

Abstract
Aplastic anemia (AA) and myelodysplasia (MDS) are forms of bone marrow failure that are often part of the same progressive underlying disorder. While most cases are simplex and idiopathic, some show a clear pattern of inheritance; therefore, elucidating the underlying genetic cause could lead to a greater understanding of this spectrum of disorders. We used a combination of exome sequencing and SNP haplotype analysis to identify causative mutations in a family with a history of autosomal-dominant AA/MDS. We identified a heterozygous mutation in SRP72, a component of the signal recognition particle (SRP) that is responsible for the translocation of nascent membrane-bound and excreted proteins to the endoplasmic reticulum. A subsequent screen revealed another autosomal-dominant family with an inherited heterozygous SRP72 mutation. Transfection of these sequences into mammalian cells suggested that these proteins localize incorrectly within the cell. Furthermore, coimmunoprecipitation of epitope-tagged SRP72 indicated that the essential RNA component of the SRP did not fully associate with one of the SRP72 variants. These results suggest that inherited mutations in a component of the SRP have a role in the pathophysiology of AA/MDS, identifying a third pathway for developing these disorders alongside transcription factor and telomerase mutations.
Main Text
Aplasia, or aplastic anemia (AA [MIM 609135]), is a condition in which bone marrow cells fail to produce a sufficient number of mature cells because of either retarded development or a cessation of regeneration. Myelodysplasia, or myelodysplastic syndrome (MDS [MIM 614286]), is the description given to a form of bone marrow failure in which immature cells in the bone marrow become malformed and dysfunctional.1 These disorders are often part of a continuum beginning with AA, progressing through MDS, and in some cases ultimately transforming into acute myeloid leukemia (AML [MIM 601626]). Most cases of AA are simplex and idiopathic and MDS is relatively common in the aged population. However, some cases of AA/MDS (that is, AA and MDS as part of a disease continuum) are shown to be familial by presentation in several members of a family through more than one generation and early onset of disease symptoms. The underlying genetic causes of AA/MDS are varied, and the genes involved in familial cases include the hematopoietic transcription factors RUNX1 (MIM 151385)2 and GATA2 (MIM 137295)3 and the telomerase components TERC (MIM 602322)4 and TERT (MIM 187270).4 A mutation in the transcription factor CEBPA (MIM 116897) has also been identified in several cases of familial AML.5 Many cases, however, remain uncharacterized at the genetic level; hence, we sought to analyze families with a history of AA/MDS for inherited mutations.
We initially chose to screen a family (designated family 1) of four individuals affected by bone marrow failure and congenital nerve deafness with an apparent autosomal-dominant mode of inheritance. The index case (II-1) was diagnosed with aplastic anemia by bone marrow biopsy, and two of her siblings (II-2 and II-3) were shown to be pancytopenic on the basis of full blood counts. One sibling with normal hearing had normal blood counts (II-4). The mother (I-2) had myelodysplasia (Figure 1A).
Figure 1.
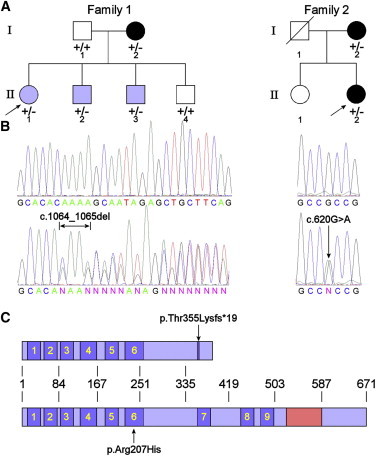
Heterozygous SRP72 Mutations Found in Two Families with Autosomal-Dominant Familial Aplastic Anemia and Myelodysplasia
(A) Pedigrees of the two families. The index case in each family is indicated by an arrow. Black symbols represent MDS; blue symbols, AA or pancytopenia. A plus sign indicates a wild-type allele; a minus sign indicates a mutant allele. The index case (II-1) in family 1 was found to have pancytopenia (hemoglobin [Hb] 11.2 g/dl, white blood cell [WBC] count 1.9 × 109/l, neutrophils 0.7 × 109/l, platelets 93 × 109/l) when she was admitted for abdominal pain in 2006 at 14 years of age. The bone marrow biopsy showed reduced cellularity without significant dysplasia and normal cytogenetics. The mother (I-2) of the index case was also found to be pancytopenic, but her BM had features of dysplasia. Her two younger brothers (II-2, 12 years old and II-3, 11 years old) were also found to be pancytpenic (II-2, Hb 12.3 g/dl, WBC 1.7 × 109/l, neutrophils 0.7 × 109/l, platelets 113 × 109/l; II-3, Hb 12.6 g/dl, WBC 2.1 × 109/l, neutrophils 0.5 × 109/l, platelets 120 × 109/l). The four affected members in this family have not required any treatment for the hematological abnormalities. The index case (II-2) in Family 2 was found to have thrombocytopenia and macrocytic anemia at 33 years of age (1987). A BM biopsy in 1993 (at 39 years of age) showed her to have trilineage dysplasia, and the trephine had a cellularity of 40%. The bone marrow karyotype showed no abnormalities. Her blood count in 2006 (at 52 years of age) showed Hb 12.3 g/dl, mean corpuscular volume (MCV) 103fl, WBC 4.6 × 109/l, and platelets 70 x109/l. The index case's mother (I-2) had been diagnosed as having MDS for several years. In 2006 (at 76 years of age) her blood count was Hb 8.2 g/dl, WBC 4.9 × 109/l, and platelets 22 × 109/l. Both affected cases in family 2 have not had any specific treatment for the hematological abnormalities.
(B) Example sequence traces showing wild-type (upper) and mutant (lower) sequences. The left panels show the 2 bp heterozygous CA deletion. The right panels show the heterozygous single-base G>A substitution.
(C) Schematic showing where the amino acid alterations lie with regard to prominent domains within SRP72. The nine predicted TPR protein-protein interaction domains are shown in blue and numbered; the 7SL RNA binding domain is depicted in red. Approximate amino acid coordinates are indicated.
The first analysis performed was a SNP typing approach. Genomic DNA samples, prepared from peripheral blood and extracted from all members of family 1 with the Puregene DNA isolation kit (Gentra, Minneapolis, MN, USA), were analyzed using the Illumina 6K SNP chip (service provided by The Genome Centre, Barts and The London School of Medicine). All samples were obtained with informed consent and the approval of our local ethics committee. Analyzing the data with Genehunter6 through the use of an autosomal-dominant inheritance pattern over 20 regions within the genome gave a positive maximum LOD score of 0.9, so this family was not powerful enough to give a unique location without large amounts of follow-up work.
With the advent of whole-exome sequencing, the four affected members of the family were reevaluated. Approximately 180,000 coding exons from 5 μg of genomic DNA from each affected individual were captured with the NimbleGen SeqCap EZ exome library (Roche NimbleGen, Madison, WI, USA). After cluster generation, the exome DNA library was then sequenced on the Illumina Genome Analyzer IIx (Illumina, San Diego, CA, USA) with the use of 76 base pair (bp) paired-end reads. Sequencing data were processed through the Illumina pipeline, and unique heterozygous changes common to all four samples were identified by filtering the resultant data set against variations reported on dbSNP and the 1000 Genomes project. Only coding or splice variants that were present in all four affected family members were analyzed further, which left 107 possible variations for analysis (Table S1 available online). Of these, one was a 2 bp deletion, three introduced a premature stop codon, and 103 were nonsynonymous coding single-nucleotide substitutions. The locations of these mutations were referred back to the SNP data previously obtained. We excluded any variation that was not in a region shared by a common haplotype between the affected individuals. The remaining variations were filtered through the Exome Variant Server (EVS) to exclude mutations already observed in the population on the assumption that, under an autosomal-dominant mode of inheritance, any change seen should be pathological. Even if reported at a low incidence on the EVS (e.g., 1 in 10,000), this would still be far higher than the frequency of AA, which is 2–7 cases per million per year.7 For the remaining variations, standard nucleotide sequencing was performed on the other family members to determine whether the variant sequences segregated with the disease. Where a variant was found in an unaffected individual or absent in an affected individual, this gene was excluded from further investigation. This left three possible gene variants of interest: SRP72 (MIM 602122) c.1064_1065del (p.Thr355Lysfs∗19) (NM_006947.3), RAB3GAP1 (MIM 602536) c.1268C>G (p.Pro423Arg) (NM_001172435.1), and PIWIL3 (MIM 610314) c.2518T>C (p.Tyr840His) (NM_001008496.3).
Mutations in RAB3GAP1 have previously been associated with Warburg micro syndrome (MIM 60018), an autosomal-recessive disorder characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe intellectual disability, spastic diplegia, and hypogonadism. This gene was therefore excluded, because this disorder has no phenotypic overlap with family 1. Heterozygous individuals are also not reported to display any disease phenotype. PIWIL3 was excluded on the basis of its expression profile, given that it is expressed exclusively in the adult testis8 and transmission in both these families is maternal. This left SRP72 c.1064_1065del (p.Thr355Lysfs∗19) as the only candidate (Figure 1B).
SRP72 is a component of the signal recognition particle (SRP), which is responsible for arresting translation of nascent proteins that are destined for the cell membrane or extracellular secretion and transferring them to the endoplasmic reticulum (ER) for correct trafficking. The SRP is composed of six protein subunits (SRP9 [MIM 600707], SRP14 [MIM 600708], SRP19 [MIM 182175], SRP54 [MIM 604857], SRP68 [MIM 604858], and SRP72) and a 7SL RNA (MIM 612177).9 The SRP72 c.1064_1065del mutation is predicted to cause a truncation of the protein, which effectively deletes the domain that has been implicated in binding of SRP72 to the 7SL RNA10,11 (Figure 1C), potentially leading to a loss of function. This region was also initially predicted to be required for binding SRP68,12 although this has been disputed by a later study.13
As this was a promising gene candidate, we subsequently selected an additional 96 individuals with bone marrow failure for screening of SRP72 (oligonucleotide primers listed in Table S2), with preference given to those with a suspected autosomal-dominant mode of disease transmission (n = 35) and/or hearing/ear abnormalities (n = 14). These were analyzed by denaturing high-performance liquid chromatography (Transgenomic, Glasgow, UK) at a single temperature at which the fragment was at least 80% helical. Any samples that gave abnormal elution patterns were reamplified and sequenced.
This secondary screen identified one additional individual with a heterozygous missense mutation in exon 6 (c.620G>A [p.Arg207His]) (Figure 1B), which was also not represented on the EVS. Both the index case (II-2) and the mother (I-2) in this family (family 2, Figure 1A) presented with MDS, and we were able to confirm that this mutation was present in both individuals. Interestingly, although no deafness was noted in the family, the index case presented with possible labyrinthitis, an inflammation of the inner ear. The p.Arg207His substitution affects an amino acid in the sixth tetratricopeptide repeat (TPR). The TPRs are predicted to form protein binding sites that may interact with other proteins of the SRP (such as SRP54 or SRP68) or ribosomal proteins11 (Figure 1C). In addition, we screened 120 healthy controls of various ethnicities and confirmed that neither of these mutations was prevalent in the general population.
To determine whether these mutations might lead to a loss of function, we subcloned SRP72 from an expression plasmid (kindly provided by Prof. Thoru Pederson, University of Massachusetts Medical School14) into plasmid pEGFP-C1 (Clontech, Saint-Germain-en-Laye, France) such that it was fused to enhanced green fluorescent protein (EGFP) at its N terminus. The resultant plasmid was designated pEGFP-SRP72. The c.1064_1065del (p.Thr355Lysfs∗19) and c.620G>A (p.Arg207His) mutations were introduced by PCR, and the constructs were resequenced to check sequence integrity. Wild-type and mutant SRP72 were transfected into human HEK293 and rat NRK cells with the use of Lipofectamine LTX (Life Technologies, Paisley, UK), and the ER was costained with ER-Tracker Red (Life Technologies). Western blotting with the SRP72 antibody, HPA034621 (Sigma-Aldrich, Dorset, UK), against transfected HEK293 cell lysates confirmed the production of a truncated protein for the p.Thr355Lysfs∗19 variant (Figure 2A). Microscopic observation of EGFP localization showed that, whereas the wild-type SRP72 was located mainly in the ER of both cell types as predicted from previous studies,14 the variant forms of SRP72 showed reduced colocalization with the ER (Figure 2B). This diffuse pattern suggests incorrect localization, perhaps due to an inability of the SRP72 variants to bind appropriately to the remainder of the SRP or translocate to the ER. Given that the 7SL RNA binding domain is absent from the p.Thr355Lysfs∗19 variant, we theorized that this mislocalization might be due to an inability of the variant proteins to bind to the SRP and its RNA component.
Figure 2.
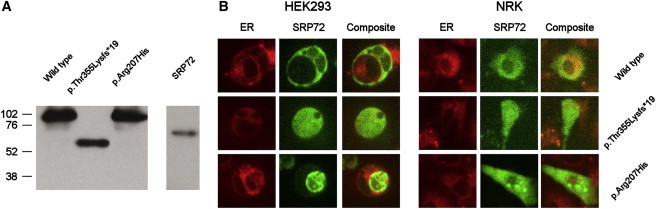
Production of EGPF-SRP72 Fusion Protein in Mammalian Cells
(A) Western blot showing EGFP-SRP72 fusion proteins including wild-type and variant SRP72 sequences. Endogenous SRP72 was detected with the same antibody but at a much lower level than exogenous protein, requiring a separate, longer exposure time. Approximate molecular weights (in kDa) are indicated.
(B) HEK293 and rat NRK cells were transfected with the EGFP-SRP72 constructs, and the EGFP was visualized by fluorescence microscopy. The ER was costained red with ER-Tracker Red. Wild-type SRP72 shows greater coincidence with the ER than either of the variant SRP72 proteins in both cell types, suggesting that the variant SRP72 proteins are not localizing correctly within the cell.
With the use of the GFP antibody ab1218 (Abcam, Cambridge, UK) and the Universal Magnetic Co-IP Kit (Active Motif, La Hulpe, Belgium), the EGFP-SRP72 was recovered from transfected cells by immunoprecipitation. Any associated 7SL RNA was isolated with an RNeasy extraction kit (QIAGEN, Crawley, UK) and reverse transcribed with SuperScript II reverse transcriptase (Life Technologies) and the 7SL RNA-specific primer 5′-GACGGGGTCTCGCTATGTTG-3′. Standard PCR was performed to amplify a ∼200 bp product representing the 7SL RNA cDNA. This showed a marked reduction in the amount of 7SL RNA coprecipitated with the p.Thr355Lysfs∗19 variant, although this was not apparent with the p.Arg207His variant (Figure 3A). Quantitative PCR (qPCR) on an ABI7500 thermal cycler (Life Technologies) using the TaqMan fluorescent probe (Hs00601540_m1; Life Technologies) indicated that the level of 7SL RNA coprecipitated with the p.Thr355Lysfs∗19 variant was around 15% compared to wild-type SRP72 (Figure 3B), as measured by a nonnormalized qPCR. Although other mRNA transcripts are largely removed by the immunoprecipitation process, there was sufficient retention of genomic DNA to allow the use of the housekeeping gene ABL1 (MIM 189980) (an average Ct value of 26.13, SD of 0.56) in an attempt to normalize the 7SL RNA reading. This produced largely the same result (Figure 3C), although it obviously negatively skewed the result for the positive control due to the much higher levels of ABL1 in a raw lysate (average Ct of 20.83, average SD of 0.34). Again, however, the p.Arg207His variant did not show any lack of 7SL RNA binding, and in fact demonstrated increased levels of 7SL RNA coprecipitation, suggesting a different mode of action for this protein.
Figure 3.
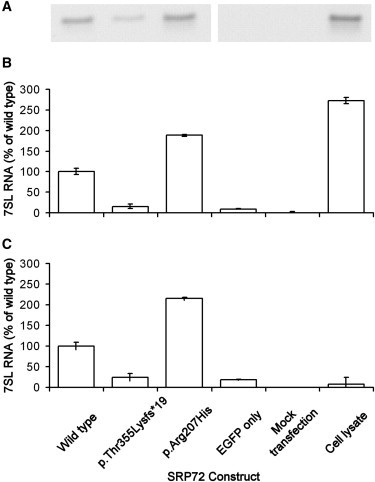
PCR Quantitation of 7SL RNA Coprecipitated from HEK293 Cells Transfected with Wild-Type and Mutant EGFP-SRP72 Constructs
(A) Standard PCR of a ∼200 bp fragment representing the 7SL RNA cDNA
(B) Quantitative real-time PCR with a fluorescent primer/probe assay. Quantitative results are shown as levels relative to those obtained by coprecipitation with the wild-type construct. An EGFP-only and a mock transfection were included as negative controls, and a nonimmunoprecipitated HEK293 lysate was included as a positive control.
(C) Quantitative real-time PCR results normalized to contaminating levels of the housekeeping gene ABL1. The result for the positive control is greatly reduced as a result of the higher levels of ABL1 in a raw cell lysate compared to one that has been depleted by coimmunoprecipitation.
Error bars represent SEM from triplicate samples.
Clearly, the p.Arg207His variant should not lack the SRP68 or SRP RNA binding domains. Given that the amino acid substitution lies near the middle of the sixth TPR domain, this change may disrupt binding to SRP68, or alternatively to SRP54, and thereby destabilize the association while still resulting in a similar overall phenotype. The finding that this variant appeared to coprecipitate more 7SL RNA is intriguing and might suggest a derangement of particle assembly resulting in a completely aberrant stoichiometry.
SRP72 has been implicated in binding to the SRP receptor and promoting the directional translocation of newly translated proteins bearing a signal sequence into the lumen of the ER.15 Failure of SRP72 to either bind to the complex or interact with the nascent polypeptide might therefore result in an inability to properly translocate peptides that are otherwise destined for the cell membrane or extracellular space. Alternatively, although SRP68 binds weakly to 7SL RNA, the binding of SRP72 significantly enhances this interaction;12 therefore, a lack of associated SRP72 might destabilize the whole particle, resulting in a complete failure to arrest cytoplasmic translation. Precisely how a defect in protein translocation might result in the AA/MDS phenotype is unclear at this point, although it is worth noting that genetic defects in protein processing have been implicated in polycystic liver disease.16 At face value, such a defect might be expected to have a global impact on cell membrane and secreted proteins; however, previous studies of SRP depletion using interfering RNA have shown that SRP can be massively reduced (< 10%) without affecting the rate of cell growth and survival, despite observable defects in protein translocation.17,18 In fact, SRP depletion seems to have very selective effects. For example, depletion of SRP in cell lines selectively reduced tumor necrosis factor (TNF)-related apoptosis-inducing ligand death receptor DR4 and not DR5, suggesting a specific role in the mediation of apoptosis via the DR4 pathway.18 Even so, the mutations identified here are present in the heterozygous state, which suggests that SRP-dependent protein trafficking, although perturbed, is unlikely to be completely nonfunctional.
The SRP is already implicated in human disease. In some cases of polymyositis and dermatomyositis, autoantigens are generated to the SRP, resulting in chronic inflammation.19 However, this report identifies human disease caused by mutation in a component of the SRP. Taken together, these data suggest a role for mutation of SRP72 in familial AA/MDS, adding to the list of genes whose mutation is known to be linked with these disorders. The genes previously identified in familial AA/MDS and AML fall into two functional categories: transcription factors (RUNX1, CEBPA, and GATA2) and telomere maintenance (TERC and TERT). The SRP72 mutations reported here represent a third category: protein translocation and processing. Further work is therefore warranted to identify additional mutations, not only in SRP72 but also in other components of the SRP and the protein-processing apparatus.
Acknowledgments
We would like to thank the families and clinicians who contributed to this research. This work was funded by the Wellcome Trust.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Browser: http://browser.1000genomes.org/
Exome Variant Server: http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM): http://www.omim.org
References
- 1.Heaney M.L., Golde D.W. Myelodysplasia. N. Engl. J. Med. 1999;340:1649–1660. doi: 10.1056/NEJM199905273402107. [DOI] [PubMed] [Google Scholar]
- 2.Song W.J., Sullivan M.G., Legare R.D., Hutchings S., Tan X., Kufrin D., Ratajczak J., Resende I.C., Haworth C., Hock R. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 3.Hahn C.N., Chong C.E., Carmichael C.L., Wilkins E.J., Brautigan P.J., Li X.C., Babic M., Lin M., Carmagnac A., Lee Y.K. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011;43:1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirwan M., Vulliamy T., Marrone A., Walne A.J., Beswick R., Hillmen P., Kelly R., Stewart A., Bowen D., Schonland S.O. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum. Mutat. 2009;30:1567–1573. doi: 10.1002/humu.21115. [DOI] [PubMed] [Google Scholar]
- 5.Smith M.L., Cavenagh J.D., Lister T.A., Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 2004;351:2403–2407. doi: 10.1056/NEJMoa041331. [DOI] [PubMed] [Google Scholar]
- 6.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 7.Young N.S., Scheinberg P., Calado R.T. Aplastic anemia. Curr. Opin. Hematol. 2008;15:162–168. doi: 10.1097/MOH.0b013e3282fa7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki T., Shiohama A., Minoshima S., Shimizu N. Identification of eight members of the Argonaute family in the human genome small star, filled. Genomics. 2003;82:323–330. doi: 10.1016/s0888-7543(03)00129-0. [DOI] [PubMed] [Google Scholar]
- 9.Nagai K., Oubridge C., Kuglstatter A., Menichelli E., Isel C., Jovine L. Structure, function and evolution of the signal recognition particle. EMBO J. 2003;22:3479–3485. doi: 10.1093/emboj/cdg337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iakhiaeva E., Iakhiaev A., Zwieb C. Identification of amino acid residues in protein SRP72 required for binding to a kinked 5e motif of the human signal recognition particle RNA. BMC Mol. Biol. 2010;11:83. doi: 10.1186/1471-2199-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iakhiaeva E., Yin J., Zwieb C. Identification of an RNA-binding domain in human SRP72. J. Mol. Biol. 2005;345:659–666. doi: 10.1016/j.jmb.2004.10.087. [DOI] [PubMed] [Google Scholar]
- 12.Lütcke H., Prehn S., Ashford A.J., Remus M., Frank R., Dobberstein B. Assembly of the 68- and 72-kD proteins of signal recognition particle with 7S RNA. J. Cell Biol. 1993;121:977–985. doi: 10.1083/jcb.121.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iakhiaeva E., Hinck C.S., Hinck A.P., Zwieb C. Characterization of the SRP68/72 interface of human signal recognition particle by systematic site-directed mutagenesis. Protein Sci. 2009;18:2183–2195. doi: 10.1002/pro.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Politz J.C., Yarovoi S., Kilroy S.M., Gowda K., Zwieb C., Pederson T. Signal recognition particle components in the nucleolus. Proc. Natl. Acad. Sci. USA. 2000;97:55–60. doi: 10.1073/pnas.97.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel V., Walter P. Each of the activities of signal recognition particle (SRP) is contained within a distinct domain: analysis of biochemical mutants of SRP. Cell. 1988;52:39–49. doi: 10.1016/0092-8674(88)90529-6. [DOI] [PubMed] [Google Scholar]
- 16.Davila S., Furu L., Gharavi A.G., Tian X., Onoe T., Qian Q., Li A., Cai Y., Kamath P.S., King B.F. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 17.Lakkaraju A.K., Luyet P.P., Parone P., Falguières T., Strub K. Inefficient targeting to the endoplasmic reticulum by the signal recognition particle elicits selective defects in post-ER membrane trafficking. Exp. Cell Res. 2007;313:834–847. doi: 10.1016/j.yexcr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Ren Y.G., Wagner K.W., Knee D.A., Aza-Blanc P., Nasoff M., Deveraux Q.L. Differential regulation of the TRAIL death receptors DR4 and DR5 by the signal recognition particle. Mol. Biol. Cell. 2004;15:5064–5074. doi: 10.1091/mbc.E04-03-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Mühlen C.A., Tan E.M. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin. Arthritis Rheum. 1995;24:323–358. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.