

Abstract
Argininosuccinate lyase (ASL) is required for the synthesis and channeling of L-arginine to nitric oxide synthase (NOS) for nitric oxide (NO) production. Congenital ASL deficiency causes argininosuccinic aciduria (ASA), the second most common urea-cycle disorder, and leads to deficiency of both ureagenesis and NO production. Subjects with ASA have been reported to develop long-term complications such as hypertension and neurocognitive deficits despite early initiation of therapy and the absence of documented hyperammonemia. In order to distinguish the relative contributions of the hepatic urea-cycle defect from those of the NO deficiency to the phenotype, we performed liver-directed gene therapy in a mouse model of ASA. Whereas the gene therapy corrected the ureagenesis defect, the systemic hypertension in mice could be corrected by treatment with an exogenous NO source. In an ASA subject with severe hypertension refractory to antihypertensive medications, monotherapy with NO supplements resulted in the long-term control of hypertension and a decrease in cardiac hypertrophy. In addition, the NO therapy was associated with an improvement in some neuropsychological parameters pertaining to verbal memory and nonverbal problem solving. Our data show that ASA, in addition to being a classical urea-cycle disorder, is also a model of congenital human NO deficiency and that ASA subjects could potentially benefit from NO supplementation. Hence, NO supplementation should be investigated for the long-term treatment of this condition.
Introduction
The urea cycle consists of a series of enzymatic reactions that catalyze the conversion of toxic-waste nitrogen to urea, a nontoxic and excretable nitrogenous compound. Deficiencies of any of the enzymes of the urea cycle (N-acetyl glutamate synthase, carbamoyl phosphate synthase 1, ornithine transcarbamylase, argininosuccinate synthase 1 [ASS1], argininosuccinate lyase [ASL], and arginase 1) result in a group of inborn hepatic-metabolism errors collectively called urea-cycle disorders (UCDs).1 Subjects with UCDs typically present with hyperammonemic episodes that can have neurological consequences. Argininosuccinic aciduria (ASA [MIM 207900]), the second most common UCD, is caused by the deficiency of ASL (MIM 608310; RefSeq NM_000048.3).1,2 ASA is biochemically characterized by elevations of argininosuccinic acid (upstream of the metabolic block) and a deficiency of arginine (downstream of the metabolic block). In addition to the hyperammonemia that is common to all UCDs, subjects with ASA might present with a more complex clinical phenotype that could include neurocognitive deficiencies, hepatic disease, and systemic hypertension (HTN).3–5 Intriguingly, these manifestations can occur even in the absence of documented hyperammonemia, suggesting that they might result from the functions of ASL beyond its role in ammonia clearance.3
ASL is the only enzyme that can generate L-arginine. Most tissues express the enzymes ASS1 and ASL, which allow for cell-autonomous synthesis of arginine from citrulline. Arginine is the precursor for the synthesis of biologically important metabolites such as nitric oxide (NO), polyamines, agmatine, creatine, proline, and glutamate.6 In the presence of ASL deficiency, the secondary deficiency of arginine could lead to a decreased production of arginine metabolites. NO has diverse physiological functions including vascular relaxation, neurogenesis, and neuronal transmission. There is significant evidence that dysregulation of NO can result in HTN and neurocognitive deficits in animal models and humans.7–9 We recently showed that ASL is required for the utilization of endogenous and exogenous arginine for NO production.10 In the absence of ASL, a NO-synthesis protein complex is lost, resulting in the loss of arginine channeling from both intracellular and extracellular sources to nitric oxide synthase (NOS) for NO synthesis. Hence, we hypothesized that the long-term complications of ASA might result, at least in part, from the deficiency of NO and that subjects with ASA might therefore benefit from NO supplementation.
Material and Methods
Generation of ASA Mice
The generation of mice hypomorphic for Asl has been described previously.10 All animal procedures were authorized by the Institutional Animal Care and Use Committee of Baylor College of Medicine (BCM). The mice were treated with benzoate (250 mg/kg per day), L- arginine (100 mg/kg per day), and sodium nitrite (10 mg/kg per day) in their drinking water, as described previously, for 5 weeks.10
Helper-Dependent Adenoviral Vector Generation and Administration
The helper-dependent adenoviral vector (HDAd-gE-mAsl) contains the liver-restricted ApoE genomic promoter with a liver-specific enhancer11 driving the expression of mouse Asl. The helper-dependent adenovirus (HDAd) was produced with the helper virus AdNG16312 and the 116 producer cell line as described in detail elsewhere.13,14 Helper-virus contamination levels were determined as described13 and were found to be <0.05% for all vector preparations. DNA analyses of HDAd genomic structure were confirmed for all vectors.13
Immunoblot
Liver sections were snap frozen. The tissue was lysed with a rotor-stator homogenizer in a mild lysis buffer (50 mM Tris-HCl [pH7.5], 150 mM NaCl, and 1% Triton-100) with a complete protease-inhibitor cocktail (Roche, USA). Tissue homogenates were centrifuged at 16,000 × g for 20 min, and the supernatant protein was quantified with a microBCA assay (Pierce, USA). Immunoblot analyses were performed with primary antibodies specific to ASL (Abnova, clone 4C5-1F2) and GAPDH (Sigma clone GAPDH-71.1, peroxidase-conjugated). After the addition of the secondary antibody (goat anti-mouse antibody, Abnova), the membranes were developed with Luminata crescendo luminescent HRP substrate (Millipore, USA).
Enzyme Activity Assay
All chemicals used were obtained from Sigma-Aldrich (St.Louis, USA). Protein lysates (20 μg) were incubated with 10 mM argininosuccinic acid in 100 μl of 10 mM Tris-HCl (pH 7.5) with 5 units of arginase so that ASL activity could be determined. After being incubated for 60 min, the reaction was stopped by being boiled for 5 min, and the urea was quantified with a modified Jung assay.15
Plasma Amino Acids
Plasma amino acids were measured at the CLIA (Clinical Laboratory Improvement Amendments)-certified Medical Genetics Laboratories of BCM.16
Stable-Isotope Studies
Mice were restrained, and a tail-vein catheter was inserted. The tail-vein catheter was connected to syringe infusion pumps (PHD2000; Harvard Apparatus, Holliston, MA). The primed-continuous infusion of [guanidino-15N2]-arginine (prime 45 μmol/kg, continuous 45 μmol/kg per hr), [5-13C,4,4,5,5 2H4]-citrulline (prime 7 μmol/kg, continuous 7 μmol/kg per hr), and [13C18O]-urea (prime 100 μmol/kg, continuous 100 μmol/kg per hr) lasted for 4 hr. We have shown previously that precursors and products reach a plateau phase within 2 hr.17 Blood samples were collected at the end of the infusion and were centrifuged at 1500 × g for 10 min. Plasma was stored at −80°C until analysis. The isotopic enrichments of arginine and citrulline in the plasma were measured by LC-MS (liquid chromatography-mass spectrometry), whereas the urea enrichment was measured by GC-MS (gas chromatography-mass spectrometry). Fluxes and interconversions were calculated as previously described.18
NO Studies
Mice were anesthetized with isoflurane. After thoracotomy, a 25G needle was inserted into the apex of the left ventricle and perfused with air-equilibrated PBS supplemented with N-ethylmaleimide (NEM)/EDTA (10/2.5 mM) for a full blood exchange. The right atrium was cut open so that the blood and buffer would have an exit port. After 30 s of perfusion, the heart and liver were excised. Excised tissues were blotted dry on filter paper, weighed, cut into small pieces and homogenized immediately in ice-cold NEM/EDTA-containing perfusion buffer. The addition of NEM/EDTA served the purpose of blocking SH-groups and inhibiting transition metal-catalyzed transnitrosation reactions, thus preventing artificial nitrosylation, as well as thiolate and ascorbate-mediated degradation of endogenous RSNOs.19 Biological specimens harvested from anesthetized mice were analyzed for nitroso species and oxidation products of NO as detailed elsewhere.20,21
BH4 Measurements by High-Pressure Liquid Chromatography
Tissue biopterins (BH4 and more oxidized species) were measured by high-pressure liquid chromatography (HPLC) as previously described.22 Mouse aortae were homogenized with lysis buffer (50 mM Tris-HCl, 1 mM DTT, and 1 mM EDTA) and oxidized by exposure to 1% I2 and 2% KI at room temperature for 1 hr under dark conditions. The reaction was stopped with the addition of ascorbic acid, and the mixture was centrifuged for 10 min at 12,000 × g. Biopterins in the supernatant were quantified by HPLC on a C18 column with fluorescence detection.
Isoprostane Measurement
Urine was collected by bladder aspiration from 30 wild-type (WT) and ten ASA mice. Blood was collected by retro-orbital bleeding, and plasma was separated. Isoprostane levels were measured by ELISA according to the manufacturer's (Cayman Chemicals, USA) instructions.
Aortic NOS and Superoxide Studies
The ratio of NOS3 dimers to monomers23 and superoxide levels in the aortae24 were assessed as previously published.
Invasive Blood-Pressure Measurement
Mice were anesthetized with 1.5% isoflurane (in 100% oxygen), which was administered at a continuous flow rate of 20 ml/min (VetEquip, Pleasanton, CA). The neck and xiphoid areas were shaved, and the anesthetized mouse was placed in a supine position in which its paws were taped to electrodes on a temperature-controlled electrocardiogram (ECG) board. The right carotid artery of the mouse was isolated and tied off distally, and the proximal end was temporarily occluded. A small cut was made in the artery, and a 1.0 F (0.33 mm) Millar pressure catheter (SPR-100, Millar Instruments, Houston, TX) was inserted and held in place with a suture loosely tied over the artery-catheter overlap region. The proximal end of the artery was then opened, and the catheter was advanced into the ascending aorta as close as possible to the aortic root. A second suture was tied over the artery-catheter overlap so that blood would not leak as the catheter was advanced. After a minute of stabilization, two segments of blood-pressure tracings along with their corresponding ECGs were recorded and stored with the Doppler Signal Processing Workstation (Indus Instruments, Houston, TX). Systolic, diastolic, and mean pressure were extracted from the stored aortic pressure files.
Wire Myography
Mice were anesthetized with isoflurane. The thoracic and abdominal aortae were exposed by a thoracotomy. A 25G syringe was inserted into the apex of the left ventricle and was perfused free of blood with oxygenated Krebs Henseleit buffer. The right atrium was cut so that the blood would have an exit port. The aorta was removed and cleaned of all fat and adventitia. The aorta was cut into 2 mm segments and mounted on a 4 channel wire myograph (AD Instruments). Vessel rings were maintained in 10 ml organ baths with oxygenated Krebs buffer (95% O2 and 5% CO2) at 37°C. Rings were allowed to equilibrate for 80 min, and the buffer in each organ bath was changed every 20 min. One gram pretension was placed on each aortic ring (the appropriate starting tension for optimal vasomotor function was determined in previous experiments). An 8 channel octal bridge (Powerlab) and data-acquisition software (Chart version 5.2.2) were used for the recording of all force measurements. After equilibration for 80 min, 1 μM phenylephrine was added to each ring for submaximal contraction. After stabilization, increasing concentrations of acetylcholine were added to each bath so that the endothelium-dependent relaxation, which was expressed as percent reversal of phenylephrine-induced constriction, could be determined.
Human Studies
All research procedures were approved by the institutional review board at Baylor College of Medicine. The subject underwent continuous monitoring of vitals and hourly BP recordings during his stay in the hospital. A 24 hr collection of urine was performed, and urinary nitrite and nitrate levels were assessed both prior to and 24 hr after initiation of nitrate therapy. A baseline echocardiogram was performed before discharge from the hospital and periodically after treatment. Blood pressure was measured with Dinamap automated blood-pressure machines (GE healthcare, USA). Neo40 (Neogenis) is registered as a dietary supplement with the FDA (reg. no. 3008524085).
Neuropsychological Evaluation
Several neuropsychological measures were administered for the assessment of a variety of cognitive domains, including intelligence, academic achievement, executive functioning, memory, visual-motor and visual-perceptual skills, fine-motor functioning, adaptive functioning, and behavioral and emotional functioning. The evaluation was performed by a board-certified clinical psychologist at Texas Children's Hospital, Houston, TX. The Wechsler Abbreviated Scale of Intelligence (WASI), California Verbal Learning Test, Children's Memory Scale, The Test of Everyday Attention for Children, and Tower of London-Drexel University measures were administered via standard techniques.
Statistical Analyses
Statistical significance was computed with the Student's two-tailed t test, ANOVA, or log-rank tests. A p value < 0.05 was considered statistically significant. Data are expressed as mean values ± standard deviation in the bar and line plots in Figures 1–4. The vertical box plots in Figure 4 depict the median along with the 25th and 75th percentiles, whereas the error bars depict the 5th and 95th percentiles.
Figure 1.
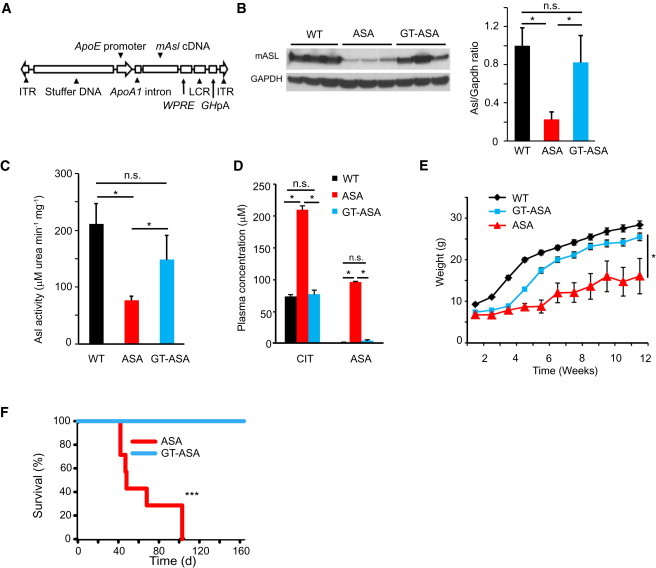
Liver-Directed Gene Transfer Corrects the Metabolic Defect in ASA Mice
A total of 23 ASA mice were randomized to receive either gene therapy (GT) or a placebo. Triple therapy was withdrawn at 5 weeks of life so that the long-term effects of GT alone could be assessed. The error bars in the bar and line graphs depict standard deviation.
(A) Structure of the helper-dependent adenoviral construct with the murine Asl cDNA under the regulation of the ApoE promoter. The following abbreviations are used: ITR, inverted terminal repeat; WPRE, Woodchuck hepatitis virus posttranscriptional regulatory element; LCR, locus control region; and GhpA, growth hormone polyA.
(B) A representative immunoblot of liver compares the ASL protein levels between three WT mice, three placebo-treated ASA mice, and three GT-treated ASA mice 2 weeks after GT. Densitometry quantification shows the correction of ASL protein levels. Asterisks indicate p < 0.05. The following abbreviation is used: n.s., not significant.
(C) Representative ASL enzyme activity. Compared with untreated mice (n = 3), ASA GT-treated mice (n = 3) show correction of enzymatic activity in the liver. Asterisks indicate p < 0.05. The following abbreviation is used: n.s., not significant.
(D) Amino acid profile of ASA mice (n = 4) versus GT-treated ASA mice (n = 4) at 8 weeks of age showing biochemical correction. Asterisks indicate p < 0.05. The following abbreviations are used: CIT, Citrulline; and ASA, argininosuccinic acid.
(E) GT-treated ASA mice (n = 15) demonstrate significantly better growth than those treated with the placebo (n = 8). Asterisks indicate p < 0.05.
(F) Kaplan-Meier survival curve. GT-treated ASA mice (n = 15) show 100% survival by week 16 of life, whereas all placebo-treated ASA mice (n = 8) expired by day 103. Asterisks indicate p < 0.001.
Figure 2.
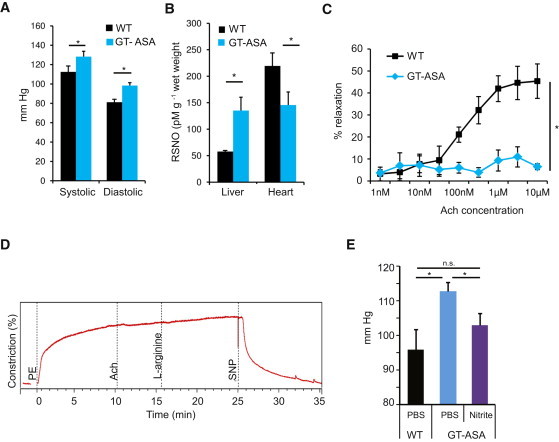
Tissue-Specific NO Deficiency and Hypertension Despite Correction of the Metabolic Defect in the Liver of GT-Treated ASA Mice
(A) Invasive BP measurements at age 12–14 weeks (n = 3 per group) showed that GT-treated ASA mice had elevated systolic and diastolic BPs when they were compared with WT mice. Asterisks indicate p < 0.05.
(B) RSNO levels measured in liver and hearts of six GT-treated ASA mice and seven WT mice showed significantly higher RSNO levels in the livers and significantly lower levels in the hearts of the GT-treated ASA mice. Asterisks indicate p < 0.05.
(C) Preconstricted WT aortic rings relaxed in a concentration-dependent fashion with acetylcholine (Ach) treatment beginning at a concentration of 10 nM, wheras aortic rings from GT-treated ASA mice were unresponsive to Ach. Data points are derived from measurements from four segments of each aorta in three WT and three GT-treated mice ASA mice. The asterisk indicates p < 0.05.
(D) Representative tracing of aortic-ring isometric tension in GT-treated ASA mice shows no relaxation in response to Ach or L-arginine, whereas NO donor sodium nitroprusside (SNP) leads to a significant relaxation. Three aortic rings from each mouse aorta were used for analysis.
(E) In vivo vascular response to nitrite infusion in 4-month-old GT-treated ASA mice (n = 3) shows normalization of BP. Asterisks indicate p < 0.05. The following abbreviation is used: n.s., not significant.
The error bars in the bar and line graphs depict standard deviation.
Figure 3.
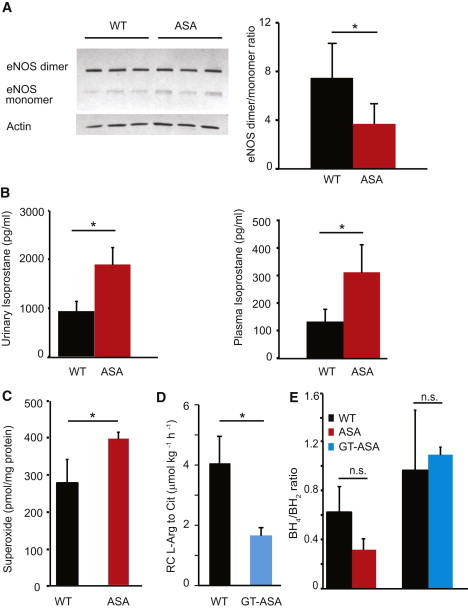
NOS Uncoupling and Increased Free-Radical Levels in ASA Mice
(A) Immunoblot and densitometric quantification from the aortae of three ASA mice and three WT mice show a decrease in the active dimer form in the ASA mice, supporting NOS3 (eNOS) uncoupling. The asterisk indicates p < 0.05.
(B) Isoprostane levels, a marker of free-radical production, are significantly increased in ASA mice. Asterisks indicate p < 0.05.
(C) Compared to those of WT mice (n = 5), superoxide levels from the aortae of ASA mice (n = 5) are elevated. The asterisk indicates p < 0.05.
(D) In vivo isotope measurement of the transfer of the guanidino-nitrogen from 15N2-arginine to 15N-citrulline, a marker of NO flux, is decreased in the GT-treated ASA mice (n = 5 per group). The asterisk indicates p < 0.05.
(E) Compared to those of WT mice, the BH4/BH2 ratios in the aortae of ASA hypomorphic mice and GT-treated ASA hypomorphic mice are not significantly different. The following abbreviation is used: n.s., not significant.
The error bars depict standard deviation.
Figure 4.
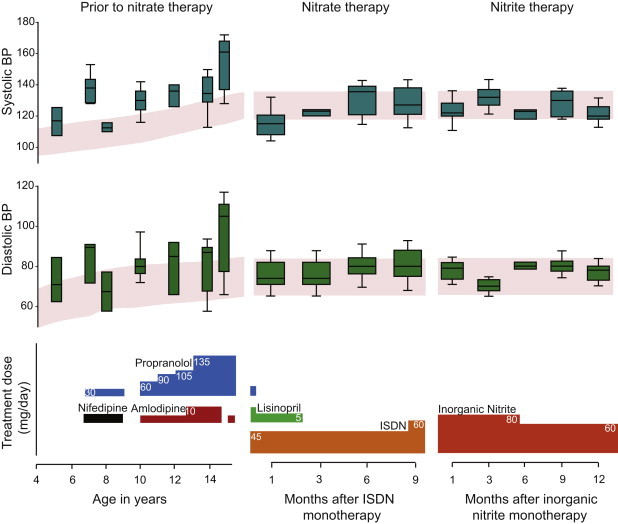
Clinical Treatment with NO Supplements Corrects Hypertension in a Subject with ASA
The vertical box plots depict the 25th and 75th percentiles along with median blood-pressure values at various time points. The error bars depict the 5th and 95th percentiles. The red shaded area depicts the 50th and 90th blood percentiles for age and stature. Median BP values that were above the 90th percentile prior to initiation of organic-nitrate therapy in spite of combination antihypertensive therapy (left panel) show sustained normalization with NO supplementation therapy; this sustained normalization allowed for the withdrawal of all other antihypertensive medications (middle and right panels).
Results
Liver-Directed Gene Therapy Results in Long-Term Correction of the Metabolic Defect in the ASA Mice
To differentiate the physiological consequences of NO deficiency from the effects of a block in the urea cycle, we set forth to correct the ureagenesis defect in the ASA mice10 by gene therapy. We and others have shown that liver-directed gene transfer with HDAd vectors can achieve long-term expression extending up to 7 years in nonhuman primates and can effectively correct a host of preclinical models of inborn metabolism errors.25 Hence, we generated a HDAd expressing Asl under the liver-specific ApoE promoter by using previously described methods (Figure 1A).11,13 Because ASA mice demonstrate reduced survival as a result of hyperammonemia, we treated these mice with triple-therapy consisting of arginine, sodium benzoate, and sodium nitrite from birth as previously described.10 At 4 weeks of life, we randomized the mice so they would receive either gene therapy (GT) or a placebo, and we withdrew triple-therapy at 5 weeks of life to assess the long-term effects of GT alone. The GT-treated ASA mice showed hepatic ASL protein levels comparable to those of WT mice; these protein levels resulted in the normalization of their hepatic ASL enzyme activity (Figures 1B and 1C). The plasma amino acid profile and growth normalized, and survival was extended beyond 5 months (Figures 1D–1F). These results show that we were able to correct the ureagenesis defect in the liver by using GT.
Hypertension and Persistent NO Deficiency in the Vasculature Despite Correction of the Metabolic Defect in the ASA Mice
Despite the long-term correction of the metabolic defect in the liver, the GT-treated ASA mice continued to have significant hypertension as measured by both tail cuff and indwelling carotid catheters (Figure 2A). To validate that the hypertension is associated with NO deficiency, we compared tissue levels of nitrosothiols (RSNO; a biomarker of NO)26 in GT-treated ASA mice to those of their WT littermates. Whereas the RSNO levels were significantly higher in the GT-treated mouse livers, the RSNO levels in the hearts were significantly lower than in WT mice (Figure 2B). This implies that normalization of hepatic ASL levels by liver-directed GT was associated with increased NO production in the liver, whereas other tissues that still lacked ASL had decreased amounts of NO, supporting a tissue-specific ASL requirement for NO production.
To evaluate whether this decreased NO production in the vascular tissue is involved in the pathogenesis of hypertension, we performed classic aortic-ring experiments and examined the relaxation response to L-arginine and NO donors. In this experiment, vessel rings from mouse aortae were preconstricted by exposure to phenylephrine, an alpha-adrenergic agonist that causes vasoconstriction. The rings were then exposed to increasing concentrations of acetylcholine (Ach), which causes vasodilatation as a result of the release of NO generated by NOS3 of the vascular endothelium.27 The aortae from WT mice showed a significant relaxation, whereas the GT-treated ASA mice showed a persistent lack of relaxation in response to either Ach or L-arginine (Figures 2C and 2D). However, the aortic rings of GT-treated mice showed a definitive relaxation in response to a NO donor, sodium nitroprusside (SNP) (Figure 2D). This ex vivo experiment demonstrates that the impaired vascular relaxation resulted from the inability of the vasculature to generate NO from arginine in a NOS-dependent manner and that a NOS-independent NO source could rescue the deficiency. To confirm this finding in vivo, we infused the GT-treated ASA mice with intravenous sodium nitrite, a NO source, and the fact that this treatment significantly decreased the mean arterial pressure corroborates the results of the aortic-ring experiments (Figure 2E).
Lack of L-Arginine-Substrate Availability to NOS Due to ASL Deficiency Is the Primary Cause of Decreased NO Production in the Vasculature
We have previously reported that the ASA mice were hypertensive and NO deficient.10 Because a lack of arginine substrate can lead to the uncoupling of NOS3 (eNOS) and result in enhanced free-radical generation along with decreased NO production,28,29 we measured the ratio of NOS3 dimers to monomers as a reflection of NOS3-coupling status in the aortae of ASA mice. Indeed, compared with their WT littermates, ASA mice had a significant reduction in this ratio, suggesting that the lack of ASL is associated with the uncoupling of NOS3 (Figure 3A). To evaluate whether NOS3 uncoupling was also associated with increased free-radical production, we measured isoprostane levels (a marker of oxidative stress) in plasma and urine. The ASA mice had significantly higher levels of isoprostane than did their WT littermates (Figure 3B). To specifically assess the free-radical production at the level of vascular tissues, we measured superoxide levels in murine aortae and found that the levels in ASA mice were significantly higher than those in their WT littermates (Figure 3C). Together, these data demonstrate increased free-radical production in the ASA mice.
In the context of increased free-radical production, NO deficiency can result either from NOS uncoupling or from its scavenging by free radicals. To evaluate whether decreased NO production or increased NO scavenging contributed to the NO deficiency, we performed stable-isotope studies in the GT-treated ASA mice. Our results show that the transfer of nitrogen from guanidino-[15N2]-arginine to ureido-[15N]-citrulline, a dynamic marker of NO production, was significantly decreased in the GT-treated ASA mice (Figure 3D). This finding supports the hypothesis that the decreased NO production in ASL deficiency predominantly results from the decreased oxidation of arginine to citrulline.
Because BH4 availability is a known regulator of NOS3 activity and enzymatic coupling,30 we further assessed the impact of ASL loss of function on BH4 availability and metabolism. We measured the ratio of BH4 levels to BH2 levels in the aortae of ASA mice, GT-treated ASA mice, and their corresponding WT littermates. We found that the BH4/BH2 ratio in ASA and GT-treated ASA mice was no different from that of the WT mice, suggesting that NOS3 uncoupling was not caused by perturbation of BH4 (Figure 3E). When free radicals are the primary cause of vascular injury, the BH4/BH2 ratio is usually dramatically reduced as a result of the oxidation of BH4 to BH2.30 All together, the lack of evidence of BH4 deficiency in both the ASA and the GT-treated ASA mice, the decreased in vivo NO production as measured by the isotope flux studies, and the response of the HTN to nitrite supplementation indicate that decreased arginine-substrate availability is the primary explanation for NOS uncoupling. As a result of NOS uncoupling in ASL deficiency, there is a decrease in NO production along with a secondary increase in free-radical production.
In summary, these results suggest that the liver-targeted GT rescues the ureagenesis defect in the liver but not the tissue-specific ASL requirement for the utilization of arginine in NO production. More importantly, our results confirm that the vascular dysfunction seen with ASL deficiency is independent of the metabolic defect in the urea cycle, is directly associated with NO deficiency in the vasculature, and can be rescued by a NOS-independent NO source in vivo.
NO Supplementation for Treatment of Hypertension in a Human Subject with ASA
HTN has been anecdotally reported as a complication in human ASA subjects.3,31 We have previously shown a significantly decreased NOS-dependent flow-mediated vasodilatation in ASA subjects while their vasodilatory response to a NOS-independent NO donor is normal.10 These results suggest that in ASL deficiency, the cellular signaling events downstream of NO are intact and that the vascular pathology and hypertension are probably due to a deficiency of NO in the vasculature.
One of the ASA subjects under our clinical care provided us with an opportunity to translate our findings in a human therapeutic context. This subject was diagnosed with ASA at 3 years of age and with idiopathic HTN at 5 years of age. His systolic and diastolic BPs were repeatedly above the 95th percentile for his age and stature for a period of over 10 years. This was in the setting of normal levels of plasma arginine due to continuous supplementation with L-arginine, a standard therapy for the prevention of hyperammonemia (data not shown). Evaluations for secondary causes of hypertension, including measurements of serum electrolytes, renal function, and plasma-renin and aldosterone levels and a renal-arterial duplex ultrasonography, were unremarkable (Table 1). Dietary salt restriction, thiazide diuretics, and enalapril were sequentially introduced but failed to normalize his BP. The subject was started on treatment with propranolol, nifedipine, and subsequently, amlodipine (Figure 4, left panel). In spite of adequate dosing and compliance (evidenced by significant bradycardia), the control of his hypertension remained suboptimal. The median systolic and diastolic BPs ranged between 130–140 and 75–90 mm of Hg, respectively, which were both above the 95th percentile for his age and stature (Figure 4, left panel).
Table 1.
Blood and Urine Analysis before and after Initiation of NO Supplementation
| Before NO Supplementation | After NO Supplementation | |
|---|---|---|
| Plasma renin | normal | normal |
| Plasma aldosterone | normal | normal |
| Plasma brain natriuretic peptide (BNP) | not tested | normal |
| Urinary protein (mg/24 h, normal 28–141) | 257 (elevated) | 51 (normal) |
| Urinary nitrite (μg/ml, normal is not detectable) | not detectable | 17 (elevated) |
| Urinary nitrate (μg/ml, normal is 0–124 μg/ml) | 140 (elevated) | 480 (elevated) |
Urinanalysis showed a decrease in protein excretion together with increased urinary nitrate, as would be expected from therapy.
Clinical mutation analysis revealed that the subject had compound heterozygous mutations in ASL: c.857A>G (p.Gln286Arg) and c.557G>A (p.Arg186Gln). Both mutations have been reported to be pathogenic.32,33 Molecular modeling of the ASL tetrameric protein with these two alterations predicts that at least one active catalytic site is reconstituted and thus allows for partial enzymatic activity (Figure S1A, available online). Indeed, study of this subject's fibroblasts showed residual protein and enzyme activity consistent with a hypomorphic state (Figures S1B and S1C).
At the age of 15 years, this subject presented with hypertensive urgency and had a systolic BP between 160–170 mm of Hg. Because of the ASL requirement for systemic NO production, the resemblance of his biochemical phenotype to the ASA mouse model, and the response to sodium-nitrite treatment in the ASA mouse, we hypothesized that the hypertension in this subject was probably due to systemic NO deficiency and that treatment with a NOS-independent NO source would be beneficial. Hence, over a 4 day period, the subject was initiated on divided doses of an organic nitrate, isosorbide dinitrate (ISDN) (starting at 0.2 mg/kg per day and titrated to 0.6 mg/kg per day). At the same time, amlodipine was discontinued, and propranolol was weaned to 0.6 mg/kg per day. The subject's systolic, diastolic, and mean arterial pressures showed dramatic normalization during this short period of time (Figure S2). Concurrent with the weaning of his β-blocker (propranolol), the subject's bradycardia resolved (Figure S2).
An echocardiogram prior to the patient's discharge from the hospital showed mild concentric left ventricular hypertrophy (LVH) with normal left ventricular systolic function (Table 2). During the subsequent 3 months, the mean BP was 120/74 mm Hg (standard error of 0.89 and 0.72 for systolic and diastolic pressures, respectively; n = 125 readings), thus allowing for the discontinuation of the remaining antihypertensive medications. On monotherapy with ISDN, the subject's BP remained within normal limits for 9 months. An echocardiogram performed after 5 months of ISDN treatment showed an improvement in left ventricular parameters and mild residual hypertrophy (Table 2). In addition, urinanalysis showed a decrease in proteinuria together with increased urinary nitrate, as would be expected from such therapy (Table 1).
Table 2.
Echocardiogram Measurements before and after Initiation of NO Supplementation
| Left Ventricle (LV) Parameters | Before NO Supplementation (Z Score) | After NO Supplementation (Z Score) |
|---|---|---|
| LV diastolic septal thickness | 2.26 | 1.33 |
| LV diastolic dimension | −2.10 | −0.36 |
| LV diastolic wall thickness | 3.59 | 2.24 |
| LV systolic septal thickness | 4.08 | 1.94 |
| LV systolic dimension | −2.08 | −0.67 |
| LV systolic wall thickness | 3.01 | 1.53 |
LV dimensions were measured before and 5 months after initiation of NO supplementation. All parameters demonstrate normalization or improvement.
After 9 months of treatment with ISDN, the subject's BP started to trend toward the upper limits of normal values (Figure 4, middle panel). The loss of efficacy of therapy with nitrate might be due to acquired tolerance resulting from decreased bioconversion to NO34 and is a common effect of chronic organic-nitrate treatment. Hence, we started treatment with a uniquely formulated dietary supplement that contains sodium nitrite together with natural products that have nitrite-reductase activity to enhance direct conversion to NO (Neogenis Labs). Indeed, this direct NO supplementation normalized the subject's BP for an additional 12 months (Figure 4, right panel). Our results suggest that NO deficiency was an important contributor to the hypertension in this ASA subject, and treatment with a NOS-independent NO source therefore resulted in the normalization of his BP.
NO Supplementation is Associated with Improvement in Neurocognitive Measures in a Human Subject with ASA
Subjects with ASA have an increased incidence of attention-deficit hyperactivity disorder, developmental disability, and seizures when they are compared with subjects diagnosed with other UCDs.2 Recent long-term-outcome studies in subjects with ASA show that these neurobehavioral abnormalities can be observed even in subjects without metabolic decompensations.4 This raises the interesting question of whether the deficiency of arginine metabolites, specifically NO, could partially contribute to these phenotypes. Hence, in this ASA subject who was treated with NO supplementation, we performed neuropsychological evaluations before and after treatment to evaluate whether NO therapy was associated with any changes in the neurocognitive parameters. We administered several neuropsychological tests to assess a variety of cognitive domains, including intelligence, academic achievement, executive functioning, memory, visual-motor and visual-perceptual skills, fine-motor functioning, adaptive functioning, and behavioral and emotional functioning. Commonly used measures that have established validity and reliability were selected (Figure 5). The subject's overall intelligence quotient (IQ) as measured by the WASI remained steadily in the impaired range (full IQ = 62, performance IQ = 72, and verbal IQ = 55) and did not show any significant changes with treatment. However, there were marked improvements in the areas of verbal memory and significant gains in the area of nonverbal problem solving. Improvements were noted in problem-solving efficiency, and there was a decrease in rule-breaking behavior (Figures 5A–5C). The subject's tendency to engage in impulsive or intrusive behavior decreased in the list-learning task, in which the number of intrusion responses decreased significantly (Figure 5A).
Figure 5.
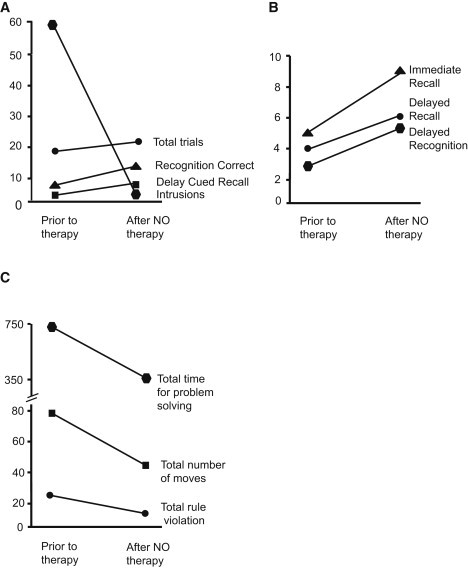
Neuropsychological Testing Results before and after NO Supplementation
(A) California Verbal Learning Test-Children's Version. Raw scores show a significant decrease in intrusions and a mild increase in the other parameters. Aside from the intrusion score, higher scores on the test indicate improved performance.
(B) Children's Memory Scale: Stories Subtest Scaled Scores show improvement in scores after treatment.
(C) Tower of London-Drexel University measurements show significant improvement in nonverbal problem solving with treatment (higher scores indicate poorer performance).
Discussion
ASA, the second most common urea cycle disorder, has a unique phenotype with features that are not observed in subjects with other defects of ureagenesis. Some of the distinguishing features include potentially severe hepatic disease, increased neurocognitive deficiencies, systemic HTN, and trichorrhexis nodosa (coarse and brittle hair). The present standard therapies, such as dietary protein restriction, arginine therapy, and nitrogen-scavenging therapy with either benzoate or phenylbutyrate, for subjects with ASA are focused on treating the defect in hepatic ureagenesis. Although this approach is effective in preventing episodes of hyperammonemia, it does not prevent the long-term complications.4,35 The facts that these complications can be observed in subjects with no significant hyperammonemic episodes and that they occur despite early initiation of L-arginine therapy imply that they might be caused by a cell-autonomous loss of ASL function rather than a block in ureagenesis. Dissecting the phenotypic consequences of ASL loss in the liver and comparing them with those of ASL loss in other tissues would not only help us to understand the molecular pathogenesis of these complications but could also lead to optimization of the treatment in subjects with ASA.
In this study, we report that treatment with a liver-specific HDAd vector encoding Asl corrects the hepatic urea-cycle defect and normalizes growth and survival in ASA mice. However, the GT-treated ASA mice remain hypertensive because of a tissue-specific ASL requirement for NOS-dependent NO production in the vasculature. We show that these GT-treated ASA mice respond to NO supplementation with the normalization of their vessel relaxation ex vivo and of their BPs in vivo. These preclinical studies show that HTN is independent of the ureagenesis defect and can be corrected with the use of NOS-independent NO supplementation. In addition, we show that although there is evidence of increased free-radical production resulting from NOS uncoupling in ASA mice, the decreased channeling of arginine to NO production is the primary cause of the NO deficiency.
Importantly, as a proof of principle, we show the translatability of our findings in the human context. We show that treatment with a NO source leads to sustained normalization of BP in an ASA subject when all other standard antihypertensive treatments had failed over a period of 10 years. Nitrates are not a standard therapy for the treatment of hypertension in the general population because of the availability of more potent antihypertensive medications. Few studies have shown the utility of nitrates for the treatment of isolated systolic hypertension in the elderly population36,37 when they are combined with other antihypertensive medications. Notably, monotherapy with nitrates has not been used as a modality for treatment of hypertension in either the pediatric or the adult populations. The hypertension in ASA is associated with an inherent deficiency of systemic NO,10 and hence, the response to monotherapy with NO supplementation was rapid in the subject with ASA. The development of tolerance to nitrates is a common finding with prolonged use of the medication,38 and we therefore substituted the organic nitrate with a specially formulated inorganic nitrite supplement that resulted in the sustained control of BP. Although the data regarding the response of BP along with the improvement of cardiac hypertrophy are encouraging, they are best considered as preliminary observations from a single subject. Evaluating whether our observations can be generalized to other subjects with ASA would require a randomized controlled trial.
Intriguingly, we observed that NO-supplementation therapy in this individual also brought about improvements in some neurobehavioral parameters pertaining to verbal memory, nonverbal problem solving, and impulsive and intrusive behavior. Although it is possible that at least some of the apparent improvement in performance reflects practice effects, the long 16 month intertest interval between evaluations makes this possibility less likely. Even though abnormalities in metabolites that are synthesized from arginine, such as creatinine and glutamate, free-radical-mediated damage, and accumulation of toxic substances like guanidinosuccinic acid might also contribute to the pathogenesis of the neurological complications, our results support a salutary effect with NO supplementation. These descriptive results, although interesting, constitute observations from a single subject and do not prove that the improvements were due to the NO supplementation. However, our results support the contention that the investigation of NO supplementation in ASA subjects should be performed in a systematic manner.
Our data show that ASA is a human genetic model of NO deficiency, and subjects might therefore benefit from long-term NO supplementation. These results, together with our previous report,10 also suggest that there might be specific genotype-phenotype correlations. Hence, the prevalence and magnitude of NO deficiency and the resulting phenotypic severity in this population will probably vary depending on the differential effects of alterations to the catalytic activity and structural integrity of ASL. This might be clinically reflected in the incomplete penetrance of hypertension and neurological abnormalities seen in ASA subjects. Ultimately, these data further support the literature regarding the fact that the nitrate-nitrite-NO pathway is a viable target for NO restoration,39 and they call for the continued exploration of NO supplementation in this disease in a randomized controlled setting and in-depth correlation with the genotype. Moreover, manipulating ASL could be a relevant therapeutic target for other diseases for which NO has been shown to play a role in disease pathogenesis.
Acknowledgments
We thank the subject and his family for their kind participation in the study. We acknowledge and thank the clinical efforts of M. Mullins, S. Carter, A. Tran, J. Stuff, and the nursing staff at the General Clinical Research Center at Texas Children's Hospital. We also thank the technical assistance of J. Zhang and H. Garg. This work was supported by the National Institutes of Health (DK54450, RR19453, RR00188, GM90310 to B. Lee, GM07526 and DK081735 to A. Erez, RR024173 to J. Marini, and HL73041 and HL22512 to A.K. Reddy), and the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (HD024064). S.C.S. Nagamani, O.A. Shchelochkov, and A. Erez are awardees of the National Urea Cycle Disorders Foundation Research Fellowship. P. Campeau is an awardee of the Canadian Institute of Health Research clinician-scientist training award. O.A. Shchelochkov and P. Campeau are awardees of the O'Malley Fellowship of the Urea Cycle Disorders Consortium Rare Diseases Clinical Research Network. K. Guse is supported by Deutsche Forschungsgemeinschaft from the German Research Foundation. N.Bryan is supported in part by subcontract TR01 GM90310. L. Salviati is supported by Telethon Italy grant GGP09207 and by Fondazione Cariplo. Neogenis Labs formulated and donated the nitric oxide dietary supplement for the study. N.S. Bryan and The University of Texas Health Science Center at Houston have financial interests in Neogenis, a company that develops, produces, and sells nitric-oxide-related products intended to improve health, develops diagnostics for nitric oxide-related metabolites, and performs commercial measurement of nitric oxide metabolites in biological samples.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Brusilow S.W. Online Metabolic and Molecular Basis of Inherited Disease. McGraw Hill Medical; New York: 2009. H. A. [Google Scholar]
- 2.Tuchman M., Lee B., Lichter-Konecki U., Summar M.L., Yudkoff M., Cederbaum S.D., Kerr D.S., Diaz G.A., Seashore M.R., Lee H.S., Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol. Genet. Metab. 2008;94:397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunetti-Pierri N., Erez A., Shchelochkov O., Craigen W., Lee B. Systemic hypertension in two patients with ASL deficiency: a result of nitric oxide deficiency? Mol. Genet. Metab. 2009;98:195–197. doi: 10.1016/j.ymgme.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ficicioglu C., Mandell R., Shih V.E. Argininosuccinate lyase deficiency: Longterm outcome of 13 patients detected by newborn screening. Mol. Genet. Metab. 2009;98:273–277. doi: 10.1016/j.ymgme.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori T., Nagai K., Mori M., Nagao M., Imamura M., Iijima M., Kobayashi K. Progressive liver fibrosis in late-onset argininosuccinate lyase deficiency. Pediatr. Dev. Pathol. 2002;5:597–601. doi: 10.1007/s10024-002-0109-7. [DOI] [PubMed] [Google Scholar]
- 6.Mori M., Gotoh T. Arginine metabolic enzymes, nitric oxide and infection. J. Nutr. 2004;134(10, Suppl):2820S–2825S. doi: 10.1093/jn/134.10.2820S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- 7.Edwards T.M., Rickard N.S. New perspectives on the mechanisms through which nitric oxide may affect learning and memory processes. Neurosci. Biobehav. Rev. 2007;31:413–425. doi: 10.1016/j.neubiorev.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Förstermann U., Münzel T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 9.Huang P.L., Huang Z., Mashimo H., Bloch K.D., Moskowitz M.A., Bevan J.A., Fishman M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 10.Erez A., Nagamani S.C., Shchelochkov O.A., Premkumar M.H., Campeau P.M., Chen Y., Garg H.K., Li L., Mian A., Bertin T.K. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011;17:1619–1626. doi: 10.1038/nm.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim I.H., Józkowicz A., Piedra P.A., Oka K., Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc. Natl. Acad. Sci. USA. 2001;98:13282–13287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer D.J., Ng P. Physical and infectious titers of helper-dependent adenoviral vectors: A method of direct comparison to the adenovirus reference material. Mol. Ther. 2004;10:792–798. doi: 10.1016/j.ymthe.2004.06.1013. [DOI] [PubMed] [Google Scholar]
- 13.Palmer D., Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Palmer D.J., Ng P. Methods for the production of first generation adenoviral vectors. Methods Mol. Biol. 2008;433:55–78. doi: 10.1007/978-1-59745-237-3_4. [DOI] [PubMed] [Google Scholar]
- 15.Zawada R.J., Kwan P., Olszewski K.L., Llinas M., Huang S.G. Quantitative determination of urea concentrations in cell culture medium. Biochem. Cell Biol. 2009;87:541–544. doi: 10.1139/o09-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McBride K.L., Belmont J.W., O'Brien W.E., Amin T.J., Carter S., Lee B.H. Heritability of plasma amino acid levels in different nutritional states. Mol. Genet. Metab. 2007;90:217–220. doi: 10.1016/j.ymgme.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Marini J.C., Didelija I.C., Castillo L., Lee B. Glutamine: Precursor or nitrogen donor for citrulline synthesis? Am. J. Physiol. Endocrinol. Metab. 2010;299:E69–E79. doi: 10.1152/ajpendo.00080.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marini J.C., Erez A., Castillo L., Lee B. Interaction between murine spf-ash mutation and genetic background yields different metabolic phenotypes. Am. J. Physiol. Endocrinol. Metab. 2007;293:E1764–E1771. doi: 10.1152/ajpendo.00525.2007. [DOI] [PubMed] [Google Scholar]
- 19.Marley R., Feelisch M., Holt S., Moore K. A chemiluminescense-based assay for S-nitrosoalbumin and other plasma S-nitrosothiols. Free Radic. Res. 2000;32:1–9. doi: 10.1080/10715760000300011. [DOI] [PubMed] [Google Scholar]
- 20.Bryan N.S., Rassaf T., Maloney R.E., Rodriguez C.M., Saijo F., Rodriguez J.R., Feelisch M. Cellular targets and mechanisms of nitros(yl)ation: An insight into their nature and kinetics in vivo. Proc. Natl. Acad. Sci. USA. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., Bryan N.S., MacArthur P.H., Rodriguez J., Gladwin M.T., Feelisch M. Measurement of nitric oxide levels in the red cell: Validation of tri-iodide-based chemiluminescence with acid-sulfanilamide pretreatment. J. Biol. Chem. 2006;281:26994–27002. doi: 10.1074/jbc.M603953200. [DOI] [PubMed] [Google Scholar]
- 22.Widder J.D., Chen W., Li L., Dikalov S., Thöny B., Hatakeyama K., Harrison D.G. Regulation of tetrahydrobiopterin biosynthesis by shear stress. Circ. Res. 2007;101:830–838. doi: 10.1161/CIRCRESAHA.107.153809. [DOI] [PubMed] [Google Scholar]
- 23.Li L., Chen W., Rezvan A., Jo H., Harrison D.G. Tetrahydrobiopterin deficiency and nitric oxide synthase uncoupling contribute to atherosclerosis induced by disturbed flow. Arterioscler. Thromb. Vasc. Biol. 2011;31:1547–1554. doi: 10.1161/ATVBAHA.111.226456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fink B., Laude K., McCann L., Doughan A., Harrison D.G., Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am. J. Physiol. Cell Physiol. 2004;287:C895–C902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 25.Brunetti-Pierri N., Ng P. Helper-dependent adenoviral vectors for liver-directed gene therapy. Hum. Mol. Genet. 2011;20(R1):R7–R13. doi: 10.1093/hmg/ddr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan N.S., Grisham M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007;43:645–657. doi: 10.1016/j.freeradbiomed.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chataigneau T., Félétou M., Huang P.L., Fishman M.C., Duhault J., Vanhoutte P.M. Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br. J. Pharmacol. 1999;126:219–226. doi: 10.1038/sj.bjp.0702300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin M.I., Fulton D., Babbitt R., Fleming I., Busse R., Pritchard K.A., Jr., Sessa W.C. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J. Biol. Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- 29.Stuehr D., Pou S., Rosen G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001;276:14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 30.Bendall J.K., Alp N.J., Warrick N., Cai S., Adlam D., Rockett K., Yokoyama M., Kawashima S., Channon K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 31.Fakler C.R., Kaftan H.A., Nelin L.D. Two cases suggesting a role for the L-arginine nitric oxide pathway in neonatal blood pressure regulation. Acta Paediatr. 1995;84:460–462. doi: 10.1111/j.1651-2227.1995.tb13673.x. [DOI] [PubMed] [Google Scholar]
- 32.Barbosa P., Cialkowski M., O'Brien W.E. Analysis of naturally occurring and site-directed mutations in the argininosuccinate lyase gene. J. Biol. Chem. 1991;266:5286–5290. [PubMed] [Google Scholar]
- 33.Trevisson E., Salviati L., Baldoin M.C., Toldo I., Casarin A., Sacconi S., Cesaro L., Basso G., Burlina A.B. Argininosuccinate lyase deficiency: Mutational spectrum in Italian patients and identification of a novel ASL pseudogene. Hum. Mutat. 2007;28:694–702. doi: 10.1002/humu.20498. [DOI] [PubMed] [Google Scholar]
- 34.Sage P.R., de la Lande I.S., Stafford I., Bennett C.L., Phillipov G., Stubberfield J., Horowitz J.D. Nitroglycerin tolerance in human vessels: Evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- 35.Batshaw M.L., MacArthur R.B., Tuchman M. Alternative pathway therapy for urea cycle disorders: Twenty years later. J. Pediatr. 2001;138(1, Suppl):S46–S54. doi: 10.1067/mpd.2001.111836. discussion S54–S55. [DOI] [PubMed] [Google Scholar]
- 36.Pickering T.G. Why don't we use nitrates to treat older hypertensive patients? J. Clin. Hypertens. (Greenwich) 2005;7:685–687. doi: 10.1111/j.1524-6175.2005.04141.x. 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stokes G.S. Systolic hypertension in the elderly: Pushing the frontiers of therapy—a suggested new approach. J. Clin. Hypertens. (Greenwich) 2004;6:192–197. doi: 10.1111/j.1524-6175.2004.03508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daiber A., Münzel T., Gori T. Organic nitrates and nitrate tolerance—state of the art and future developments. Adv. Pharmacol. 2010;60:177–227. doi: 10.1016/B978-0-12-385061-4.00007-6. [DOI] [PubMed] [Google Scholar]
- 39.Bryan N.S., Loscalzo J., editors. Nitrite and Nitrate in Human Health and Disease. First Edition. Humana Press; New York: 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.