

Abstract
AIM
To predict the ceftazidime dosage regimen as a function of the glomerular filtration rate expressed by the modification of the diet in renal disease (MDRD), reason for admission and mechanical ventilation in intensive care unit (ICU) patients to treat Pseudomonas aeruginosa pneumonia.
METHOD
A published and qualified population pharmacokinetic model was used to perform Monte Carlo simulations of ceftazidime concentrations. The serum target of 40–100 mg l−1 was defined based on the minimal inhibitory concentration (MIC), the European break point (EBP), the pulmonary drug diffusion and toxicity. The recommended dosage regimens were based on the maximum percentile of the patients with simulated steady state concentrations reaching the target.
RESULTS
Steady-state was reached at 72 h whatever the MDRD. The simulations of serum concentrations generated higher percentiles of the population reaching the target after continuous administration. We recommend a 4 g continuous dose after the usual 2 g loading dose for patients with MDRD from 10 to 30 ml min−1, 6 g for MDRD between 40 and 80 ml min−1, 8 g for MDRD from 90 to 110 ml min−1, 10 g for MDRD from 120 to 190 ml min−1 and 12 g day−1 for patients with MDRD higher than 200 ml min−1.
CONCLUSION
Our study demonstrated that in ICU patients for a given MDRD, steady-state takes longer to reach in polytrauma patients than in patients with medical or post surgery reasons for admission. Continuous infusion ensures that a higher percentage of patients reaches the target than the same dose given by discontinuous administration and this only depends on MDRD.
Keywords: ceftazidime, dosage regimen, intensive care patients, pharmacokinetics, pharmacodynamics, simulation
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The large variability in drug pharmacokinetic disposition has already been described in ICU patients leading to important variations in drugs concentrations.
The usual recommended dosage of ceftazidime is not adapted for all ICU situations and ceftazidime should be monitored closely. New recommendations have to be given for some specific cases.
WHAT THIS STUDY ADDS
Our results propose individual therapeutic drug monitoring taking into account:
For the patient: the reason of admission in the ICU, the mechanical ventilation status and the creatinine clearance calculated by the modified diet in enal disease (MDRD).
For the antibiotics: the lung distribution, the minimal inhibitory concentration (MIC) of the strain to eradicate and the potential toxicity.
Introduction
In daily clinical practice, for beta-lactams, the initiation of antimicrobial treatment begins with a standard dosage regimen [1], sometimes adjusted to the body weight. The dose adjustment is essentially used for patients suffering from renal disease or cystic fibrosis [2, 3]. In most of the cases, there is no therapeutic drug monitoring and the concentrations in blood and at the site of infection are unknown [4]. The main risk consists of inefficacy with the potential emergence of antibiotic resistance and toxicity [5]. This risk is particularly high in intensive care unit (ICU) patients due to the large variability in antibiotic pharmacokinetics [6–10]. For some bacteria like Pseudomonas aeruginosa, with increased minimal inhibitory concentrations (MIC), this risk has to be managed very carefully. In these cases, ceftazidime could be an interesting drug of choice due to less risk of resistance compared with most of the beta-lactams [11]. Continuous infusion regimens of some beta-lactams have the greatest likelihood of achieving pharmacodynamic targets [12, 13]. However a 4 g ceftazidime dose by continuous infusion in critically ill patients with severe nosocomial pneumonia did not provide concentrations in excess of the MIC. Therefore, higher doses of ceftazidime needed to be administered [6, 14, 15]. Rather than using a dosage regimen mainly based on body weight, pharmacokinetic and pharmacodynamic principles, that predict antimicrobial efficacy, can be used to set targets for antimicrobial regimes and optimization [16]. For clinical practice, we performed and qualified a population model of ceftazidime disposition in ICU patients [8]. The results of that study combined the relationship between ceftazidime pharmacokinetic parameters and patient covariates that may be useful for dose adjustment, i.e. glomerular filtration rate, mechanical ventilation status and reason for admission [8].
The aim of our present work was to predict in ICU patients the best a priori adapted dosage regimen as a function of these covariates taking into account ceftazidime pharmacodynamic indexes and to give to the patient the maximal chance that his/her steady-state ceftazidime serum concentration was in the therapeutic zone as soon as possible.
Methods
Our work was only performed in silico, so no patients were included and no informed consent was required.
Data
This work was based on a NONMEM population pharmacokinetic model constructed during a previously published study [8]. The final model was qualified with the following equations and the corresponding program can be obtained from the authors.
where: TVCL, TVV1, TVQ, TVV2 are typical values, respectively, for ceftazidime clearance (l h−1), central volume of distribution (l), inter-compartmental clearance (l h−1) and peripheral volume of distribution (l).
This model was based on the following design. Briefly, a prospective, open and randomized population pharmacokinetics study of ceftazidime in 72 ICU patients was carried out, following the agreement of the Toulouse Ethics Committee. Patients were suffering from Pseudomonas aeruginosa nosocomial pneumonia. They received ceftazidime as a 6 g continuous infusion with or without a 2 g loading dose or by 2 g × 3 discontinuous injections per day. Sixty patients were mechanically ventilated. Admitting diagnosis was poly-traumatism (n = 27), post surgery (n = 19) and medical reason (n = 26). The mean glomerular filtration rate estimated by the MDRD formula [17] was 121 ± 55 ml min−1. The data from 49 patients were used for the model building and those of 23 patients for the qualification. The model was qualified by a comparison of the predicted and observed concentrations in the 23 patients. A final model was elaborated from the whole population.
Table 1 presents the typical value for the central, peripheral and total volume of distribution fitted for the six sub groups as a function of mechanical ventilation status and reason for admission.
Table 1.
Typical values of the volume of distribution (l) in the sub-populations defined by their mechanical ventilation status (without = I0 or with = I1) and their reason of admission (P0 = polytrauma, P1 = post surgery and P2 = medical reason)
| V1+ V2 (l) | V1 (l) | V2 (l) | |
|---|---|---|---|
| I0P0 | 76.0 | 18.9 | 57.1 |
| I0P1 | 44.6 | 18.9 | 25.7 |
| I0P2 | 32.5 | 18.9 | 13.6 |
| I1P0 | 66.1 | 9.0 | 57.1 |
| I1P1 | 34.7 | 9.0 | 25.7 |
| I1P2 | 22.6 | 9.0 | 13.6 |
Simulation
Typical patients were defined as a function of the combination of the selected covariates of the model:
Glomerular filtration rate estimated by MDRD from 10 to 200 ml min−1 with a step of 10 ml min−1 (20 cases). This range is included into the range of the population used to elaborate the model (7.9–253 ml min−1).
Ventilation status: I0 without mechanical ventilation and I1 with mechanical ventilation (two cases).
Admitting diagnoses: P0 = polytrauma, P1 = post surgery and P2 = medical reason (three cases).
In total, 120 (20 × 2 × 3) typical patients were studied.
Several ceftazidime dosage regimens were simulated: discontinuous administration of 2 g × 3 day−1 and continuous administration of 4, 6, 8, 10 and 12 g day−1 after a 2 g loading dose and 6 g day−1 after a 3 g loading dose.
For each studied dosage regimen and each of the 120 typical patients, 1000 simulated pharmacokinetic profiles were obtained by a NONMEM Monte Carlo procedure.
Interpretation
Steady-state was considered as reached when the variation of the concentration (continuous infusion) or trough concentrations (discontinuous injections) were lower than 10% from one day to another. This corresponds with the usual 90% target steady-state concentration.
For efficiency, the target serum concentration at steady-state, whatever the dosage regimen, was defined as a steady-state concentration (continuous infusion) or a trough concentration (discontinuous injections) higher than 40 mg l−1[18]. On the one hand, this 40 mg l−1 target corresponds to five-fold the European break point (8 mg l−1) [19, 20]. On the other hand, in our ICU patients, the most frequently observed MIC for Pseudomonas aeruginosa was 2 mg l−1. The 40 mg l−1 target corresponds to the combination of two parameters, a local concentration at four-fold the MIC (i.e. 8 mg l−1) and a ceftazidime disposition in the alveolar film evaluated to 20% of the blood concentration [14, 21]. For safety reasons, a maximum limit was defined as a 100 mg l−1 concentration by assimilation to the described toxicity of cefepime [22].
For the 1000 simulations of each typical patient dataset, we determined the percentiles of the concentrations which reached the goals at 24, 48 and 72 h to investigate the time required to reach steady-state.
The dosage regimen recommendations were established on the base of the maximum percentile of patients reaching the target interval of 40–100 mg l−1 after a 2 g loading dose followed by continuous infusion of ceftazidime.
Results
In the ICU population of our pivotal study, glomerular filtration rate estimated by MDRD was 141 ± 34 ml min−1 in the P0 subgroup (polytrauma), 92 ± 48 ml min−1 in the P1 subgroup (post surgery) and 120 ± 69 ml min−1 in the P2 (medical reason).
Table 2 shows the time required to reach steady-state as a function of the three covariates including in the model during discontinuous administration and continuous infusion.
Table 2.
Time required to reach steady-state (tss) AS A function of the glomerular filtration rate estimated by MDRD clearance (ml min−1) in the sub-populations defined by their mechanical ventilation status (without = I0 or with = I1) and their reason for admission (P0 = polytrauma, P1 = post surgery and P2 = medical reason) after discontinuous administration of 2 g × 3 day−1 and a 2 g loading dose followed by 6 g day−1 continuous infusion
| 2 g × 3 day−1 | 2 g loading dose + 6 g infusion | |||||||
|---|---|---|---|---|---|---|---|---|
| MDRD (ml min−1) | tss (h) | MDRD (ml min−1) | tss (h) | MDRD (ml min−1) | tss (h) | MDRD (ml min−1) | tss (h) | |
| I0P0 | ≤60 | 72 h | >60 | 48 h | ≤120 | 48 h | >120 | 24 h |
| I0P1 | ≤140 | 48 h | >140 | 24 h | ≤30 | 48 h | >30 | 24 h |
| I0P2 | ≤70 | 48 h | >70 | 24 h | – | – | ≥10 | 24 h |
| I1P0 | ≤50 | 72 h | >50 | 48 h | ≤90 | 48 h | >90 | 24 h |
| I1P1 | ≤100 | 48 h | >100 | 24 h | – | – | ≥10 | 24 h |
| I1P2 | ≤20 | 48 h | >20 | 24 h | – | – | ≥10 | 24 h |
Figure 1 presents the percentile of the ceftazidime simulated concentrations included in the target interval of 40–100 mg l−1. To ensure multiple comparisons, this figure has been divided into four parts corresponding to a 2 g × 3 g day−1 on the left (A and C) and a continuous infusion of 6 g day−1 after a 2 g loading dose on the right (B and D). The upper graphs (A and B) correspond to the results at 24 h and the lower graphs (C and D) at 72 h.
Figure 1.
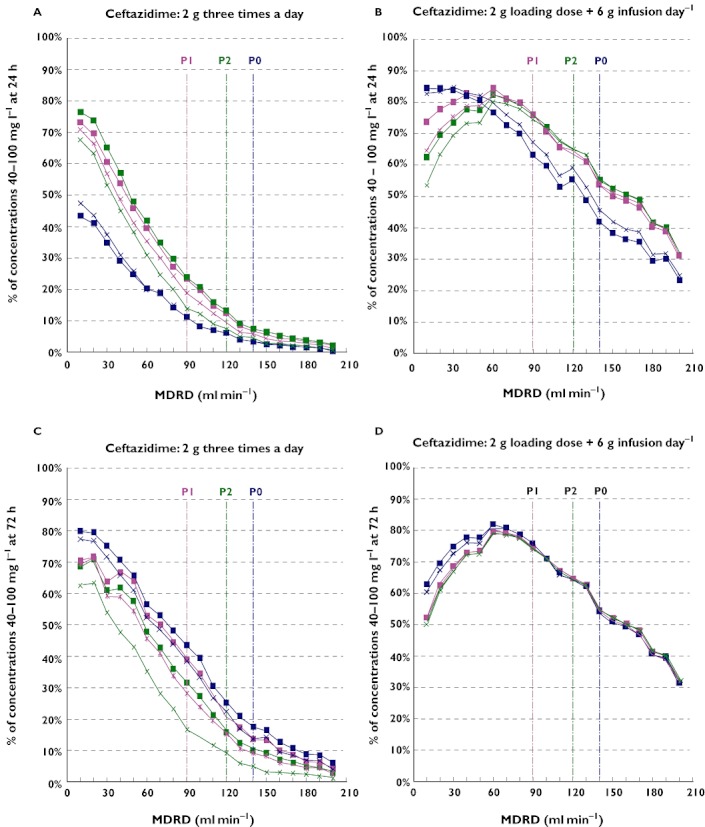
Percentage of the ceftazidime simulated concentrations in the 40–100 mg l−1 target interval after 24 h (A and B) and 72 h (C and D) for a 2 g x 3 day–1 (A and C) and a 2 g loading dose + 6 g continuous infusion (B and D) in patients with (I1) or without (I0) mechanical ventilation as a function of MDRD in ml min−1. The admitting diagnoses were: P0 = polytrauma, P1 = post surgical and P2 = medical reason. The vertical colour lines correspond to the mean glomerular filtration rate estimated by MDRD in these three subpopulations. I0P0 ( ); I0P1 (
); I0P1 ( ); I0P2 (
); I0P2 ( ); I1P0 (
); I1P0 ( ); I1P1 (
); I1P1 ( ); I1P2 (
); I1P2 ( )
)
Comparison between the upper and lower graphs shows that steady-state is not reached at 24 h for all patients as demonstrated in Table 2. Particularly, polytrauma patients (P0) with a MDRD lower than 60 ml min−1 required 72 h to be at steady-state and other polytrauma patients had a 48 h delay. To take this point into account, all the following conclusions have been based on 72 h simulations.
When comparing the graphs in Figure 1C and D, the simulations generated higher percentiles after continuous administration than discontinuous injections for all subpopulations with MDRD higher than 60 ml min−1. On the contrary, for patients with a MDRD lower than 60 ml min−1, the percentage of patients in the target interval was lower due to higher concentrations.
At steady-state, after continuous administration (Figure 1D) all the subpopulations with a MDRD higher than 60 ml min−1 presented exactly the same results due to the exclusive dependency of the steady-state concentrations on ceftazidime clearance and therefore to the MDRD. For patients with renal insufficiency, the percentage of patients in the target interval was higher for polytrauma patients since the simulated concentrations were lower. This is the consequence of the larger impact of the greater volume of distribution on ceftazidime disposition. No influence of the mechanical ventilation status was observed.
On another hand, the trough concentrations at steady-state observed after discontinuous administrations (Figure 1C) vary between subgroups due to their partial dependency to the volume of distribution including admitting diagnosis and mechanical ventilation as covariates.
Simulations were performed with 2 g and 3 g loading doses followed by a 6 g day−1 continuous infusion. After 24 h for patients with a MDRD higher than 60 ml min−1, the loading dose had no influence on the percentile of patients' concentrations in the target interval. For patients with MDRD lower than 60 ml min−1, the percentage of patients with C24 included in the 40–100 mg l−1 interval was lower with a 3 g loading dose than with 2 g dose due to a greater number of patients with concentrations higher than 100 mg ml−1. Therefore, the 2 g loading dose was chosen.
Figure 2 presents the percentile of simulated concentrations included in the 40–100 mg l−1 goal after a 2 g loading dose followed by various increasing continuous doses in polytrauma patients. The coloured arrows show the dosage regimen recommendations as a function of MDRD.
Figure 2.
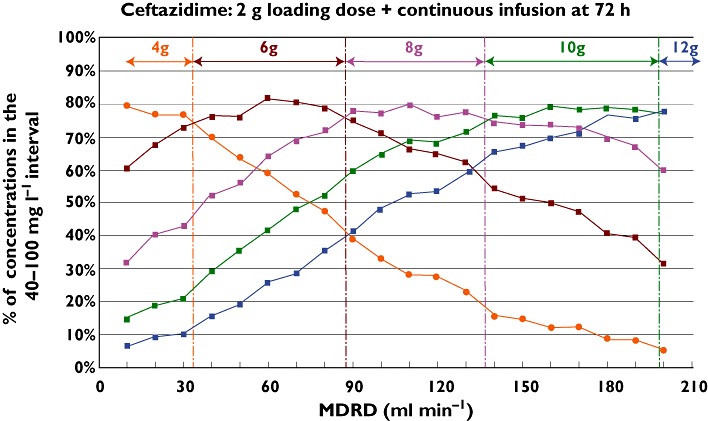
Percentage of simulated patients with 72 h ceftazidime concentrations included in the 40–100 mg l−1 target interval after a 2 g loading dose + different continuous infusion doses as a function of glomerular filtration rate estimated by MDRD in ml min−1 whatever the admitting diagnosis and the mechanical ventilation status. The coloured arrows present the recommended dosage as a function of MDRD. 4 g ( ); 6 g (
); 6 g ( ); 8 g (
); 8 g ( ); 10 g (
); 10 g ( ); 12 g (
); 12 g ( )
)
As previously demonstrated, the results of the different subgroups stratified by admitting diagnosis, with or without mechanical ventilation, are totally superimposable for the same dosage regimen given as a continuous infusion in patients with a MDRD higher than 60 ml min−1.
As the maximal percentile target has been chosen to determine the recommended dosage regimen, this figure could help everyone to define it. For example, a patient with a MDRD of 90 ml min−1 roughly requires a 8 g continuous dose after the usual 2 g loading dose; another patient with a MDRD at 120 ml min−1 would be also treated by a 8 g dosage and for a patient with a MDRD at 140 ml min−1 a 10 g day−1 dose would be recommended. Figure 2 leads to the dosage regimen recommendations presented in Table 3. Even if the polytrauma patients with a MDRD lower than 60 ml min−1 presented a small difference (less than 5%) in the maximal percentage of patients reaching the goal, the recommended dosage regimens are the same as in patients hospitalized for other reasons.
Table 3.
Recommended dosage regimen to reach the maximum percentage of ICU patients with a 40–100 mg l−1 target interval concentration at steady-state after a 2 g loading dose and continuous infusion expressed as a function of glomerular filtration rate estimated by MDRD whatever the reason for admission and the mechanical ventilation status
| MDRD (ml min−1) | Recommended dosage: 2 g loading dose followed by |
|---|---|
| 10–30 | 4 g day−1 |
| 40–80 | 6 g day−1 |
| 90–110 | 8 g day−1 |
| 120–190 | 10 g day−1 |
| ≥200 | 12 g day−1 |
Discussion
A prompt initiation of the right antibiotic therapy is the cornerstone to maximize the successful outcome of treatment. In ICU patients, due to the high pharmacokinetic variability, a standard dosage regimen of ceftazidime is not the best predictor of successful treatment and this could lead to therapeutic failure and/or to the emergence of resistance [6, 23, 24].
Efficient therapeutic drug monitoring requires evaluating not only the pharmacokinetic disposition of the drug, but also the antibiotic susceptibility of the bacteria so that the target dose can be determined. The antibiotic susceptibility may be defined by the actual MIC of the strain or by the European break point (EBP) if the MIC is not available [25].
Several approaches have been developed for prediction purposes by the combination of factors influencing pharmacokinetics and pharmacodynamics [26–28]. The pharmacodynamic relationship, historically thought to be predictive of beta-lactam efficacy, is the percentage of the dosing interval that the free drug concentration remains above the MIC of the infecting organism [29]. When the time higher than the MIC for total drug was evaluated, targets of 90 to 100% were required for predictable microbiological success in clinical practice [30–33]. AUIC (ratio of the area under the concentration curve : MIC) has been evaluated for beta-lactams and must be higher than 125 [16], which corresponds to a 40 mg l−1 target serum concentration for a MIC of 8 mg l−1. The pharmacokinetic/pharmacodynamic index values derived from concentrations measured in serum over time are not sufficient to infer similar effects from tissue concentrations or local concentrations [34]. The infected tissue concentrations would be the most relevant target but in clinical practice, these data are not available [5, 35]. Consequently, therapeutic drug monitoring has to be based on blood concentrations which are the easiest measurements to assess [28, 36]. Measurements in specific compartments or determining free concentrations in interstitial fluid by microdialysis contribute to the understanding of concentrations at the site of infection [37]. According to Bergogne-Berezin & Boselli [14, 21, 38], ceftazidime lung concentrations correspond to 20% of the serum concentration. Since beta-lactams have effective local concentrations about four-fold the MIC [19], the serum concentration probably may be 20-fold the MIC to ensure the effective treatment of pneumonia.
From another point of view, even if the relationship between ceftazidime concentration and toxicity is poorly described [39], a maximum concentration had to be fixed. We have chosen 100 mg l−1 by assimilation to the described toxicity of cefepime [22] which is chemically and pharmacokinetically very close to ceftazidime [40].
Our study evidenced that steady-state is not reached at the same time for the different groups of patients and the various modes of administration as shown in Table 2. As it was predictable, the loading dose of 2 g usually used in clinical practice, followed by continuous infusion provided a quicker time to steady-state than discontinuous injections [41]. In polytrauma patients with a normal or low glomerular filtration rate estimated by MDRD, steady-state is never reached before the 48 h whatever the schedule of administration and this could increase the risk of inefficacy [42]. The time required to reach steady-state (five-fold half-life) is always proportional to the volume of distribution of the drug and antiproportional to the drug clearance. When comparing profiles determined for different glomerular filtration rates estimated by MDRD and therefore different drug clearances, the influence of the volume of distribution is more important in polytrauma patients than in the two others groups. This can easily be explained since in polytrauma patients the total volume of distribution (57 l) is very high compared with the other groups (13 l and 26 l) as shown in Table 1[8, 43, 44].
Since beta lactams are time dependent antibiotics, another way to increase potentially the luck of success could be by increasing the loading dose. In fact, the comparison of a 2 g vs. 3 g loading dose leads to comparable concentrations at 24 h for patients with a MDRD higher than 60 ml min−1. For a MDRD lower than 60 ml min−1, a greater number of patients with concentrations higher than 100 mg ml−1 was observed with the 3 g loading dose.
For continuous infusion, when steady-state is reached, for a given glomerular filtration rate estimated by MDRD and higher than 60 ml min−1, whatever the subpopulation, pharmacokinetic profiles are super-imposable for a same dosage regimen. As it was pharmacokinetically predictable, at steady-state, only the MDRD, which is the best predictor of GFR [17], has an influence on ceftazidime disposition.
The second point evidenced by our study is that the target of 40–100 mg l−1 is reached in very few cases after the standard dose of 2 g × 3 day−1. For the mean glomerular filtration rate estimated by MDRD in our sub-populations (ranging from 90 to 140 ml min−1) the percentage reaching the target interval is always lower than 25% at 24 h and lower than 45% at steady-state. This is due to low plasma concentrations and therefore this could lead to inefficacy as previously described [6, 45]. These observations may be improved by changing the mode of administration and giving a loading dose followed by a continuous infusion, as it has already been suggested for different beta-lactams [12, 40, 41, 46–48]. For the mean glomerular filtration rates estimated by MDRD in our sub-populations (ranging from 90 to 140 ml min−1), 24 h after a 6 g continuous infusion, the target interval is reached in 75% of the post surgical patients and 65% of medical patients. For polytrauma patients, only 45% of the subpopulation reached the goal. When polytrauma patients' concentrations are at steady-state, only 55% satisfy to the efficacy criteria for a mean glomerular filtration rate estimated by MDRD of 140 ml min−1.
For patients with a MDRD higher than 60 ml min−1, the only solution to satisfy to the defined criteria (a steady-state concentration in the 40–100 mg l−1 interval reached by a maximum of patients), is to increase the dosage regimen [49, 50]. For patients with renal insufficiency, the low percentage of patients reaching the target interval is essentially due to concentrations higher than 100 mg l−1, so the dose must be decreased.
As shown in Figure 2, after a 2 g loading dose, the recommended dosage regimen is a function of the MDRD whatever the subpopulation. The usually recommended 6 g continuous dose is only valuable for MDRD between 30 and 90 ml mn−1. For a MDRD lower than 30 ml min−1 a 4 g dose is sufficient but higher than the dose recommended in the summary of the characteristics of the product. For patients with normal renal function with a MDRD higher than 90 ml min−1 the required doses are from 8 g to 12 g day−1. These observations are totally in accordance with those reported by Taconne et al. and Seyler et al. in other populations [6, 15].
However, if the uncertainty of the pharmacokinetic parameters of the model is taken into account, an overlap of the borders of the decision-making interval for the recommended dose is observed[8]. This leads to a risk of under/over estimation for the MDRD at the edges of the range for each recommended dose. That is why these recommendations must be systematically followed by a measurement of the steady-state serum concentration.
Our study demonstrated that in ICU patients for a given glomerular filtration rate estimated by MDRD, steady-state takes longer to be reached in polytrauma patients than in patients with medical or post surgery reasons for admission due to a higher volume of distribution. Continuous infusion ensures a higher percentile of patients reaching the serum target concentration interval of 40–100 mg l−1 than the same dose given by discontinuous administration. At steady-state after continuous administration, the mechanical ventilation status and the reason for admission have no influence on the drug disposition and the dosage regimen must only be determined as a function of the glomerular filtration rate estimated by MDRD.
Acknowledgments
We acknowledge the help of Professor John Woodley with the English language.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Sun HK, Kuti JL, Nicolau DP. Pharmacodynamics of antimicrobials for the empirical treatment of nosocomial pneumonia: a report from the OPTAMA Program. Crit Care Med. 2005;33:2222–7. doi: 10.1097/01.ccm.0000181528.88571.9b. [DOI] [PubMed] [Google Scholar]
- 2.Vinks AA, Mouton JW, Touw DJ, Heijerman HG, Danhof M, Bakker W. Population pharmacokinetics of ceftazidime in cystic fibrosis patients analyzed by using a nonparametric algorithm and optimal sampling strategy. Antimicrob Agents Chemother. 1996;40:1091–7. doi: 10.1128/aac.40.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulitta JB, Landersdorfer CB, Huttner SJ, Drusano GL, Kinzig M, Holzgrabe U, Stephan U, Sorgel F. Population pharmacokinetic comparison and pharmacodynamic breakpoints of ceftazidime in cystic fibrosis patients and healthy volunteers. Antimicrob Agents Chemother. 2010;54:1275–82. doi: 10.1128/AAC.00936-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taccone FS, Laterre PF, Spapen H, Dugernier T, Delattre I, Layeux B, De Backer D, Wittebole X, Wallemacq P, Vincent JL, Jacobs F. Revisiting the loading dose of amikacin for patients with severe sepsis and septic shock. Crit Care. 2010;14:R53. doi: 10.1186/cc8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts JA, Roberts MS, Semark A, Udy AA, Kirkpatrick CM, Paterson DL, Roberts MJ, Kruger P, Lipman J. Antibiotic dosing in the ‘at risk’ critically ill patient: linking pathophysiology with pharmacokinetics/pharmacodynamics in sepsis and trauma patients. BMC Anesthesiol. 2011;11:3. doi: 10.1186/1471-2253-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taccone FS, Laterre PF, Dugernier T, Spapen H, Delattre I, Wittebole X, De Backer D, Layeux B, Wallemacq P, Vincent JL, Jacobs F. Insufficient beta-lactam concentrations in the early phase of severe sepsis and septic shock. Crit Care. 2010;14:R126. doi: 10.1186/cc9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meagher AK, Forrest A, Rayner CR, Birmingham MC, Schentag JJ. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob Agents Chemother. 2003;47:548–53. doi: 10.1128/AAC.47.2.548-553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georges B, Conil JM, Seguin T, Ruiz S, Minville V, Cougot P, Decun JF, Gonzalez H, Houin G, Fourcade O, Saivin S. Population pharmacokinetics of ceftazidime in intensive care unit patients: influence of glomerular filtration rate, mechanical ventilation and reason for admission. Antimicrob Agents Chemother. 2009;53:4483–9. doi: 10.1128/AAC.00430-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joukhadar C, Frossard M, Mayer BX, Brunner M, Klein N, Siostrzonek P, Eichler HG, Muller M. Impaired target site penetration of beta-lactams may account for therapeutic failure in patients with septic shock. Crit Care Med. 2001;29:385–91. doi: 10.1097/00003246-200102000-00030. [DOI] [PubMed] [Google Scholar]
- 10.Roberts JA, Lipman J. Antibacterial dosing in intensive care: pharmacokinetics, degree of disease and pharmacodynamics of sepsis. Clin Pharmacokinet. 2006;45:755–73. doi: 10.2165/00003088-200645080-00001. [DOI] [PubMed] [Google Scholar]
- 11.Georges B, Conil JM, Dubouix A, Archambaud M, Bonnet E, Saivin S, Lauwers-Cances V, Cristini C, Cougot P, Decun JF, Mathe O, Chabanon G, Marty N, Seguin T, Houin G. Risk of emergence of Pseudomonas aeruginosa resistance to beta-lactam antibiotics in intensive care units. Crit Care Med. 2006;34:1636–41. doi: 10.1097/01.CCM.0000215517.51187.CA. [DOI] [PubMed] [Google Scholar]
- 12.Girardi C, Tonnellier M, Goldstein I, Sartorius A, Wallet F, Rouby JJ. Lung deposition of continuous and intermittent intravenous ceftazidime in experimental Pseudomonas aeruginosa bronchopneumonia. Intensive Care Med. 2006;32:2042–8. doi: 10.1007/s00134-006-0272-9. [DOI] [PubMed] [Google Scholar]
- 13.Frei CR, Burgess DS. Continuous infusion beta-lactams for intensive care unit pulmonary infections. Clin Microbiol Infect. 2005;11:418–21. doi: 10.1111/j.1469-0691.2005.01106.x. [DOI] [PubMed] [Google Scholar]
- 14.Boselli E, Breilh D, Rimmele T, Poupelin JC, Saux MC, Chassard D, Allaouchiche B. Plasma and lung concentrations of ceftazidime administered in continuous infusion to critically ill patients with severe nosocomial pneumonia. Intensive Care Med. 2004;30:989–91. doi: 10.1007/s00134-004-2171-2. [DOI] [PubMed] [Google Scholar]
- 15.Seyler L, Cotton F, Taccone FS, De Backer D, Macours P, Vincent JL, Jacobs F. Recommended beta-lactam regimens are inadequate in septic patients treated with continuous renal replacement therapy. Crit Care. 2011;15:R137. doi: 10.1186/cc10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinnon PS, Paladino JA, Schentag JJ. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents. 2008;31:345–51. doi: 10.1016/j.ijantimicag.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–70. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 18.Aubert G, Carricajo A, Coudrot M, Guyomarc'h S, Auboyer C, Zeni F. Prospective determination of serum ceftazidime concentrations in intensive care units. Ther Drug Monit. 2010;32:517–9. doi: 10.1097/FTD.0b013e3181e60ca6. [DOI] [PubMed] [Google Scholar]
- 19.Mouton JW, den Hollander JG. Killing of Pseudomonas aeruginosa during continuous and intermittent infusion of ceftazidime in an in vitro pharmacokinetic model. Antimicrob Agents Chemother. 1994;38:931–6. doi: 10.1128/aac.38.5.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouton JW, Punt N, Vinks AA. A retrospective analysis using Monte Carlo simulation to evaluate recommended ceftazidime dosing regimens in healthy volunteers, patients with cystic fibrosis, and patients in the intensive care unit. Clin Ther. 2005;27:762–72. doi: 10.1016/j.clinthera.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Bergogne-Berezin E, Pierre J, Berthelot G, Kafe H, Even P, Gibert C, Safran D, Stern M. [Bronchial diffusion of new anti-pseudomonal beta-lactams. Clinical significance] Pathol Biol. 1984;32:421–5. [PubMed] [Google Scholar]
- 22.Jallon P, Fankhauser L, Du Pasquier R, Coeytaux A, Picard F, Hefft S, Assal F. Severe but reversible encephalopathy associated with cefepime. Neurophysiol Clin. 2000;30:383–6. doi: 10.1016/s0987-7053(00)00234-3. [DOI] [PubMed] [Google Scholar]
- 23.Andes D, Anon J, Jacobs MR, Craig WA. Application of pharmacokinetics and pharmacodynamics to antimicrobial therapy of respiratory tract infections. Clin Lab Med. 2004;24:477–502. doi: 10.1016/j.cll.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 24.DeRyke CA, Kuti JL, Nicolau DP. Pharmacodynamic target attainment of six beta-lactams and two fluoroquinolones against Pseudomonas aeruginosa, Acinetobacter baumannii, Escherichia coli, and Klebsiella species collected from United States intensive care units in 2004. Pharmacotherapy. 2007;27:333–42. doi: 10.1592/phco.27.3.333. [DOI] [PubMed] [Google Scholar]
- 25.Cavallo JD, Merens A. [Pseudomonas aeruginosa and beta-lactam antibiotics at the time of Europe] Pathol Biol. 2008;56:435–8. doi: 10.1016/j.patbio.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 26.Bressolle F, Gomeni R. Predictive performance of a semiparametric method to estimate population pharmacokinetic parameters using NONMEM. J Pharmacokinet Biopharm. 1998;26:349–61. doi: 10.1023/a:1023289527297. [DOI] [PubMed] [Google Scholar]
- 27.Craig WA. Proof of concept: performance testing in models. Clin Microbiol Infect. 2004;10(Suppl. 2):12–7. doi: 10.1111/j.1470-9465.2004.00865.x. [DOI] [PubMed] [Google Scholar]
- 28.Tam VH, Louie A, Lomaestro BM, Drusano GL. Integration of population pharmacokinetics, a pharmacodynamic target, and microbiologic surveillance data to generate a rational empiric dosing strategy for cefepime against Pseudomonas aeruginosa. Pharmacotherapy. 2003;23:291–5. doi: 10.1592/phco.23.3.291.32110. [DOI] [PubMed] [Google Scholar]
- 29.Turnidge JD. The pharmacodynamics of beta-lactams. Clin Infect Dis. 1998;27:10–22. doi: 10.1086/514622. [DOI] [PubMed] [Google Scholar]
- 30.Lee SY, Kuti JL, Nicolau DP. Cefepime pharmacodynamics in patients with extended spectrum beta-lactamase (ESBL) and non-ESBL infections. J Infect. 2007;54:463–8. doi: 10.1016/j.jinf.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 31.Tam VH, McKinnon PS, Akins RL, Rybak MJ, Drusano GL. Pharmacodynamics of cefepime in patients with Gram-negative infections. J Antimicrob Chemother. 2002;50:425–8. doi: 10.1093/jac/dkf130. [DOI] [PubMed] [Google Scholar]
- 32.Crandon JL, Bulik CC, Kuti JL, Nicolau DP. Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2010;54:1111–6. doi: 10.1128/AAC.01183-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolau DP, Onyeji CO, Zhong M, Tessier PR, Banevicius MA, Nightingale CH. Pharmacodynamic assessment of cefprozil against Streptococcus pneumoniae: implications for breakpoint determinations. Antimicrob Agents Chemother. 2000;44:1291–5. doi: 10.1128/aac.44.5.1291-1295.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O. Tissue concentrations: do we ever learn? J Antimicrob Chemother. 2008;61:235–7. doi: 10.1093/jac/dkm476. [DOI] [PubMed] [Google Scholar]
- 35.Kiem S, Schentag JJ. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother. 2008;52:24–36. doi: 10.1128/AAC.00133-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieft H, Hoepelman AI, Knupp CA, van Dijk A, Branger JM, Struyvenberg A, Verhoef J. Pharmacokinetics of cefepime in patients with the sepsis syndrome. J Antimicrob Chemother. 1993;32(Suppl. B):117–22. doi: 10.1093/jac/32.suppl_b.117. [DOI] [PubMed] [Google Scholar]
- 37.Theuretzbacher U. Tissue penetration of antibacterial agents: how should this be incorporated into pharmacodynamic analyses? Curr Opin Pharmacol. 2007;7:498–504. doi: 10.1016/j.coph.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Bergogne-Berezin E. Antibiotic tissue concentrations revisited in lungs. Crit Care Med. 2003;31:2242–3. doi: 10.1097/01.CCM.0000069735.58871.26. [DOI] [PubMed] [Google Scholar]
- 39.Jackson GD, Berkovic SF. Ceftazidime encephalopathy: absence status and toxic hallucinations. J Neurol Neurosurg Psychiatry. 1992;55:333–4. doi: 10.1136/jnnp.55.4.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conil JM, Georges B, Lavit M, Seguin T, Tack I, Samii K, Chabanon G, Houin G, Saivin S. Pharmacokinetics of ceftazidime and cefepime in burn patients: the importance of age and creatinine clearance. Int J Clin Pharmacol Ther. 2007;45:529–38. doi: 10.5414/cpp45529. [DOI] [PubMed] [Google Scholar]
- 41.Roberts JA, Kirkpatrick CM, Roberts MS, Dalley AJ, Lipman J. First-dose and steady-state population pharmacokinetics and pharmacodynamics of piperacillin by continuous or intermittent dosing in critically ill patients with sepsis. Int J Antimicrob Agents. 2010;35:156–63. doi: 10.1016/j.ijantimicag.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Zubert S, Funk DJ, Kumar A. Antibiotics in sepsis and septic shock: like everything else in life, timing is everything. Crit Care Med. 2010;38:1211–2. doi: 10.1097/CCM.0b013e3181d69db7. [DOI] [PubMed] [Google Scholar]
- 43.Gomez CM, Cordingly JJ, Palazzo MG. Altered pharmacokinetics of ceftazidime in critically ill patients. Antimicrob Agents Chemother. 1999;43:1798–802. doi: 10.1128/aac.43.7.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zagli G, Tarantini F, Bonizzoli M, Di Filippo A, Peris A, De Gaudio AR, Geppetti P. Altered pharmacology in the intensive care unit patient. Fundam Clin Pharmacol. 2008;22:493–501. doi: 10.1111/j.1472-8206.2008.00623.x. [DOI] [PubMed] [Google Scholar]
- 45.Lipman J, Gomersall CD, Gin T, Joynt GM, Young RJ. Continuous infusion ceftazidime in intensive care: a randomized controlled trial. J Antimicrob Chemother. 1999;43:309–11. doi: 10.1093/jac/43.2.309. [DOI] [PubMed] [Google Scholar]
- 46.Nicolau DP, McNabb J, Lacy MK, Quintiliani R, Nightingale CH, Banevicius MA, Fu Q. Continuous versus intermittent administration of ceftazidime in intensive care unit patients with nosocomial pneumonia. Int J Antimicrob Agents. 2001;17:497–504. doi: 10.1016/s0924-8579(01)00329-6. [DOI] [PubMed] [Google Scholar]
- 47.Roberts JA, Paratz J, Paratz E, Krueger WA, Lipman J. Continuous infusion of beta-lactam antibiotics in severe infections: a review of its role. Int J Antimicrob Agents. 2007;30:11–8. doi: 10.1016/j.ijantimicag.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Roberts JA, Kirkpatrick CM, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Meropenem dosing in critically ill patients with sepsis and without renal dysfunction: intermittent bolus versus continuous administration? Monte Carlo dosing simulations and subcutaneous tissue distribution. J Antimicrob Chemother. 2009;64:142–50. doi: 10.1093/jac/dkp139. [DOI] [PubMed] [Google Scholar]
- 49.Udy AA, Roberts JA, Boots RJ, Paterson DL, Lipman J. Augmented renal clearance: implications for antibacterial dosing in the critically ill. Clin Pharmacokinet. 2010;49:1–16. doi: 10.2165/11318140-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 50.Udy AA, Putt MT, Shanmugathasan S, Roberts JA, Lipman J. Augmented renal clearance in the Intensive Care Unit: an illustrative case series. Int J Antimicrob Agents. 2010;35:606–8. doi: 10.1016/j.ijantimicag.2010.02.013. [DOI] [PubMed] [Google Scholar]