclase, which leads to synthesis and insertion of aquaporin-2 water channels in the luminal plasma membrane of collecting tubule principle cells, allowing water reabsorption in the hypertonic medulla [2]. The absence of AVP or the inhibition of AVP activity by V2-receptor antagonists leads to excretion of large volumes of water (aquaresis). Single ascending dose (SAD) trials of oral tolvaptan doses from 60 mg up to 480 mg in healthy subjects reported significant aquaresis without significantly changing total electrolyte excretion, serum potassium concentrations or markers of renal function (e.g. creatinine clearance). Saturation of response in urine excretion rate and urine volume was observed, with all doses having about the same mean 0 to 12 h urine volume (∼7 l) and maximal urine excretion rate (∼12 ml min−1). Therefore, doses greater than 60 mg produce a longer effect but have the same maximal excretion rate [3].
Non-clinical studies with cDNA expressed human cytochrome P450 (CYP) experiments and human liver microsome experiments (by microsomes derived from human AHH-1 TK+/−cells expressing human cytochrome P450), indicated that CYP3A4 was the only enzyme involved in tolvaptan metabolism (data on file). Urinary excretion is less than 1% of the dose [3]. Following doses of 120 mg and higher, the terminal phase elimination half-life of tolvaptan was determined to be about 12 h [3]. However, tolvaptan concentrations did not accumulate following multiple oral doses of 30 mg [4] or 60 mg [5], suggesting a dominant half-life somewhat shorter than 12 h. There is marked inter-subject variation in peak and average exposure to tolvaptan with a percent coefficient of variation ranging between 30 and 60%. The mean absolute bioavailablity of tolvaptan is reported to be 56% [6].
Tolvaptan is approved in the United States for the treatment of clinically significant hypervolaemic and euvolaemic hyponatraemia including patients with heart failure, cirrhosis and syndrome of inappropriate antidiuretic hormone (SIADH). It is also approved in Europe for the treatment of adults with hyponatraemia secondary to SIADH and in Japan for volume overload in heart failure when adequate response is not obtained with other diuretics (e.g. loop diuretics) [7, 8]. Dosing for the treatment of hyponatraemia is once daily to begin at 15 mg with up-titration to 30 mg or 60 mg once daily, depending on the rate and magnitude of increase observed in serum sodium concentrations. A rapid increase in serum sodium, >12 mEq l−1 in the first 24 h, is undesirable and this has been associated with osmotic demyelination [9]. Understanding the effects of potential large drug–drug interactions mediated by CYP3A in the pharmacokinetics (PK) and pharmacodynamics (PD) of tolvaptan is therefore important for the safe and efficacious initiation of tolvaptan.
Separate clinical trials in healthy men and women were conducted to determine the effects of ketoconazole, a strong systemic acting inhibitor of CYP3A4, and rifampicin (rifampin), an inducer of CYP3A4 on tolvaptan PK and on the PD endpoints of urine excretion rate and urine volume.
Methods
Subject recruitment and general trial conduct
Each trial was conducted in healthy male and female subjects, 18–45 years of age, with body weights within 15% of ideal body weight. No more than 75% of subjects in each trial were to be of one gender. Screening evaluations were performed within 21 days prior to administration of the trial medication. A complete physical examination including vital signs, 12-lead electrocardiograms (ECG) and laboratory tests (serum chemistry, haematology, urinalysis, urine drug screen, serum alcohol, and, for women, a serum or urine pregnancy test) was done. All subjects abstained from xanthine-containing food and drinks, Seville oranges, grapefruit and grapefruit juice, and alcohol for 72 h before admission and for the duration of the trial. No medications or herbal supplements were permitted from 14 days prior to dosing and for the duration of the trial. Subjects were not allowed to consume tobacco products for the duration of the trial.
Subjects abstained from food from midnight on the evening before tolvaptan dose administration until a meal at 4 h. Water was allowed at all times except for the ±2 h interval around dosing. All doses were given with 240 ml of room temperature water (see individual trial designs below for specifics on ketoconazole and rifampicin administration).
Safety evaluations were based on physical examinations, recording of adverse events, clinical laboratory tests, vital signs (sitting ≥ 3 min for ketoconazole trial otherwise supine ≥3 min) and 12-lead ECGs (see Table 1 for a summary of subject demographic characteristics for both trials).
Table 1.
Summary of subject demographics
| Demographics | Ketoconazole trial 30 mg TLV (n = 19) | Placebo (n = 5) | Rifampicin trial (n = 15) |
|---|---|---|---|
| Age (years) [Mean (range)] | 31 (19–45) | 27 (20–39) | 26 (18–42) |
| Weight (kg) [Mean (range)] | 75.0 (59.0–89.4) | 66.1 (54.2–77.5) | 72.0 (52.0−91.0) |
| Gender, n (%) | |||
| Male | 12 (63.1) | 2 (40.0) | 9 (60.0) |
| Female | 7 (36.8) | 3 (60.0) | 6 (40.0) |
| Race (n) | |||
| Caucasian | 14 (73.7) | 3 (60.0) | 10 (66.7) |
| Other | 5 (26.3) | 2 (40.0) | 5 (33.3) |
TLV, tolvaptan.
Trial designs
Ketoconazole (inhibitor) trial
The trial was a double-blind, placebo-controlled, randomized (five tolvaptan : one placebo), trial in healthy subjects reviewed by the institutional review board of and conducted at SFBC International (Miami, FL). Twenty-four subjects were enrolled into the trial; one subject who checked in on day −1 had elevated liver enzymes and was not dosed.
Nineteen subjects were randomly assigned to receive tolvaptan and five subjects were assigned to receive placebo. On day 1, subjects received a 30 mg dose of tolvaptan (as two 15 mg tablets) or placebo (as two matching placebo tablets). On days 4, 5 and 6, subjects received a single daily 200 mg tablet of ketoconazole (Janssen Pharmaceutica, Titusville, NJ). On day 5, either 30 mg tolvaptan or placebo was co-administered with the ketoconazole. Previously it was determined that 1 day of pre-exposure to ketoconazole was sufficient to produce a maximal inhibition of midazolam pharmacokinetics [10]. Ketoconazole dosing was continued on day 6 to ensure inhibition was sustained through out the tolvaptan washout period.
Blood samples for PK analysis on day 1 were taken at pre dose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, 24, 48 and 72 h post dose. Daily (24 h) urine samples were collected for 72 h post dose. Individual voids were pooled, the volume determined and an aliquot taken for determination of tolvaptan concentrations. On day 5, urine and blood samples for PK analysis were taken following the same schedule as day 1. In addition, subjects received intravenous (i.v.) [14C-N-methyl]erythromycin (3 mCi) for an erythromycin breath test on day −1 and at 2 h after dose administration on days 1, 4 and 5. On days 2, 3, 4, 6 and 7, breakfast was provided after the morning blood sample had been taken and, for days 4 and 6, 1 h after ketoconazole dosing.
Rifampicin (inducer) trial
This was an open-label, three-period, sequential treatment trial reviewed by the Independent Investigational Review Board (Plantation, FL) and conducted at Northwest Kinetics (Tacoma, WA). Fifteen healthy subjects were enrolled and completed the trial.
On day 1, a single dose of 240 mg (four 60 mg oral tablets) tolvaptan was administered in the fasted state with 240 ml room temperature water. On days 3 to 8 and 10, rifampicin 600 mg (two 300 mg oral capsules, Rifadin®, Eon Labs, Lake Success, NY) were given once daily, 1 h prior to a standard breakfast. On day 9, tolvaptan 240 mg and rifampicin 600 mg were simultaneously administered to subjects in the fasted state. Rifampicin administration has been reported to decrease the concentrations of weak CYP3A4 substrates such as midazolam by 96–98% [11]. Therefore, a tolvaptan dose of 240 mg was chosen so that tolvaptan plasma concentrations would still be above the lower limit of quantification (LLOQ, 5.00 ng ml−1) of the assay if concentrations dropped 95%.
Tolvaptan plasma concentrations were measured from blood samples taken on days 1 and 9. Blood samples were taken within 30 min of dosing for the pre dose measurement and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 16, 24, 30, 36 and 48 h post dose. Urine volumes were determined for 0–2, 2–4, 4–6, 6–8, 8–12, 12–16, 16–24, 24–26, 26–28, 28–30, 30–32, 32–36, 36–40 and 40–48 h post dose.
Rifampicin plasma concentrations were measured from blood samples taken at pre dose on day 1 and pre dose and 2 h post dose on days 7 and 8.
Erythromycin breath testing
The [14C-N-Methyl] erythromycin i.v. injection was obtained from Metabolic Solutions (Merrimack, NH). The 3 µCi of [14C-N-methyl] erythromycin was diluted in 3 ml of saline and administered as a bolus i.v. injection. Breath samples were collected pre dose and every 10 min until 60 min after injection.
Breath samples were collected as follows: in a glass beaker, 21 ml of benzethonium hydroxide (approximately 1 m in methanol) was combined with 21 ml of absolute ethanol. Four millilitres of this solution plus 0.5 ml of a 1% phenophthalein solution were aliquoted into scintillation vials. A mouth piece with two one-way valves and tubing was set up to allow easy exhalation through the tubing, assuring that the breath could be bubbled through the mixture in the scintillation vial, and that aspiration of the mixture could not occur. Subjects blew into the mouthpiece until the pink color in the vials disappeared. At that time, 10 ml of scintillation fluid was added to the scintillation vials, mixed, and then the vials were stored refrigerated until counted. The samples were counted for radioactivity using a scintillation counter over a 10 min programme period. The sample results were then reported as disintegrations min−1.
Bioanalytical methods
Sample handling
Blood samples for tolvaptan (10 ml) were collected in sodium heparin tubes. Samples were centrifuged for 10 min at 2500 rev min−1 to obtain plasma. Plasma was then pipetted into polypropylene tubes and stored at −20°C or colder.
Individual urine voids in a collection interval were pooled and kept refrigerated until the end of the collection interval when the total volume was determined and 20 ml aliquots were taken and stored at −20°C or colder.
Blood samples for rifampicin (10 ml) were collected in sodium heparin tubes. Rifampicin in blood and plasma is light sensitive so samples were processed under yellow light. The tubes were gently inverted three to four times and then centrifuged at 2500 rev min−1 for at least 10 min at 4°C. Plasma was then pipetted into polypropylene tubes and stored at −20°C or colder.
Bioanalytical assays
Ketoconazole trial
Plasma and urine concentrations of tolvaptan were analyzed using reverse-phase high performance liquid chromatography with tandem mass spectrometric detection (HPLC-MS/MS) as described previously; LLOQ of tolvaptan was 5.00 ng ml−1[3]. The presence of ketoconazole did not affect the quantitation of tolvaptan. For tolvaptan quality control (QC), samples in the range of 8.00 to 800 ng ml−1, the percent coefficient of variation (%CV) values ranged from 2.85 to 4.67% in plasma and 4.99 to 6.12% in urine.
Rifampicin trial
Plasma concentrations of tolvaptan were analyzed as described previously; the LLOQ of tolvaptan was 5.00 ng ml−1[3]. The presence of rifampicin did not affect the quantitation of tolvaptan. For QC samples in the range of 15.0 to 800 ng ml−1, the %CV values ranged from 5.72 to 6.88%.
Plasma concentrations of rifampicin were determined at Prevalere Life Sciences (Whitesboro, NY) using a proprietary assay. In brief, rifampicin and an internal standard were extracted from human plasma using a liquid-liquid extraction procedure, followed by reversed-phase HPLC with isocratic elution. The eluant was monitored at 340 nm using an ultraviolet absorbance detector. The method was linear over the range of 0.100 to 20.0 µg ml−1 for rifampicin. The %CV of QC samples was 11.5%.
Pharmacokinetic methods
For each subject, the tolvaptan plasma concentration–time data were analyzed using a noncompartmental method [12]. Noncompartmental analysis was also used to determine the area under the curve of the ERBT data. Subjects who did not receive tolvaptan were excluded from the analysis. Concentration values below the LLOQ were assigned a value of ‘0’ for the determination of mean concentrations and values following the last sample with a measurable concentration were excluded from the PK analysis. Actual blood sample times were used for PK calculations. Values for peak plasma concentrations (Cmax) and time to Cmax (tmax) were taken directly from the observed data. Where possible, the terminal phase elimination rate constant (λz) was estimated by a log-linear regression of at least three non-zero concentrations. The area under the curve from time zero to the time of the last measurable concentration (AUC(0,tlast)) was calculated using the linear trapezoidal rule. Values of area under the curve from time zero to infinity (AUC(0,∞)) and apparent total body clearance (CL/F) were determined using standard methods [12]. In the ketoconazole trial, the amount (Ae,u) of tolvaptan excreted in the urine and fraction of dose excreted unchanged (%fe,u) in urine for tolvaptan were determined.
WinNonlin Professional Edition (version 2.1 for ketoconazole trial and version 4.0 for rifampicin trial, Pharsight Corp., Mountain View, CA) was used for the analyses. Descriptive statistics were determined for the ketoconazole trial using Excel 97 (version SR-2) and for the rifampicin trial using S-Plus® (Insightful Corporation, version 6.1). Plots were created using Sigma Plot 2002 (SPSS Inc., version 10.0).
Statistical methods
Ketoconazole trial
The primary assessment was based on a paired t-test of tolvaptan pharmacokinetic parameters. Adherence to normality assumptions was tested for both log transformed and raw data. Log transformed data were used in cases where they better satisfied the assumption of normality. If neither raw nor log transformed data adequately satisfied the normality assumption, the Wilcoxon signed rank test was used. For tmax, only the Wilcoxon signed rank test was used. Data analysis was performed with Statistical Analysis System (SAS, Cary, NC) software, version 6.12.
Rifampicin trial
The primary endpoints were the values of Cmax and AUC(0,∞) for tolvaptan following a single oral dose of tolvaptan with rifampicin vs. tolvaptan alone. As AUC(0,∞) could not be determined for subjects following the tolvaptan + rifampicin treatment, AUC(0,tlast) was used. A paired t-test, with two-sided alpha of 0.05, was used to determine if rifampicin treatment produced a significant change in tolvaptan Cmax or AUC(0,tlast). Data analysis was performed with SAS, version 8.2.
Results
Tolvaptan pharmacokinetics
Ketoconazole trial
Twenty-two subjects completed the trial. Two subjects in the tolvaptan-treated group withdrew for personal reasons after dosing on day 1. Therefore, 17 subjects had evaluable concentration data on both day 1 and day 5. A plot of mean (SD) tolvaptan plasma concentrations following 30 mg tolvaptan alone or with ketoconazole is presented in Figure 1. A summary of tolvaptan plasma PK parameters is presented in Table 2. While the mean Cmax and AUC(0,∞) ratios were 3.48 and 5.30, respectively, the range of responses was 2.08 to 7.39 for Cmax and 3.50 to 7.71 for AUC(0,∞). The mean (SD) values for Ae,u were 29.7 (28.7) µg and 242 (135) µg for tolvaptan alone and tolvaptan + ketoconazole, respectively. The corresponding values for %fe,u were 0.099 (0.096) and 0.81 (0.45) percent. Except for tmax, all PK parameters following tolvaptan + ketoconazole were statistically significantly different (P < 0.05) from values following tolvaptan alone.
Figure 1.
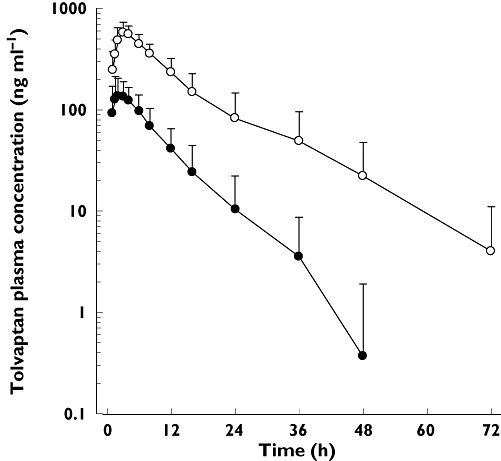
Tolvaptan (TLV) plasma concentrations following administration of 30 mg alone or with 200 mg ketoconazole to 17 healthy subjects. TLV 30 mg ( ); TLV + ketoconazole (
); TLV + ketoconazole ( )
)
Table 2.
Mean (SD) pharmacokinetic parameters of tolvaptan
| Trial and treatment | n | Cmax (ng ml−1) | tmax (h) | t½,z (h) | AUC(0,tlast) (ng h ml−1) | AUC(0,∞) (ng h ml−1) | CL/F (ml min kg−1) | Mean Cmax ratio | Mean AUC(0,∞) ratio |
|---|---|---|---|---|---|---|---|---|---|
| 30 mg tolvaptan | 17 | 174 (65) | 3.00 (1.00–6.00) | 6.9 (3.3) | 1 360 (634) | 1 460 (653) | 5.63 (2.68) | – | – |
| 30 mg tolvaptan + 200 mg ketoconazole | 17 | 606 (141) | 3.02 (1.50–6.00) | 10.5 (2.7)* | 7 670 (3 090) | 7 880 (3 150)* | 0.97 (0.33)* | 3.48 | 5.40 |
| 240 mg tolvaptan | 15 | 1000 (436) | 2.52 (1.00–12.00) | 7.7 (3.9)† | 11 600 (4 060) | 12 100 (4 140)† | 5.23 (1.97)† | – | – |
| 240 mg tolvaptan + 600 mg rifampicin | 15 | 168 (73.5) | 3.00 (1.50–6.02) | ND | 1 470 (686) | ND | ND | 0.17 | 0.13 |
n = 9.
n = 12.
Values for tmax are median (range). ND, Not able to be determined.
Rifampicin trial
All 15 subjects completed the trial and were included in the PK analysis. A plot of mean (SD) tolvaptan plasma concentrations following 240 mg tolvaptan alone or with rifampicin is presented in Figure 2. A summary of the PK parameters is presented in Table 2. While the mean Cmax and AUC(0,tlast) ratios were 0.17 and 0.13, respectively, the range of responses was 0.11 to 0.31 for Cmax and 0.05 to 0.19 for AUC(0,tlast) The values of Cmax and AUC(),tlast) were statistically significantly lower. The large decrease in concentrations meant that values of AUC(),∞), t1/2,z and CL/F could not be determined for tolvaptan following co-administration with rifampicin and therefore a statistical comparison could not be performed.
Figure 2.
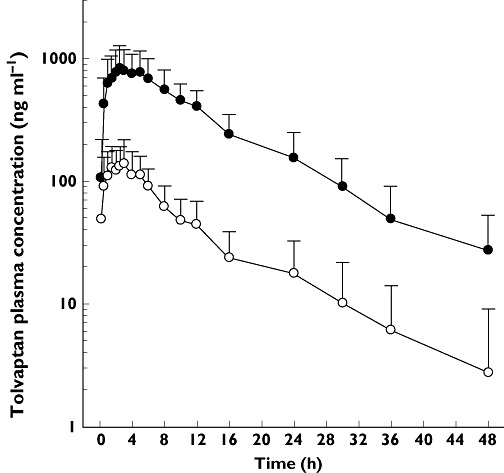
Tolvaptan (TLV) plasma concentrations following administration of 240 mg alone or with 600 mg rifampicin to 15 healthy subjects. TLV 240 mg ( ); TLV + rifampicin (
); TLV + rifampicin ( )
)
All pre dose concentrations of rifampicin were less than 0.100 µg ml−1. The mean (SD) rifampicin concentrations at 2 h post dose were 9.33 (3.24) and 9.92 (3.94) µg ml−1 for days 7 and 8, respectively.
Breath erythromycin pharmacokinetics
The mean (SD) area under the disintegrations min−1 curve from 0 to 60 min (AUC(0,60 min)) post dose is presented in Table 3. A single dose of 30 mg tolvaptan alone produced no change in AUC(0,60 min) while a single dose of 200 mg ketoconazole decreased AUC(0,60 min) by about 49%. The AUC(0,60 min) was reduced further, to 63% of control, following the second 200 mg dose of ketoconazole.
Table 3.
Mean (SD) erythromycin breath test results
| Treatment (trial day) | AUC(0,60 min) (disintegrations min−1 min) n = 17 | Change from baseline n = 17 |
|---|---|---|
| Baseline (day −1) | 26 500 (7580) | – |
| 30 mg tolvaptan (day 1) | 26 700 (5580) | +3.5% |
| 200 mg ketoconazole (day 4) | 12 900 (3890)* | −49.4% |
| 30 mg tolvaptan + 200 mg ketoconazole (day 5) | 9 410 (3040)* | −63.3% |
P < 0.05 when compared with baseline.
Pharmacodynamics
Ketoconazole trial
Mean (SD) daily urine volume is presented in Table 4. Compared with the placebo group on day 1, a 30 mg dose of tolvaptan increased the 0–24 h urine volume by 2.7 l and when 30 mg tolvaptan was co-administered with ketoconazole, urine output was about 4.4 l higher.
Table 4.
Mean (SD) daily urine volume (ml) following tolvaptan or placebo alone or following co-administration with ketoconazole or rifampicin
| Collection interval | ||
|---|---|---|
| Treatment (n) | 0–24 h | 24–48 h |
| Placebo, day 1 (5) | 3 250 (810) | 2360 (1390) |
| Placebo + ketoconazole, day 5 (5) | 2 820 (1070) | 2840 (2030) |
| Tolvaptan 30 mg, day 1 (17) | 5 950 (2110) | 2140 (880) |
| Tolvaptan 30 mg + ketoconazole 200 mg, day 5 (17) | 7 670 (3630) | 3340 (1400) |
| Tolvaptan 240 mg, day 1 (15) | 12 335 (2233) | 6133 (2571) |
| Tolvaptan 240 mg + rifampicin 600 mg, day 9 (15) | 8 790 (1935) | 2968 (2076) |
Rifampicin trial
Mean (SD) daily urine volume is presented in Table 4 in order to provide a direct comparison with the ketoconazole trial results. Following 240 mg tolvaptan, mean 0–24 h urine volume was greater than 12 l, a 4-fold increase compared with the placebo group in the ketoconazole trial. Urine output in the 24–48 h interval was increased two-fold compared with placebo. When co-administered with rifampicin, the 0–24 h urine volume was about 8 l and, for the 24–48 h interval, urine output returned to the same level as that observed in placebo subjects.
Figure 3 presents plots of mean tolvaptan concentrations overlaid on plots of mean urine excretion rates following 240 mg tolvaptan alone (A) or with rifampicin (B).
Figure 3.
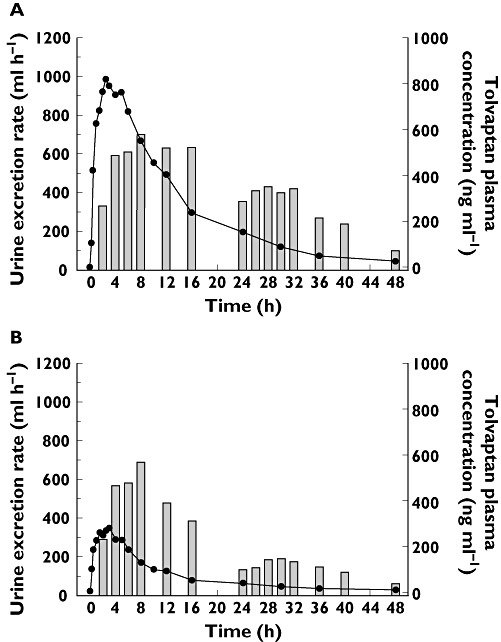
Mean urine excretion rate at the end time of the collection interval and mean tolvaptan plasma concentration vs. time following a single oral 240 mg dose of tolvaptan alone (A) or with rifampicin at steady-state (B) to 15 healthy subjects. Urine excretion rate ( ); Tolvaptan Concentration (
); Tolvaptan Concentration ( )
)
Safety
In both trials, all treatment emergent adverse events (TEAE) were mild to moderate in severity with none reported as severe or resulting in discontinuation from the trial. No clinically meaningful changes were observed in ECGs, vital signs or clinical laboratory tests in either trial. In the ketoconazole trial eight of 19 subjects who received tolvaptan/ketoconazole and three out of five subjects who received placebo/ketoconazole had TEAEs. In tolvaptan- and placebo-treated subjects, only headache (3/19 [15.8%] and 1/5 [20.0%]) and dizziness (1/19 [5.3%] and 1/5 [20.0%]) occurred in more than one subject. In the rifampicin trial, only nausea (1/15 [6.7%] in each arm) and headache (2/15 [13.3%] in each arm) were reported in more than one subject.
Discussion
The pharmacokinetic results confirm that tolvaptan is a sensitive CYP3A4 substrate. Co-administration with ketoconazole, a strong inhibitor of CYP3A4, increased Cmax and AUC(0,∞) to 3.48-fold and 5.30-fold compared with tolvaptan alone values, respectively. Even in the presence of ketoconazole, tolvaptan is extensively metabolized. Urine excretion of unchanged tolvaptan was increased about 8-fold compared with tolvaptan alone but the total amount excreted unchanged was still less than 1% of the dose. Co-administration of tolvaptan with rifampicin, a strong inducer of CYP3A4, decreased Cmax and AUC(0,tlast) to 0.13-fold and 0.17-fold of tolvaptan alone values, respectively.
In comparison, when 6 mg midazolam was given orally with 200 mg ketoconazole, midazolam Cmax and AUC(0,∞) were increased 4-fold and 13.6-fold compared with midazolam alone [13]. When oral midazolam was given with rifampicin at steady-state, the midazolam AUC was decreased 96–98% [11].
The results of the ERBT support that ketoconazole administration of 200 mg once daily did inhibit CYP3A activity and that tolvaptan administration of 30 mg did not produce any significant changes in CYP3A activity. While ERBT values were not directly correlated with the PK parameters of CYP3A substrates such as midazolam [14] or alfentanil [15] changes in the ERBT have been shown to be correlated with CYP3A4 inhibition (troleandomycin [16], ketoconazole [17, 18]) and induction (glucocorticoids [16], rifampicin [16]) and to show no change following midazolam [18], a sensitive CYP3A4 substrate, or paclitaxel [17], not a substrate for CYP3A4. When 200 mg ketoconazole was given 2 h prior to the ERBT on day 3, a 49% decrease from baseline was observed, a result very similar to the 43% decrease observed by McCrea et al. [18] using the same regimen. A 30 mg dose of tolvaptan produced no change in the ERBT.
Following co-administration of 30 mg tolvaptan with ketoconazole, 24 h urine volume was only increased 1.3-fold, a much smaller change than the 5.4-fold increase seen in tolvaptan AUC(0,∞). Previously, it was reported that a 30 mg dose of tolvaptan produces a maximal increase in urine output for 8 h post dose [19]. Therefore, increased tolvaptan plasma concentrations during this time period would not be expected to increase total urine output due to the saturation of response in urine excretion rate. The increased 24 h urine output observed with co-administration of ketoconazole is likely due to longer exposure to effective concentrations of tolvaptan beyond the 8 h timepoint.
This dynamic is important to understand better the impact of co-administration of CYP3A4 inhibitors when treating patients with tolvaptan. The initial dose of tolvaptan for the currently approved indications is 15 mg once daily. For a 15 mg dose administered with 200 mg ketoconazole, the PK/PD response would be expected to be similar to that produced by a 60 mg dose, as tolvaptan concentrations would be increased about 4-fold. The overall effect of tolvaptan and ketoconazole co-administration would be similar to the effects observed following high doses of tolvaptan, namely that as plasma concentrations remain elevated for longer periods of time, the duration that urine excretion rate will remain elevated is increased but the maximum urine excretion rate will not be increased. Strong and moderate CYP3A4 inhibitors may increase tolvaptan concentrations and duration of effect beyond that of the recommended 15 mg initial dose. As such, current labelling for tolvaptan contraindicates concomitant use with strong CYP3A4 inhibitors and recommends avoiding use with moderate inhibitors.
In vitro studies have shown that rifampicin can inhibit CYP3A activity so there is a theoretical possibility that the simultaneous administration of rifampicin with tolvaptan could have interfered with the determination of the true extent of CYP3A induction. However, the reported Ki for CYP3A inhibition was 18.5 µm or 15.6 µg ml−1[20]. Mean rifampicin concentrations determined at 2 h post dose (reported tmax) on days 7 and 8 were less than 10 µg ml−1. Rifampicin is also reported to be 80% plasma protein bound [21] so the mean free amount of rifampicin at 2 h would be less than 2 µg ml−1. Therefore, the interaction results of this trial largely represent the effect of rifampicin on CYP3A4 induction.
In the rifampicin trial, the maximal urine excretion rate and 0–12 h urine volume following 240 mg tolvaptan was similar to that reported previously [3], suggesting that a maximal response in these PD endpoints had been produced. Following tolvaptan + rifampicin in this report, mean urine excretion rates in the 0–8 h period post dose were similar to those following 240 mg tolvaptan alone, suggesting that urine output was maximized in this period even though tolvaptan concentrations only ranged from 48.3 to 296 ng ml−1 in the 0.5 to 8 h time period. It can therefore be inferred that tolvaptan concentrations in this range may be sufficient to produce a maximal inhibition of AVP binding at the V2 receptor and produce a maximal response in urine output. However, at the doses of tolvaptan used in currently approved indications, concomitant administration with CYP3A4 inducers would require an approximate 7-fold increase in tolvaptan dose to offset the decrease in concentrations seen with co-administration of rifampicin.
In the 24 to 48 h period following tolvaptan + rifampicin, the mean 24 h urine volume was similar to the 24 h urine volumes following placebo plus or minus ketoconazole in the ketoconazole trial. It appears that tolvaptan concentrations in this period are too low to have a measurable effect. At 24 h, the mean (SD) tolvaptan plasma concentration was 18 (15) ng ml−1. These data indicate that there is a narrow range of concentrations between where tolvaptan has a measurable effect (>20 ng ml−1) and the maximum effect is achieved. They also indicate that the onset and offset of tolvaptan action is rapid.
Co-administration of single doses of tolvaptan with either ketoconazole or rifampicin in healthy subjects resulted in no clinically meaningful changes observed in ECGs, vital signs or clinical laboratory tests and very few subjects reporting TEAEs in any treatment arm. The TEAEs of headache, nausea and dizziness have been reported previously [3].
In summary, tolvaptan is a sensitive CYP3A4 substrate with no apparent inhibitory activity at CYP3A4. Co-administration of 30 mg tolvaptan with a strong CYP3A4 inhibitor increased tolvaptan concentrations by approximately five-fold with approximately 30% greater urine output over 24 h. Co-administration of 240 mg tolvaptan in the presence of CYP3A4 induction reduced tolvaptan concentrations and effects by approximately 85%. Tolvaptan produces a maximal effect on urine excretion rate as concentrations approach 100 ng ml−1 and appears to have no effect on urine excretion rate when concentrations fall below 20 ng ml−1. Tolvaptan has a rapid onset and offset of action. Due to the saturable nature of the effect of tolvaptan on urine excretion rate, changes in the PK profile of tolvaptan do not produce proportional changes in urine output.
Acknowledgments
We would like to thank Holly Krasa, also an employee at OPDC, for her review and help with revisions of the manuscript. Upside Endeavors provided professional writing assistance, which was supported by Otsuka America Pharmaceutical, Inc.
Competing Interests
All authors were employees of Otsuka Pharmaceutical Development & Commercialization (OPDC) at the time the trials were conducted. These trials were supported by OPDC.
REFERENCES
- 1.Yamamura Y, Nakamura S, Itoh S, Hirano T, Onogawa T, Yamashita T, Yamada Y, Tsujimae K, Aoyama M, Kotosai K, Ogawa H, Yamashita H, Kondo K, Tominaga M, Tsujimoto G, Mori T. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther. 1998;287:860–7. [PubMed] [Google Scholar]
- 2.King LS, Agre P. Pathophysiology of the aquaporin water channels. Annu Rev Physiol. 1996;58:619–48. doi: 10.1146/annurev.ph.58.030196.003155. [DOI] [PubMed] [Google Scholar]
- 3.Shoaf SE, Wang Z, Bricmont P, Mallikaarjun S. Pharmacokinetics, pharmacodynamics, and safety of tolvaptan, a nonpeptide AVP antagonist, during ascending single-dose studies in healthy subjects. J Clin Pharmacol. 2007;47:1498–507. doi: 10.1177/0091270007307877. [DOI] [PubMed] [Google Scholar]
- 4.Hauptman PJ, Zimmer C, Udelson J, Shoaf SE, Mallikaarjun S, Bramer SL, Orlandi C. Comparison of two doses and dosing regimens of tolvaptan in congestive heart failure. J Cardiovasc Pharmacol. 2005;46:609–14. doi: 10.1097/01.fjc.0000180899.24865.b6. [DOI] [PubMed] [Google Scholar]
- 5.Shoaf SE, Ohzone Y, Ninomiya SI, Furukawa M, Bricmont P, Kashiyama E, Mallikaarjun S. In vitro P-glycoprotein interactions and steady-state pharmacokinetic interactions between tolvaptan and digoxin in healthy subjects. J Clin Pharmacol. 2010;51:761–9. doi: 10.1177/0091270010376193. [DOI] [PubMed] [Google Scholar]
- 6.Samsca® (Tolvaptan): EU Summary of Product Characteristics. Uxbridge: Otsuka Pharmaceutical Europe Ltd; 2009. [Google Scholar]
- 7.Tolvaptan (Samsca) for hyponatremia. Med Lett Drugs Ther. 2009;51:95–6. [PubMed] [Google Scholar]
- 8.Thompson CA. FDA approves oral vasopressin antagonist. Am J Health Syst Pharm. 2009;66:1154. doi: 10.2146/news090054. [DOI] [PubMed] [Google Scholar]
- 9.Fried LF, Palevsky PM. Hyponatremia and hypernatremia. Med Clin North Am. 1997;81:585–609. doi: 10.1016/s0025-7125(05)70535-6. [DOI] [PubMed] [Google Scholar]
- 10.Stoch SA, Friedman E, Maes A, Yee K, Xu Y, Larson P, Fitzgerald M, Chodakewitz J, Wagner JA. Effect of different durations of ketoconazole dosing on the single-dose pharmacokinetics of midazolam: shortening the paradigm. Clin Pharmacol. 2009;49:398–406. doi: 10.1177/0091270008331133. [DOI] [PubMed] [Google Scholar]
- 11.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin : clinical relevance. Clin Pharmacokinet. 2003;42:819–50. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 12.Jusko WJ. Guidelines for collection and analysis of pharmacokinetic data. In: Evans WE, Jusko WJ, Schentag JJ, editors. Applied Pharmacokinetics: Principles of Therapeutic Drug Monitoring. Vancouver, WA: Lippincott Williams & Wilkins; 1992. p. 1202. [Google Scholar]
- 13.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66:461–71. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- 14.Kinirons MT, O'Shea D, Kim RB, Groopman JD, Thummel KE, Wood AJJ, Wilkinson GE. Failure of erythromycin breath test to correlate with midazolam clearance as a probe of cytochrome P4503A. Clin Pharmacol Ther. 1999;66:224–31. doi: 10.1016/S0009-9236(99)70029-9. [DOI] [PubMed] [Google Scholar]
- 15.Krivoruk Y, Kinirons MT, Wood AJJ, Wood M. Metabolism of cytochrome P4503A substrates in vivo administered by the same route: lack of correlation between alfentanil clearance and erythromycin breath test. Clin Pharmacol Ther. 1994;56:608–14. doi: 10.1038/clpt.1994.185. [DOI] [PubMed] [Google Scholar]
- 16.Watkins PB, Murray SA, Winkelman LG, Heuman DM, Wrighton SA, Guzelian PS. Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J Clin Invest. 1989;83:688–97. doi: 10.1172/JCI113933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamis-Dow CA, Pearl ML, Watkins PB, Blake DS, Klecker RW, Collins JM. Predicting drug interactions in vivo from experiments in vitro. Human studies with paclitaxel and ketoconazole. Am J Clin Oncol. 1997;20:592–9. doi: 10.1097/00000421-199712000-00013. [DOI] [PubMed] [Google Scholar]
- 18.McCrea J, Prueksaritanont T, Gertz BJ, Carides A, Gillen L, Antonello S, Brucker MJ, Miller-Stein C, Osborne B, Waldman S. Concurrent administration of the erythromycin breath test (EBT) and oral midazolam as in vivo probes for CYP3A activity. J Clin Pharmacol. 1999;39:1212–20. doi: 10.1177/00912709922012015. [DOI] [PubMed] [Google Scholar]
- 19.Shoaf SE, Bramer SL, Bricmont P, Zimmer CA. Pharmacokinetic and pharmacodynamic interaction between tolvaptan, a non-peptide AVP antagonist, and furosemide or hydrochlorothiazide. J Cardiovasc Pharmacol. 2007;50:213–22. doi: 10.1097/FJC.0b013e318074f934. [DOI] [PubMed] [Google Scholar]
- 20.Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT. Metabolism of repaglinide by CYP2C8 and CYP3A4 in vitro: effect of fibrates and rifampicin. Basic Clin Pharmacol Toxicol. 2005;97:249–56. doi: 10.1111/j.1742-7843.2005.pto_157.x. [DOI] [PubMed] [Google Scholar]
- 21.Rifadin (rifampin) [Package Insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; revised July 2008. Available at http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=036ab68e-5085-4edc-bd83-784b43d64eab (last accessed 27 October 2011) [Google Scholar]