

Abstract
Congenital heart disease (CHD) is the most common birth defect in humans. It is a leading infant mortality factor worldwide, caused by defective cardiac development. Mutations in transcription factors, signalling and structural molecules have been shown to contribute to the genetic component of CHD. Recently, mutations in genes encoding myofibrillar proteins expressed in the embryonic heart have also emerged as an important genetic causative factor of the disease, which implies that the contraction of the early heart primordium contributes to its morphogenesis. This notion is supported by increasing evidence suggesting that not only contraction but also formation, mechanosensing, and mechanotransduction of the cardiac myofibrillar proteins influence heart development. In this paper, we summarize the genetic clues supporting this idea.
1. Introduction
The heart is the first organ to form in vertebrates during embryogenesis, and cardiac contraction initiates shortly after the cardiac tube is formed. The ever increasing needs of the peripheral tissues of the growing embryo for nutrients and oxygen are met gradually by the blood circulation induced by early cardiac contraction. The heart primordium has to perform its function for most of its own development, and this unique feature has profound implications in the relationship between form and function during cardiogenesis: factors that compromise early cardiac function result in altered morphology, and heart malformation usually has an important impact over the function of the organ. Defective heart development results in congenital heart disease (CHD), the commonest congenital defect in humans and a leading cause of infant mortality worldwide. CHD is a complex disease, as multiple genetic and environmental factors have been implicated in its pathogenesis. As in most complex diseases, most cases are sporadic, although a relatively small fraction of cases show familial segregation, following Mendelian patterns of inheritance. The study and identification of the genes causing the familial forms of the disorder can also give important information about the genetic component of the more common, complex variety.
Genetic linkage is a technique that identifies genes responsible for inherited traits solely based in their position on chromosomes without prior knowledge of their product or its function. More than two decades ago, by genetic linkage analysis, a mutation of a gene encoding a myofibrillar protein (β-cardiac myosin heavy chain, MYH7) was shown to cause another cardiac disease, hypertrophic cardiomyopathy (HCM) [1]. Since then, mutations in multiple genes encoding myofibrillar proteins have been identified as responsible for other cases of this disease and other types of cardiomyopathy: dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), and restrictive cardiomyopathy (RCM) (for a recent review, see Ghosh and Haddad, 2011) [2].
The first genes shown to be responsible for familial cases of CHD (NKX2-5 and GATA4) encode transcription factors. Subsequently, mutations in genes that encode other types of protein, like signalling and structural molecules, have been described in families with Mendelian CHD, and common variants have been identified as predisposing factors for non-Mendelian CHD in large cohorts of sporadic cases (for a review, see Wessels and Willems, 2010) [3].
Some relatively recent developments in the search for the genetic origins of CHD were initiated by an unexpected observation: a causative point mutation in chromosome 14 was identified in a large family with an autosomal dominant atrial septal defect (ASD), one of the most common forms of CHD [4]. This mutation is located in a gene that encodes a myofibrillar protein (α-cardiac myosin heavy chain, MYH6). Subsequently, we and others have shown that mutations in other genes that encode other myofibrillar proteins can cause defective cardiac development [5–10]. Traditionally, compromised contractility of the cardiac muscle by the presence of dominant-negative mutant myofibrillar proteins or their haploinsufficiency (for nonsense mutations) was expected to cause only functional phenotypes during postnatal life which in turn induce a maladaptive response, as occurs in the different types of cardiomyopathy. The unexpected observation that mutations in these genes can also affect the functionality of the heart during its early development and thus induce cardiac malformation is supported by analysis of spontaneous and induced mutations in model organisms.
In this paper, we discuss recent findings that indicate the importance of the functionality of the myofibrils, specifically their formation, contractility and mechanotransduction, in several aspects of cardiogenesis.
2. Overview of Cardiac Development
Cardiac development is a complex process, by which an initial primordium consists of a straight tube of cardiac muscle (myocardium), lined by a single layer of endothelial cells (endocardium) [11], bends to the right (looping) in order to define the prospective chamber that receives blood through the veins (primitive atrium) from the chamber that expel blood to the arteries (primitive ventricle) [12]. The extracellular matrix that separates myocardium and endocardium (cardiac jelly) expands to form the endocardial cushions, located between the prospective atrium and ventricle and between this and the prospective outflow vessel [13]. The myocardium and endocardium forming the wall of the future chambers expand (ballooning) [14] and penetrate the interior of the chambers with ramified ridge-like structures called trabeculae [15]. Both the primitive atrium and ventricle are divided into right and left chambers by atrial and ventricular septation, respectively, resulting in the adult configuration of a right and left atria and a right and left ventricle, and each ventricle is aligned with its atrial counterpart [16]. Both the endocardial cushions and the trabeculae contribute to both atrial and ventricular septation. Additionally, the endocardial cushions condense to form the valves between atria and ventricles and between the ventricles and the prospective outflow vessels. Trabeculae also form the papillary muscles that anchor the valves to the inner surface of the ventricles [17].
3. Defective Myofibrillogenesis in the Heart Causes Cardiac Malformation
In vivo studies in chicken and mouse have shown that tropomodulin is an essential component of the myofibrils required for its proper formation [18, 19]. This protein is located in the membrane of the differentiating myocardial cell, closely associated with spectrin [18]. Once tropomyosin localizes within the thin filament, tropomodulin localizes in the pointed end of the actin filament, where it is thought to act as length stabilizer (Figure 1) [20].
Figure 1.
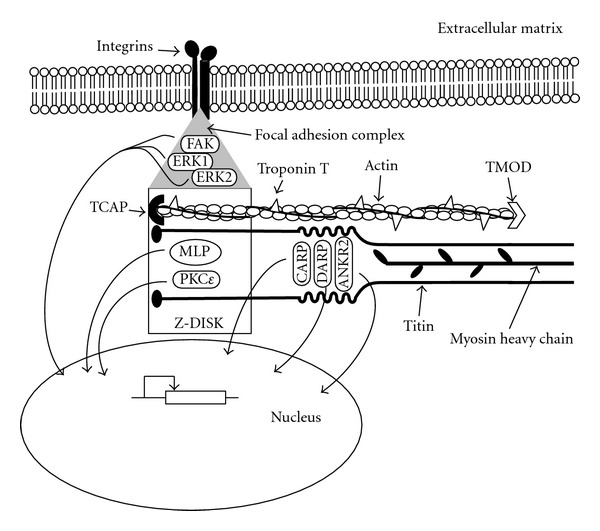
Schematic representation of the structures of the myofibrils responsible of contraction, sensation, and transduction of mechanical stimuli in the developing heart. Force is generated by the thick filaments formed by myosin heavy chain (amhc, MYH6, MYH7) associated with myosin regulatory (Myl7) and essential light chains and their interaction with the thin filaments, formed by actin (ACTC1, cfk) and troponin T (Tnnt2), TCAP and TMOD, amongst others. Titin spans a half of the sarcomere, from the Z-Disc (where it interacts with TCAP and MLP) to the M-band. One of its intermediate segments, the N2A-region, acts as a mechanotransducer and binds CARP, DARP, and ANKR2. The peripheral myofibrils of the cardiomyocytes are linked to integrins embedded in the cell membrane (sarcolemma) by means of the focal adhesion complexes of the costameres, formed by FAK ERK1 and ERK2, amongst others. In response to mechanical stimulation, proteins bound to titin (CARP, DARP, and ANKR2), proteins of the Z-Disc (MLP, PKCε), as well as FAK, ERK1, and ERK2, located to the focal adhesion complexes, translocate to the nucleus, where they interact with cardiac transcription factors to modify gene expression.
Mice with targeted mutations of Tmod1 exhibit disruption of myofibrillogenesis, absence of cardiac contraction, looping defects, trabeculation defects, thinning of the ventricular wall, and impaired chamber formation [19, 21, 22].
4. Mutant Thick and Thin Filament Proteins and Cardiogenesis: Lessons from Animal Models
Analysis of spontaneous and experimental mutations in animal models has highlighted the role of myofibrillar proteins in cardiogenesis. In vertebrates, contraction of the cardiac primordium through interaction between the thick and thin filaments of the sarcomere occurs very early in development, well before the circulation of blood is needed to sustain the tissues of the embryo. It has been proposed that this initial activity of the cardiac myofibril apparatus functions as a stimulus for adequate heart development [23]. Deficiency of the genes encoding proteins of both the thick and thin filaments of the sarcomere has been implicated in cardiac malformation. For example, in the case of proteins of the thin filament, mutations in the genes encoding cardiac troponin t (tnnt2) and sarcomeric actin (cfk) in zebrafish induce endocardial cushion and valve formation defects [24]. Targeted disruption of the cardiac troponin T gene (Tnnt2) causes looping and endocardial cushion defects in mice [25]. In the case of genes encoding proteins of the thick filament, mutation of the atrial myosin heavy chain gene (amhc) results in deficient ventricular development [26]. Defective atrial myosin heavy chain causes inadequate atrial septation in chicken (MYH7) [27] and malformation of the valves and trabeculae in the amphibian X. tropicalis (myh6) [28]. Abnormal cardiac chamber morphology, possibly due to inadequate looping of enlarged and amorphous heart tubes, was detected in mice with mutant atrial myosin regulatory light chain (Myl7) gene [29].
5. Mutations of Myofibrillar Protein Genes in CHD
The aforementioned finding by linkage that a mutation in the α-myosin heavy chain gene (MYH6) causes a dominant form of ASD [4] has been supported by reports of additional mutations in the same gene in cases of ASD and other types of CHD by us [6, 7] an others [10]. Recently, by array-based sequence of 13 sarcomeric genes in 31 familial cases of ASD, MYH6 was identified as the predominant sarcomeric disease gene for ASD [10]. Additionally, mutations of the α-cardiac actin gene (ACTC1) have been identified as causative in other familial and sporadic cases of ASD [8]. Different mutations of the ACTC1 gene have been found in cases of septal defects associated with ventricular noncompaction [9] (expansion of the trabeculated component of the ventricular wall at the expense of its compact layer). In cases of another type of CHD (Ebstein's anomaly) associated with ventricular noncompaction, mutations of the β-myosin heavy chain gene (MYH7) have been identified [5]. Other mutations of MYH6, ACTC1, and MYH7 also cause hypertrophic and dilated cardiomyopathy [2, 30–32].
6. Mechanotransduction of Myofibrils and Heart Development
Besides their function in contraction, specific myofibrillar and associated proteins can also act as sensors and transducers of the strains imposed by their own activity and the resulting fluid dynamics. The function of some of these mechanosensors and transducers could be relevant for cardiac development.
Titin, the largest myofibrillar protein, spans half of the sarcomere as it links its outer limit (the Z-band) with its centre (the M-band). This protein is composed of a series of springs of variable resistance that provide elasticity to the myofibrils and also contains segments that act as mechanosensors [33, 34]. The Ig-domains 180 and 181 in the N2A-region of titin interact with three proteins of the muscle ankyrin-repeat family, CARP (also known as ANKRD1 and a known target of NKX2.5 [35] and other cardiac transcription factors [36]), DARP, and Ankrd2 [37, 38]. When stretch is applied to cardiomyocytes, CARP translocates to the nucleus, where it interacts with transcription factors to modify gene expression [39]. A mutation on the ANKRD1 gene that enhances the stability and potentiates the transcriptional repression of the cardiac specific ANF promoter was described in a patient with total anomalous pulmonary venous return (TAPVR, another type of CHD, where the pulmonary vein empties in a vessel or cardiac chamber different from the right atria). A chromosomal translocation with a breakpoint near the ANKRD1 gene was discovered in another patient with the same abnormality [40].
Mechanical stimuli are first transmitted to the membrane of the cardiomyocytes (sarcolemma) through the extracellular matrix. Myofibrils are bound to the membrane of cardiomyocytes via specialized focal adhesion complexes concentrated in the costameres. These structures link the sarcolemma to the Z-bands of the sarcomeres that form the peripheral myofibrils [41]. The mechanotransduction of the focal adhesion complexes at the costameres includes phosphorylation of focal adhesion kinase (FAK), extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2), and paxillin (PXN) (Figure 1) [42].
It has been shown that the focal adhesion complexes at the costameres can react differentially according to the rate and direction of the strain applied to the cell. Prolonged static strain over cardiomyocytes causes FAK to translocate from the focal adhesion complex to the nucleus [42]. Transverse strain (perpendicular to the sarcomere) increases the phosphorylation of FAK, ERK1, and ERK2 in comparison with nonstrained cells, whereas longitudinal strain (parallel to the sarcomere) induces the phosphorylation of ERK1 and ERK2 but does not increase phosphorylation of FAK [42]. It has been proposed that transverse strain extends the FAK molecule, promoting its activation by phosphorylation [43].
Deficiency of FAK in produces a general defect of mesoderm development in mice [44] and, specifically in the developing heart, results in compromised migration of neural crest cells to the cardiac outflow tract, which in turn induces malformation of conotruncus [45]. However, myocardium-specific ablation of the FAK gene induces ventricular septal defects (VSDs) and deficient cardiomyocyte proliferation in mice, which suggests that the role of FAK as a mechanotransducer is also important for cardiogenesis and specifically for ventricular septation [46]. Knockdown of FAK transcripts by antisense morpholino induced looping defects in X. laevis [47]. Upon activation by phosphorylation, ERK1 and 2 translocate to the nucleus [48] and are known to participate in several processes important for heart development, including FGF, BMP, and VEGF signalling [49]. Moreover, a 1Mb microdeletion in chromosome 22q11.2 spanning the ERK2 gene has been described in patients with VSD and PTA [50]. Ablation of the PXN gene in mice results in abnormal heart structure [51]. These data suggest a possible mechanism by which cardiac contraction and the resulting circulation of blood in early development can, in turn, promote heart development by inducing cardiomyocyte proliferation.
In contrast, animal models of mutations of genes encoding other proteins known to act as mechanosensors in the cardiac Z-band and the costameres do not display cardiac malformation. In response to variations in mechanical stress, muscle LIM protein (MLP) translocates to the nucleus [52], where it has been proposed to bind cardiac transcription factors like GATA4 [53]. Also, it has been hypothesised that MLP forms a complex with telethonin (TCAP) and the N-terminal Z1 and Z2 domains of titin that acts as a stretch sensor in cardiomyocytes [54]. Protein kinase C epsilon (PKCε) is necessary and sufficient to induce FAK autophosphorylation [55], and it is known to localize to the Z-band in cardiomyocytes [56] and to translocate to the nucleus in response to pressure overload [57], where it has been suggested to regulate the transcription of α-skeletal actin [58]. Humans or mice harbouring mutations of the genes encoding MLP [54, 59], TCAP [60, 61], or PKCε [62] do not show cardiac malformation. Further research is required to establish why defective specific mechanosensing pathways of the myofibrils and associated structures have a deleterious effect in cardiogenesis and others do not.
7. Conclusions
Mutations in many genes that encode proteins involved in the formation, contraction as well as force sensing and transduction of the myofibrils produce phenotypes that include impaired cardiac development in animal models and humans. It is being increasingly recognized that the early activity of the embryonic heart has a significant impact in the morphogenesis of the organ. The sensing and transduction of the forces it generates modify the gene expression of the early myocardium, which elicits remodelling, proliferation, and possibly apoptosis. Thus, we suggest that gene candidate approaches designed to discover new genetic determinants of Mendelian CHD or common variants predisposing to the nonfamilial CHD should consider genes involved in these signalling pathways.
Acknowledgments
Work in the JDB lab is supported by the British Heart Foundation (Grant RG/07/010). J. T. Granados-Riveron held a scholarship from the Mexican Council of Science and Technology (CONACYT).
References
- 1.Geisterfer-Lowrance AAT, Kass S, Tanigawa G, et al. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh N, Haddad H. Recent progress in the genetics of cardiomyopathy and its role in the clinical evaluation of patients with cardiomyopathy. Current Opinion in Cardiology. 2011;26(2):155–164. doi: 10.1097/HCO.0b013e3283439797. [DOI] [PubMed] [Google Scholar]
- 3.Wessels MW, Willems PJ. Genetic factors in non-syndromic congenital heart malformations. Clinical Genetics. 2010;78(2):103–123. doi: 10.1111/j.1399-0004.2010.01435.x. [DOI] [PubMed] [Google Scholar]
- 4.Ching YH, Ghosh TK, Cross SJ, et al. Mutation in myosin heavy chain 6 causes atrial septal defect. Nature Genetics. 2005;37(4):423–428. doi: 10.1038/ng1526. [DOI] [PubMed] [Google Scholar]
- 5.Budde BS, Binner P, Waldmuller S, et al. Noncompaction of the ventricular myocardium is associated with a de novo mutation in the β-myosin heavy chain gene. PLoS ONE. 2007;2(12) doi: 10.1371/journal.pone.0001362. Article ID e1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Granados-Riveron JT, Ghosh TK, Pope M, et al. α-Cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Human Molecular Genetics. 2010;19(20):4007–4016. doi: 10.1093/hmg/ddq315. Article ID ddq315. [DOI] [PubMed] [Google Scholar]
- 7.Granados-Riveron JT, Pope M, Bu’Lock FA, et al. Combined mutation screening of NKX2-5, GATA4 and TBX5 in congenital heart disease: multiple heterozygosity and novel mutations. Congenital Heart Disease. 2012;7(2):151–159. doi: 10.1111/j.1747-0803.2011.00573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsson H, Eason J, Bookwalter CS, et al. alpha-Cardiac actin mutations produce atrial septal defects. Human Molecular Genetics. 2008;17(2):256–265. doi: 10.1093/hmg/ddm302. [DOI] [PubMed] [Google Scholar]
- 9.Monserrat L, Hermida-Prieto M, Fernandez X, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. European Heart Journal. 2007;28(16):1953–1961. doi: 10.1093/eurheartj/ehm239. [DOI] [PubMed] [Google Scholar]
- 10.Posch MG, Waldmuller S, Muller M, et al. Cardiac alpha-myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0028872. Article ID e28872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Cruz MV, Gómez CS, Cayre R. The developmental components of the ventricles: their significance in congenital cardiac malformations. Cardiology in the Young. 1991;1(2):123–128. [Google Scholar]
- 12.Manner J. Cardiac looping in the chick embryo: a morphological review with special reference to terminological and biomechanical aspects of the looping process. The Anatomical Record. 2000;259(3):248–262. doi: 10.1002/1097-0185(20000701)259:3<248::AID-AR30>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 13.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. International Review of Cytology. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 14.Christoffels VM, Habets PEMH, Franco D, et al. Chamber formation and morphogenesis in the developing mammalian heart. Developmental Biology. 2000;223(2):266–278. doi: 10.1006/dbio.2000.9753. [DOI] [PubMed] [Google Scholar]
- 15.Contreras-Ramos A, Sanchez-Gomez C, Garcia-Romero HL, Cimarosti LO. Normal development of the muscular region of the interventricular septum—I. The significance of the ventricular trabeculations. Journal of Veterinary Medicine Series C. 2008;37(5):344–351. doi: 10.1111/j.1439-0264.2008.00852.x. [DOI] [PubMed] [Google Scholar]
- 16.Anderson RH, Webb S, Brown NA, Lamers W, Moorman A. Development of the heart: (2) septation of the atriums and ventricles. Heart. 2003;89(8):949–958. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirby ML. Cardiac Development. Oxford, UK: Oxford University Press; 2007. An overview of cardiac morphogenesis: getting from a muscle-covered tube to a four-chmaberes pump; pp. 3–8. [Google Scholar]
- 18.Ehler E, Fowler VM, Perriard JC. Myofibrillogenesis in the developing chicken heart: role of actin isoforms and of the pointed end actin capping protein tropomodulin during thin filament assembly. Developmental Dynamics. 2004;229(4):745–755. doi: 10.1002/dvdy.10482. [DOI] [PubMed] [Google Scholar]
- 19.Fritz-Six KL, Cox PR, Fischer RS, et al. Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. Journal of Cell Biology. 2003;163(5):1033–1044. doi: 10.1083/jcb.200308164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregorio CC, Fowler VM. Mechanisms of thin filament assembly in embryonic chick cardiac myocytes: tropomodulin requires tropomyosin for assembly. Journal of Cell Biology. 1995;129(3):683–695. doi: 10.1083/jcb.129.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKeown CR, Nowak RB, Moyer J, Sussman MA, Fowler VM. Tropomodulin1 is required in the heart but not the yolk sac for mouse embryonic development. Circulation Research. 2008;103(11):1241–1248. doi: 10.1161/CIRCRESAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu X, Chen J, Reedy MC, Vera C, Sung KLP, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. American Journal of Physiology. 2003;284(5):H1827–H1838. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- 23.Burggren WW. What is the purpose of the embryonic heart beat? Or how facts can ultimately prevail over physiological dogma. Physiological and Biochemical Zoology. 2004;77(3):333–345. doi: 10.1086/422230. [DOI] [PubMed] [Google Scholar]
- 24.Bartman T, Walsh EC, Wen KK, et al. Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biology. 2004;2(5, article E129) doi: 10.1371/journal.pbio.0020129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishii K, Morimoto S, Minakami R, et al. Targeted disruption of the cardiac troponin T gene causes sarcomere disassembly and defects in heartbeat within the early mouse embryo. Developmental Biology. 2008;322(1):65–73. doi: 10.1016/j.ydbio.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Berdougo E, Coleman H, Lee DH, Stainier DYR, Yelon D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development. 2003;130(24):6121–6129. doi: 10.1242/dev.00838. [DOI] [PubMed] [Google Scholar]
- 27.Rutland C, Warner L, Thorpe A, et al. Knockdown of alpha myosin heavy chain disrupts the cytoskeleton and leads to multiple defects during chick cardiogenesis. Journal of Anatomy. 2009;214(6):905–915. doi: 10.1111/j.1469-7580.2009.01079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abu-Daya A, Sater AK, Wells DE, Mohun TJ, Zimmerman LB. Absence of heartbeat in the Xenopus tropicalis mutation muzak is caused by a nonsense mutation in cardiac myosin myh6. Developmental Biology. 2009;336(1):20–29. doi: 10.1016/j.ydbio.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C, Sheikh F, Hollander M, et al. Embryonic atrial function is essential for mouse embryogenesis, cardiac morphogenesis and angiogenesis. Development. 2003;130(24):6111–6119. doi: 10.1242/dev.00831. [DOI] [PubMed] [Google Scholar]
- 30.Carniel E, Taylor MRG, Sinagra G, et al. α-Myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112(1):54–59. doi: 10.1161/CIRCULATIONAHA.104.507699. [DOI] [PubMed] [Google Scholar]
- 31.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circulation. 2010;3(2):155–161. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niimura H, Patton KK, McKenna WJ, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105(4):446–451. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 33.Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270(5234):293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 34.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovascular Research. 2008;77(4):637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 35.Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development. 1997;124(4):793–804. doi: 10.1242/dev.124.4.793. [DOI] [PubMed] [Google Scholar]
- 36.Mikhailov AT, Torrado M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. International Journal of Developmental Biology. 2008;52(7):811–821. doi: 10.1387/ijdb.082655am. [DOI] [PubMed] [Google Scholar]
- 37.Miller MK, Bang ML, Witt CC, et al. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of Molecular Biology. 2003;333(5):951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 38.Witt CC, Ono Y, Puschmann E, et al. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2. 5 pathway in the MDM mouse with impaired titin-based signaling. Journal of Molecular Biology. 2004;336(1):145–154. doi: 10.1016/j.jmb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 39.Kojic S, Nestorovic A, Rakicevic L, et al. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Archives of Biochemistry and Biophysics. 2010;502(1):60–67. doi: 10.1016/j.abb.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 40.Cinquetti R, Badi I, Campione M, et al. Transcriptional deregulation and a missense mutation define ANKRD1 as a candidate gene for total anomalous pulmonary venous return. Human Mutation. 2008;29(4):468–474. doi: 10.1002/humu.20711. [DOI] [PubMed] [Google Scholar]
- 41.Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. American Journal of Physiology. 2005;289(6):H2291–H2301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- 42.Senyo SE, Koshman YE, Russell B. Stimulus interval, rate and direction differentially regulate phosphorylation for mechanotransduction in neonatal cardiac myocytes. The FEBS Letters. 2007;581(22):4241–4247. doi: 10.1016/j.febslet.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell B, Curtis MW, Koshman YE, Samarel AM. Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. Journal of Molecular and Cellular Cardiology. 2010;48(5):817–823. doi: 10.1016/j.yjmcc.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilic D, Furuta Y, Kanazawa S, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 45.Vallejo-Illarramendi A, Zang K, Reichardt LF. Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. Journal of Clinical Investigation. 2009;119(8):2218–2230. doi: 10.1172/JCI38194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng X, Wu X, Druso JE, et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(18):6638–6643. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doherty JT, Conlon FL, Mack CP, Taylor JM. Focal adhesion kinase is essential for cardiac looping and multichamber heart formation. Genesis. 2010;48(8):492–504. doi: 10.1002/dvg.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lidke DS, Huang F, Post JN, et al. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. The Journal of Biological Chemistry. 2010;285(5):3092–3102. doi: 10.1074/jbc.M109.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiological Reviews. 2010;90(4):1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newbern J, Zhong J, Wickramasinghe SR, et al. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(44):17115–17120. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hagel M, George EL, Kim A, et al. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Molecular and Cellular Biology. 2002;22(3):901–915. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boateng SY, Senyo SE, Qi L, Goldspink PH, Russell B. Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. Journal of Molecular and Cellular Cardiology. 2009;47(4):426–435. doi: 10.1016/j.yjmcc.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buyandelger B, Ng KE, Miocic S, et al. MLP (muscle LIM protein) as a stress sensor in the heart. Pflugers Archiv European Journal of Physiology. 2011;462(1):135–142. doi: 10.1007/s00424-011-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knoll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111(7):943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 55.Heidkamp MC, Bayer AL, Scully BT, Eble DM, Samarel AM. Activation of focal adhesion kinase by protein kinase Cε in neonatal rat ventricular myocytes. American Journal of Physiology. 2003;285(4):H1684–H1696. doi: 10.1152/ajpheart.00016.2003. [DOI] [PubMed] [Google Scholar]
- 56.Robia SL, Ghanta J, Robu VG, Walker JW. Localization and kinetics of protein kinase C-epsilon anchoring in cardiac myocytes. Biophysical Journal. 2001;80(5):2140–2151. doi: 10.1016/S0006-3495(01)76187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu X, Bishop SP. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circulation Research. 1994;75(5):926–931. doi: 10.1161/01.res.75.5.926. [DOI] [PubMed] [Google Scholar]
- 58.Wu G, Toyokawa T, Hahn H, Dorn GW. ε Protein kinase C in pathological myocardial hypertrophy. Analysis by combined transgenic expression of translocation modifiers and Gα(q) The Journal of Biological Chemistry. 2000;275(39):29927–29930. doi: 10.1074/jbc.C000380200. [DOI] [PubMed] [Google Scholar]
- 59.Arber S, Hunter JJ, Ross J, Jr., et al. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88(3):393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 60.Markert CD, Meaney MP, Voelker KA, et al. Functional muscle analysis of the Tcap knockout mouse. Human Molecular Genetics. 2010;19(11):2268–2283. doi: 10.1093/hmg/ddq105. Article ID ddq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nature Genetics. 2000;24(2):163–166. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 62.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase Cε is required for macrophage activation and defense against bacterial infection. Journal of Experimental Medicine. 2001;194(9):1231–1242. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]