

Abstract
Auriculocondylar syndrome (ACS) is a rare, autosomal-dominant craniofacial malformation syndrome characterized by variable micrognathia, temporomandibular joint ankylosis, cleft palate, and a characteristic “question-mark” ear malformation. Careful phenotypic characterization of severely affected probands in our cohort suggested the presence of a mandibular patterning defect resulting in a maxillary phenotype (i.e., homeotic transformation). We used exome sequencing of five probands and identified two novel (exclusive to the patient and/or family studied) missense mutations in PLCB4 and a shared mutation in GNAI3 in two unrelated probands. In confirmatory studies, three additional novel PLCB4 mutations were found in multigenerational ACS pedigrees. All mutations were confirmed by Sanger sequencing, were not present in more than 10,000 control chromosomes, and resulted in amino-acid substitutions located in highly conserved protein domains. Additionally, protein-structure modeling demonstrated that all ACS substitutions disrupt the catalytic sites of PLCB4 and GNAI3. We suggest that PLCB4 and GNAI3 are core signaling molecules of the endothelin-1-distal-less homeobox 5 and 6 (EDN1-DLX5/DLX6) pathway. Functional studies demonstrated a significant reduction in downstream DLX5 and DLX6 expression in ACS cases in assays using cultured osteoblasts from probands and controls. These results support the role of the previously implicated EDN1-DLX5/6 pathway in regulating mandibular specification in other species, which, when disrupted, results in a maxillary phenotype. This work defines the molecular basis of ACS as a homeotic transformation (mandible to maxilla) in humans.
Main Text
Auriculocondylar syndrome (ACS; MIM 602483; also known as “question-mark ear syndrome” or “dysgnathia complex”) is an autosomal-dominant craniofacial malformation syndrome characterized by highly variable mandibular anomalies ranging from mild to severe micrognathia, often with temporomandibular joint (TMJ) ankylosis, cleft palate, and a distinctive ear malformation that consists of separation of the lobule from the external ear, giving the appearance of a question mark (Figure 1). Other frequently described features include prominent cheeks, cupped and posteriorly rotated ears, preauricular tags, and microstomia. ACS was first described in a mother and her two affected children just over thirty years ago,1 and nine well-characterized families with multiple affected individuals2–8 (and at least one linkage peak mapping to 1p21-1q236,9) as well as several simplex cases4,10–12 have been reported to date.
Figure 1.
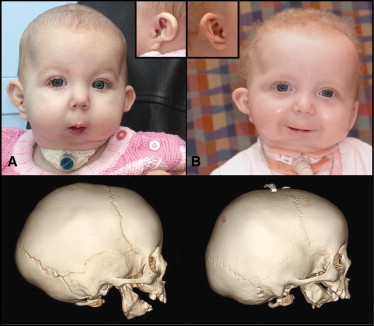
PLCB4 and GNAI3 Mutations Lead to Severe Mandibular Hypoplasia and Ear Dysplasia
(A) Facial photos of proband S001 (PLCB4 p.Tyr623Cys)
(B) S008 (GNAI3 p.Gly40Arg) with severe micrognathia and the classic “question mark” ear (inset). Below each photo is the respective three-dimensional computed tomography (3D CT) scan, which shows the severe mandibular hypoplasia associated with mandibular body and ramus dysplasia.
Our index case (S011) and her affected mother (S012P; kindred D, individuals I-1 and II-1, respectively, in Figure S1, available online) had severe mandibular abnormalities:3 congenital mandibular ankylosis with lateral fusion of the mandible to the temporozygomatic suture and medial fusion to the skull base (medial and lateral pterygoid plates) (Figure 2), resulting in airway obstruction requiring tracheostomy. Intraoperative evaluation of S011 was notable for the presence of severe microglossia, elongated soft-tissue masses attached at the posterior floor of the mouth, and excessive soft tissue protruding from the medial alveolus of the lower jaw, giving the appearance of a “mandibular palate” (Figure 2). We identified four additional kindreds with dysgnathia complex (i.e., ACS),3 as well as two simplex case child-parent trios (S001 and S008 [II-1 individuals in kindreds B and C, respectively, in Figure S1]), a family with two affected siblings (S004 [proband] and S005 [kindred A, individuals II-1 and II-2 in Figure S1]) whose father had mild mandibular hypoplasia, and an additional singleton proband (A001; Figure S1). We obtained approval for this study from our institutional review board (Seattle Children's Hospital), written consent was obtained for all participants, and consent for photography was obtained from all individuals whose images are included in this manuscript. In each case, mandibular ankylosis was progressive, of variable severity, and characterized by inconsistent fusion to the medial and lateral pterygoid plates. All cases demonstrated a similar phenotype, consisting of a lateral mandibular bony prominence with or without TMJ ankylosis, and had features consistent with classic ACS. The anatomic features of these cases led us to hypothesize that the malformations observed in individuals with ACS were due to a homeotic transformation, with the mandible assuming a maxillary phenotype (Figures 2E–2G).
Figure 2.
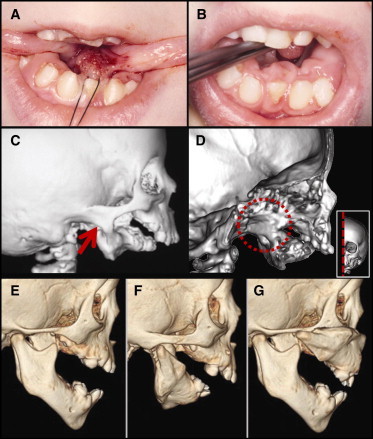
Homeotic Transformation of the Mandible to a Maxillary Phenotype
(A) Microglossia and lateral soft-tissue projections at the tongue base as seen intraoperatively in our index case S011 (GNAI3 p.Gly40Arg).
(B) Excessive soft-tissue projections emanating from the medial surface of the lower jaw.
(C) 3D CT demonstrates severe mandibular hypoplasia, with fusion of the lateral mandibular process to the temporozygomatic suture (red arrow).
(D) Medial view of a parasagittal section of the skull reveals complete fusion of the left mandibular ramus with the medial and lateral pterygoid plates (within red dashed circle); inset depicts the plane of the section.
(E) Control CT image of an age- and sex-matched individual showing normal mandible anatomy.
(F) CT image of individual S001 with a PLCB4 p.Tyr623Cys substitution.
(G) A composite CT image generated with photo-editing software in which the mandible from panel F was copied, inverted, and placed over the control maxilla in panel G. The resultant image shown here depicts the remarkable alignment of the ACS mandible with the control maxilla, zygomatic arch, and midfacial structures, demonstrating transformation to a maxillary identity (a homeotic transformation).
Other reports of mandibular patterning mutations in mice and zebrafish have implicated the endothelin-1 (EDN1) receptor A (EDNRA) and its downstream targets, distal-less homeobox 5 and 6 (DLX5 and DLX6).13–16 Sanger sequencing of endothelin-1 pathway candidates EDNRA, DLX5, and DLX6 in our probands was negative. We performed exome sequencing on all five probands and parents (excluding A001 [no parental DNA available] and S012-P [inadequate sample]) to identify gene-based coding variants present in individuals with ACS. Genomic libraries were prepared and underwent exome capture with the use of a 28 Mb target derived from the consensus coding sequence (CCDS) database (version 20080902)17 or the ∼32 Mb target from Roche Nimblegen SeqCap EZ (version 1.0). Exome-enriched libraries were sequenced on Illumina GAIIx or HiSeq 2000 platforms with paired-end 50 or 76 base reads. Each individual generated ∼75 million unique reads mapping to the exome target and nearby flanking regions. Over 93% of the exome positions had a coverage depth > 8×, and the average overall coverage depth was 103×. Data from each individual were processed from real-time base calls and aligned to a human reference (hg19) through the use of the Burrows-Wheeler Aligner.18 Data was processed with the use of the Genome Analysis Toolkit19 (GATK reference version 1.0.2905), and variant detection and genotyping were performed with the use of the Unified Genotyper (GATK) tool. We flagged variant sites by using the filtration walker (GATK) to mark sites that were of lower quality and more likely to be false positives. Each individual generated an average of ∼22,000 total variants, with a final-pass filter set of ∼18,900 variants annotated for variant function (i.e., missense, synonymous, splicing, insertion or deletion [in/del], etc.) (Genome Variation Server).
Given the consistency among the phenotypic features, we first analyzed exome data from all five probands to identify any genes with novel variants shared among them. Proband variant data (i.e., missense, in/dels, and splice acceptor and donor sites) were initially filtered against a smaller database of ∼1,200 control exomes drawn from a subset of individuals from the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) to identify novel variants, those passing GATK filtering, and those evident at a robust coverage level (>20x). Second, we performed discrete filtering to identify any gene in which all probands shared novel missense, insertion/deletion, nonsense, or splice variants consistent with either an autosomal-dominant or -recessive model. Under these models and criteria, no genes were detected in common between all probands.
We expanded our search for de novo mutations in probands (S001, S004, and S008), using data from parental individuals along with stringent parameters for GATK pass filters and a sequence depth of >20× in both parents and probands. This identified six putative genes (ASPSCR1, C2orf16, HERC1, PAIP1, PLCB4, and TCHP) within these three individuals. The PLCB4 (MIM 600810) variant c.1868A>G (NM_000933.3; p.Tyr623Cys) had the highest conservation Genomic Evolutionary Rate Profiling (GERP)20,21 score (6.17) and was considered the top candidate on the basis of the core signaling role of its ortholog plcb3 in mandibular patterning of zebrafish.22,23 Sanger sequencing of parents S002-P and S003-P (kindred B, individuals I-1 and I-2 in Figure S1) and the proband (S001) confirmed it as a de novo mutation (Table 1). On the basis of this finding, we searched the remaining proband exome data for additional PLCB4 variants and identified, in siblings S004 and S005, a second missense mutation at c.986A>C, which changed the amino acid at position 329 from asparagine to serine (p.Asn329Ser; Table 1). In this kindred, the mutation was transmitted from the mildly affected father (S006-P [kindred A, individual I-1 in Figure S1]), a transmission which supports the variable expressivity of ACS.
Table 1.
PLCB4 and GNAI3 Mutations in Probands with Auriculocondylar Syndrome
| Individual/ Pedigree | Gene | Amino Acid | Nucleotide | GERP | GS | Control Frequency | Inheritance |
|---|---|---|---|---|---|---|---|
| S001 | PLCB4 | Tyr623Cys | c.1868A>G | 6.17 | 194 | 0/10758 | de novo |
| S004a | PLCB4 | Asn329Ser | c.986A>C | 5.93 | 46 | 0/10758 | transmitted (IPb) |
| M001 | PLCB4 | Arg621His | c.1862G>A | 6.17 | 29 | 0/10758 | transmitted |
| M002 | PLCB4 | Asn650His | c.1948A>C | 5.92 | 68 | 0/10758 | transmitted |
| M003 | PLCB4 | Arg621Cys | c.1861C>T | 6.17 | 180 | 0/10758 | transmitted (IPb) |
| S008, S011 | GNAI3 | Gly40Arg | c.118G>C | 5.82 | 125 | 0/10758 | transmitted |
Exome sequencing and Sanger confirmation identified five missense mutations that lead to rare substitutions in the catalytic domain of PLCB4 in five families and a single GNAI3 catalytic domain substitution in two unrelated families. These mutations are in highly conserved domains as indicated by GERP score and Grantham score (GS), and they are not found in a control set of human exomes from the NHLBI ESP data release ESP5400, accessed on December 10, 2011. M001 and M002 are families 1 and 2, respectively, as described in Storm et al.5
Proband has a similarly affected sibling and mildly affected father with the same mutation.37
IP, incomplete penetrance.
The discovery of putative coding-sequence, disease-causing PLCB4 mutations in isolated ACS kindreds led us to use Sanger sequencing to screen the gene coding regions containing the conserved catalytic site (exons 11–26) in two additional multigeneration ACS pedigrees5 (family 1 [M001] and family 2 [M002] in Figure S2). We identified two additional novel PLCB4 missense mutations at c.1862G>A (M001) and c.1948A>C (M002) (with resultant protein changes, p.Asn650His and p.Arg621His, respectively) that segregated in affected individuals (Table 1 and Figure S2). We have also identified a family with an ACS proband (family 3 [M003], individual IV-1 in Figure S2; with micrognathia, cleft palate, glossoptosis, and a constriction between the helix and the lobule of the left ear) and his “unaffected” father (individual III-1) who presented with asymmetric ear lobes. The family history is significant for four paternal relatives with ear malformations with or without micrognathia (Table 1; family 3 in Figure S2). Sanger sequencing confirmed a PLCB4 missense mutation at c.1862G>A (p.Arg621Cys) in the proband and his unaffected father, the same amino-acid position (621) as noted in family M001 but a different amino-acid change. The phenotypic variability of ACS in these families was confirmed by the variable expressivity and apparent incomplete penetrance associated with the father (S006-P) of a proband with a PLCB4 mutation.5 None of the PLCB4 mutations we discovered had been identified previously in a large set of control exomes containing 10,758 chromosomes (Table 1). Each of the mutations led to substitutions at highly conserved nucleotide positions (those with a GERP score > 5.8 make up the top 0.006% of coding nucleotides21) and evolutionary invariant amino-acid positions (Figure 3).
Figure 3.
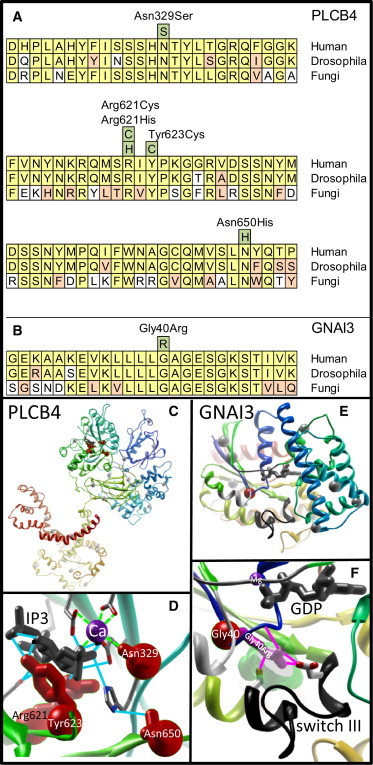
Conservation and Substitution Modeling of PLCB4 and GNAI3
(A) PLCB4 sequence alignment at the highly conserved catalytic domains. Alignments were performed between human, Drosophila, and fungus protein sequences with the use of ClustalW2. Conservation among vertebrates, invertebrates, and fungi suggests evolutionary importance for these residues. Identified substitutions are depicted in green boxes; residues conserved between humans, Drosophila, and fungi are depicted in yellow; conservative substitutions are in orange; and divergent residues are in white. Protein sequences used for alignment of PLCB4 include Homo sapiens (NP_877949.2), Drosophila (no receptor potential A, isoform D, NP_001014720.1), and Aspergillus clavatus NPRL 1 (1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase 1 [XP_001268930.1]).
(B) Sequence alignment (similar to panel A) of the GNAI3 catalytic region with the p.Gly40Arg ACS subsitution. Protein sequences used for alignment of GNAI3 include Human (NP_006487.1), Drosophila (G protein αi subunit 65A, NP_477502.1), and Saccharomyces cerevisiae (Gpa2p, NP_010937.1).
(C) PLCB4 structure depicting the four missense substitutions (red residues). In this panel and in panel E, other missense variants observed in controls are shown as light gray spheres.
(D) Magnification of the active site of PLCB4 demonstrates variant residues that contribute directly to catabolic release of inositol triphosphate (IP3, gray molecule) and disrupt catalytic function without destabilizing protein folding. These residues are presumed to inhibit MAPK signaling without affecting other protein interactions. Calcium ion is depicted in purple. Hydrogen bonds between amino-acid side chains, calcium ion, and IP3 are shown as blue lines, and ionic metal bonds are shown as green dashed lines.
(E) GNAI3 structure showing the p.Gly40Arg mutant (red) residue.
(F) Magnification of the GNAI3 active site shows the Gly40Arg mutant residue (pink lines) reaching across a previously undescribed cavity (transparent green), consistent in all three GNAI3 crystal structures, to stabilize the RapGapII-recognized active conformation of guanosine triphosphate-activated switch III (black ribbon; switch I in gray ribbon, switch II in white ribbon) by forming a hydrogen bond network (pink lines) between the Gly40Arg residue, a nearby main chain carbonyl (blue ribbon), and stabilizing the active confirmation of switch III through interaction with the key switch residues (red and white sticks). Switch III is recognized by RapGapII and would lead to inhibition of the MAPK pathway activation through Rap (Figure 4).
Discrete filtering of exome data from the PLCB4-mutation-negative probands (S008, S011, and A001) revealed a Sanger-confirmed single-missense mutation in an inhibitory G protein (GNAI3; MIM 139370) in exon 1 (c.118G>C [NM_006496.2; p.Gly40Arg]) shared by individuals S008 and S011 (Table 1). In both kindreds, the p.Gly40Arg variant was inherited; in one case (S011) it was inherited from an affected mother (S012-P), and in the other (S008), from an unaffected father (S009-P), documenting incomplete penetrance. Using the exome data, we confirmed that these individuals did not share a haplotype region encompassing this mutation, and all other measures of global genomic relatedness, determined by analyzing all exome variants or specific high-frequency variants, were consistent with unrelatedness. These data suggest the possibility of a recurrent mutation at this position. The p.Gly40Arg substitution also was not observed in 10,758 control chromosomes, and it occurred at a highly conserved amino-acid residue (and the Grantham score [125] suggested a moderately radical amino-acid replacement24) within the guanosine diphosphate (GDP)-binding catalytic domain of this G protein (Table 1; Figure 3). Furthermore, GNAI3 maps in the vicinity of the region on chromosome 1 previously linked with the ACS phenotype.6 Given this, we considered the GNAI3 p.Gly40Arg change to be an ACS candidate because of the known role of phospholipase C (PLC) in G protein-coupled receptor signaling, a core component of the endothelin-DLX pathway14 (reviewed in Clauthia et al.25).
To further evaluate the significance of variants in PLCB4 (p.Asn329Ser, p.Arg621His, p.Arg621Cys, p.Tyr623Cys, p.Asn650His) and GNAI3 (p.Gly40Arg), we performed detailed structural protein modeling of specific substitutions found in ACS probands. All missense variants observed in cases, controls, and accessible databases were mapped to PLCB4 and GNAI3 protein structures and visualized with UCSF Chimera.26 None of the variants identified in controls occurred in the catalytic domains of these highly conserved proteins.
In PLCB4, modeling each mutated side chain resulted in minimal overall structural impact27 but removed hydrogen and ionic bonds essential to the catalysis of phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-trisphosphate and diacylglycerol (Figures 3C and 3D). Thus, these ACS-associated PLCB4 substitutions are localized within the PLCB4 catalytic substrate binding domain (Figure 3D) and would be expected to directly affect function.
The GNAI3 p.Gly40Arg variant side chain was modeled with each of the three currently available GNAI3 structures.28,29 Effects of the substitution on GNAI3 stability were measured with multiple knowledge-based scoring functions.27 Minimal to positive influence on stability suggested further study and led us to examine why this large arginine substitution was tolerated in the conserved core of the G-alpha protein.27 A detailed assessment30 revealed a cavity large enough for an organic cationic metabolite in each of the three crystal structures (Figures 3E and 3F, semi-transparent green surface) and large enough to hold and electrostatically balance the guanidinium group. Our protein modeling suggested the presence of hydrogen bonds between the Arg40 side-chain nitrogens and (1) the P loop Gly42 backbone carbonyl, (2) the Asp229 side chain, and (3) the Ser246 side chain (Figure 3F, pink lines). Modeling the p.Gly40Arg variant suggests stabilization of GNAI3 in the active conformation (switch III).25 Examination of potential interactions with the downstream signaling partner RapGAPII through analysis of the common structural fold between Rap and GNAI331 revealed close recognition of the constitutively activated switch III. Thus, modeling the GNAI3 p.Gly40Arg substitution suggests a gain of function, with stabilization of the active conformation recognized by RapGapII triggering inhibition of the mitogen-activated protein kinase (MAPK) pathway activation through Rap.
As a proxy for assessing the effects of the substitution on GNAI3 activity, we chose to assay the expression of two known downstream targets of the G protein-coupled endothelin receptor pathway, DLX5 and DLX6, which play a key role in mandibular patterning.12–15 The importance of PLC and G protein signaling in this pathway led us to hypothesize that a potential dominant-negative effect of PLCB4 mutations and a gain-of-function GNAI3 mutation would lead to downregulation of DLX5 and DLX6. We measured gene expression levels for DLX5 and DLX6 in available cultured mandibular osteoblasts from our two GNAI3 probands (p.Gly40Arg), one PLCB4 proband (p. Try623Cys), and one unresolved ACS proband (A001). Using quantitative, real-time RT-PCR of steady-state mRNA, we identified a 6-fold reduction of DLX5 (p < 0.005; probands versus controls) and an 8-fold reduction of DLX6 expression (p < 0.001) in mandibular osteoblasts from selective probands relative to 17 randomly selected calvarial osteoblasts from controls. Preliminary RNA sequencing experiments showed similar and relatively strong levels of expression of PLCB4 and GNAI3 in control calvarial osteoblasts (data not shown), and at levels 2–4 fold greater than DLX5 and DLX6. These data support a final common pathway leading to DLX5/6 regulation through suppression of the endothelin pathway as a unifying cause. These data also provide a basis for examining other candidate genes with similar functional effects in our single unresolved case (A001), negative for mutations in PLCB4 and GNAI3. To do this we looked at our initial exome results for A001 to identify singleton exon sequence variants in candidate genes from the PLC gene family (PLCD1, PLCXD1, PLCH1), PLC pathway candidate genes (PI4KA, DOCK1, DOCK6, MIOX, HRASLS, RAC1), or known endothelin-DLX5/6 pathway genes (BMPER, IGF2BP2, WNT7A). Although sequence variants were detected in five candidate genes (from the list above), upon the application of information from our large control set, only DOCK1 and DOCK6 sequence variants were not present in controls (Table S1). As members of the DOCK-C subfamily of the DOCK family of guanine nucleotide exchange factors, both genes have a suggestive function as an activator of small G proteins.32 Although we are hesitant to draw conclusions from a single proband without parental data, it is intriguing to have identified an exceptionally rare mutation in a G protein activator in a disorder known to be caused by disruption of signaling through a G protein-dependent signaling pathway. Further functional studies will be required to test the putative role of these DOCK-related genes in the endothelin-DLX5/DLX6 pathway and mandibular patterning.
Our primary observation of a de novo mutation in PLCB4 led us to identify other mutations in a second kindred and in three confirmatory pedigrees. Further analysis of PLCB4-negative probands identified a second (GNAI3) mutation in two probands and a DOCK6 sequence variant in another proband, indicating a role for endothelin-1 pathway genes in this heterogeneous condition. In each of the families with transmission of the PLCB4 or GNAI3 mutations, we observed incomplete penetrance and/or variable expressivity of the ACS phenotype. Although variable expression is common among dominant craniofacial disorders, the nature of phenotypic variability in ACS suggests that common craniofacial disorders such as micrognathia, Pierre Robin sequence (MIM 261800), and microtia may also be caused by mutations in this pathway.
The interpretation of our data was supported by knowledge of classic developmental model systems. In zebrafish, mutations in edn1 result in a reduction in lower jaw size and fusion to dorsal structures with loss of the intervening joints.25,33,34 Mutations of schmerle (she), the zebrafish ortholog of PLCB3 (and ortholog of PLCB4), result in a near phenocopy of edn1 mutations, with reduction of the ventral facial skeleton and fusion to dorsal skeletal elements.22 Two she alleles have been identified in the highly conserved catalytic domains of plcb3, the same domains in which we have defined substitutions in PLCB4. she is required for edn1-dependent dlx5a, dlx6a, and dlx3a expression. Furthermore, when plcb3−/− neural crest cells were transplanted into wild-type embryos, they contributed to dorsal, but not ventral or intermediate, jaw structures, suggesting that plcb3 may be necessary for the determination of ventral branchial arch fates and functions downstream of edn1. In another animal model, knockdown of PLCB3 in Xenopus results in reduction of cartilage in the first pharyngeal arch and loss of the jaw joint, in a proportion of treated embryos.35 The lack of a craniofacial phenotype in the Plcb4 knockout mouse36 suggests that the catalytic domain substitutions identified in our cases and in zebrafish schmerle may result in dominant negative effects.37 A similar mechanism has been suggested for a splice variant affecting the catalytic domain of PLCD4, which has a dominant negative effect on the function of other wild-type PLC enzymes.38 Although studies to confirm this hypothesis are necessary, these data would suggest that the loss of PLC catalytic function, in the absence of effects on protein stability, may confer dominant negative effects.
The observed conversion from a mandibular to a maxillary phenotype, as we have described for ACS, represents a homeotic transformation and is supported by several mouse knockout models (Ednra39 and Edn140 annotated in Figure 4). Mice with targeted mutations of endothelin-converting enzyme 1 (Ece-1), Edn1, Ednra, and G protein alpha subunits (Gαq, Gα11) each exhibit a reduction in ventral jaw size and fusion to dorsal skeletal components.13,14,40–43 Similarly, mice lacking expression of Dlx5 and Dlx6 demonstrate transformation of the lower jaw into an upper jaw phenotype.13,15,16,25 This homeotic transformation was the first described in any gene other than the classic Hox genes.15 Furthermore, selective reduction of another member of the endothelin-Dlx5/Dlx6 pathway, Hand2, in the cranial neural crest results in duplication of palatine bones in the mouse mandible and aglossia.44 These findings support a role for Edn1-Dlx5/Dlx6 as a prototypical signaling pathway (Figure 4) in cranial neural crest cell proliferation in the first branchial arch that is necessary for mandibular specification. As described above, this pathway is further supported by our quantitative real-time PCR analysis in osteoblasts from four ACS cases, which demonstrates a significant reduction in the downstream expression of DLX5 and DLX6 (Figure S3).
Figure 4.
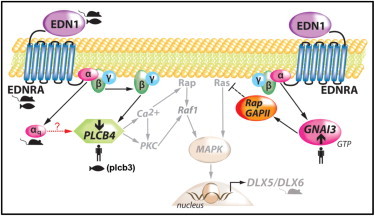
Hypothesized Pathway for Mandibular Patterning Associated with Mutations in the Endothelin-DLX5/6 Pathway
Several reports support the premise that mutations in the endothelin pathway cause homeotic transformation of the lower (ventral) jaw to a maxillary (dorsal) identity. Mutations in mice of Edn1, Ednra, the G protein αq subunit, Dlx5/Dlx6, and most recently Hand2, each result in a small ventral jaw with dorsal jaw features. Mutations in zebrafish plcb3 lead to the same homeotic transformation. In this figure, cartoons of fish, mice, and humans indicate gene family members in this pathway that, when mutated, result in a homeotic transformation of the dorsal and ventral jaws (left: PLCB4 loss-of-function, right: GNAI3 gain-of-function). The arrow with a dashed line indicates hypothetical signaling between αq and PLCB4. Figure adapted from Marinissen and Gutkind.45
We report the identification of extremely rare, highly conserved mutations (both de novo and inherited) in two endothelin pathway signaling enzymes, PLCB4 and GNAI3, as the molecular causes of ACS and further define the role of the endothelin-distal-less pathway in the specification and patterning of the lower jaw of vertebrates. The ACS phenotype is consistent with the transformation of the mandible to a maxillary identity and constitutes a molecularly defined homeotic transformation in humans. The phenotypic variability of ACS suggests that mutations in this pathway, especially those affecting core signaling molecules such as PLCB4 and GNAI3, should be considered as potential candidates for other ear and jaw malformations. The identification of phenotypic variability and incomplete penetrance underscores the utility of these results in genetic counseling of families affected by ACS.
Acknowledgments
This study was supported by grants from the NIH-NHGRI Next Generation Mendelian Genetics (HG005608, M.J.R., D.A.N., M.J.B.), the Jean Renny Endowment for Craniofacial Medicine, the Laurel Foundation Center for Craniofacial Research (M.L.C.), NIH-NIDCD (P30-DC05188, G.E.G.), and the E-Rare CRANIRARE project (S.L.).
The authors would like to thank the participating families for their interest in this work and participation in these studies. The authors wish to thank Orapin Horst and Henry Bourne for illuminating discussions on the roles of the variant genes in G protein signaling, Marianne L. Seto for her early scientific contributions, Hitesh Kapadia for 3DCT graphics and manuscript review, and Eden Palmer for the graphic arts. We further acknowledge the NHLBI GO Exome Sequencing Project; i.e., the Lung GO Sequencing Project (HL102923), the WHI Sequencing Project (HL102924), the Broad GO Sequencing Project (HL102925), the Seattle GO Sequencing Project (HL102926), and the Heart GO Sequencing Project (HL103010), which produced and provided the control exome variant calls for comparison, and the Northwest Genomics Center for exome preparation, sequencing, and initial analysis. J.M.J. would like to dedicate his contributions to this project to Georgia Floyd-Smith.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Genome Variation Server, http://gvs.gs.washington.edu/GVS/
NHLBI Exome Sequencing Project (ESP) Exome Variation Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
UCSF Chimera, http://www.cgl.ucsf.edu/chimera/
References
- 1.Uuspää V. Combined bilateral external ear deformity and hypoplastic mandible. Case report. Scand. J. Plast. Reconstr. Surg. 1978;12:165–167. doi: 10.3109/02844317809012989. [DOI] [PubMed] [Google Scholar]
- 2.Jampol M., Repetto G., Keith D.A., Curtin H., Remensynder J., Holmes L.B. New syndrome? Prominent, constricted ears with malformed condyle of the mandible. Am. J. Med. Genet. 1998;75:449–452. doi: 10.1002/(sici)1096-8628(19980217)75:5<449::aid-ajmg1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 3.Erlich M.S., Cunningham M.L., Hudgins L. Transmission of the dysgnathia complex from mother to daughter. Am. J. Med. Genet. 2000;95:269–274. [PubMed] [Google Scholar]
- 4.Guion-Almeida M.L., Zechi-Ceide R.M., Vendramini S., Kokitsu-Nakata N.M. Auriculo-condylar syndrome: additional patients. Am. J. Med. Genet. 2002;112:209–214. doi: 10.1002/ajmg.10631. [DOI] [PubMed] [Google Scholar]
- 5.Storm A.L., Johnson J.M., Lammer E., Green G.E., Cunniff C. Auriculo-condylar syndrome is associated with highly variable ear and mandibular defects in multiple kindreds. Am. J. Med. Genet. A. 2005;138A:141–145. doi: 10.1002/ajmg.a.30883. [DOI] [PubMed] [Google Scholar]
- 6.Masotti C., Oliveira K.G., Poerner F., Splendore A., Souza J., Freitas Rda.S., Zechi-Ceide R., Guion-Almeida M.L., Passos-Bueno M.R. Auriculo-condylar syndrome: mapping of a first locus and evidence for genetic heterogeneity. Eur. J. Hum. Genet. 2008;16:145–152. doi: 10.1038/sj.ejhg.5201955. [DOI] [PubMed] [Google Scholar]
- 7.Ozturk S., Sengezer M., Isik S., Gul D., Zor F. The correction of auricular and mandibular deformities in auriculo-condylar syndrome. J. Craniofac. Surg. 2005;16:489–492. doi: 10.1097/01.scs.0000147655.94656.0d. [DOI] [PubMed] [Google Scholar]
- 8.Shkalim V., Eliaz N., Linder N., Merlob P., Basel-Vanagaite L. Autosomal dominant isolated question mark ear. Am. J. Med. Genet. A. 2008;146A:2280–2283. doi: 10.1002/ajmg.a.32452. [DOI] [PubMed] [Google Scholar]
- 9.Kokitsu-Nakata N.M., Zechi-Ceide R.M., Vendramini-Pittoli S., Romanelli Tavares V.L., Passos-Bueno M.R., Guion-Almeida M.L. Auriculo-condylar syndrome. Confronting a diagnostic challenge. Am. J. Med. Genet. A. 2011 doi: 10.1002/ajmg.a.34337. in press. Published online November 21, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Guion-Almeida M.L., Kokitsu-Nakata N.M., Zechi-Ceide R.M., Vendramini S. Auriculo-condylar syndrome: further evidence for a new disorder. Am. J. Med. Genet. 1999;86:130–133. doi: 10.1002/(sici)1096-8628(19990910)86:2<130::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Priolo M., Lerone M., Rosaia L., Calcagno E.P., Sadeghi A.K., Ghezzi F., Ravazzolo R., Silengo M. Question mark ears, temporo-mandibular joint malformation and hypotonia: auriculo-condylar syndrome or a distinct entity? Clin. Dysmorphol. 2000;9:277–280. doi: 10.1097/00019605-200009040-00009. [DOI] [PubMed] [Google Scholar]
- 12.Gerkes E.H., van Ravenswaaij C.M., van Essen A.J. Question mark ears and post-auricular tags. Eur. J. Med. Genet. 2008;51:264–267. doi: 10.1016/j.ejmg.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Vieux-Rochas M., Mantero S., Heude E., Barbieri O., Astigiano S., Couly G., Kurihara H., Levi G., Merlo G.R. Spatio-temporal dynamics of gene expression of the Edn1-Dlx5/6 pathway during development of the lower jaw. Genesis. 2010;48:262–373. doi: 10.1002/dvg.20625. [DOI] [PubMed] [Google Scholar]
- 14.Ivey K., Tyson B., Ukidwe P., McFadden D.G., Levi G., Olson E.N., Srivastava D., Wilkie T.M. Galphaq and Galpha11 proteins mediate endothelin-1 signaling in neural crest-derived pharyngeal arch mesenchyme. Dev. Biol. 2003;255:230–237. doi: 10.1016/s0012-1606(02)00097-0. [DOI] [PubMed] [Google Scholar]
- 15.Depew M.J., Lufkin T., Rubenstein J.L. Specification of jaw subdivisions by Dlx genes. Science. 2002;298:381–385. doi: 10.1126/science.1075703. [DOI] [PubMed] [Google Scholar]
- 16.Beverdam A., Merlo G.R., Paleari L., Mantero S., Genova F., Barbieri O., Janvier P., Levi G. Jaw transformation with gain of symmetry after Dlx5/Dlx6 inactivation: mirror of the past? Genesis. 2002;34:221–227. doi: 10.1002/gene.10156. [DOI] [PubMed] [Google Scholar]
- 17.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker M.B., Miller C.T., Swartz M.E., Eberhart J.K., Kimmel C.B. phospholipase C, beta 3 is required for Endothelin1 regulation of pharyngeal arch patterning in zebrafish. Dev. Biol. 2007;304:194–207. doi: 10.1016/j.ydbio.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker M.B., Miller C.T., Coffin Talbot J., Stock D.W., Kimmel C.B. Zebrafish furin mutants reveal intricacies in regulating Endothelin1 signaling in craniofacial patterning. Dev. Biol. 2006;295:194–205. doi: 10.1016/j.ydbio.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 24.Li W.H., Wu C.I., Luo C.C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 25.Clouthier D.E., Garcia E., Schilling T.F. Regulation of facial morphogenesis by endothelin signaling: insights from mice and fish. Am. J. Med. Genet. A. 2010;152A:2962–2973. doi: 10.1002/ajmg.a.33568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27.Ngan S.C., Hung L.H., Liu T., Samudrala R. Scoring functions for de novo protein structure prediction revisited. Methods Mol. Biol. 2008;413:243–281. doi: 10.1007/978-1-59745-574-9_10. [DOI] [PubMed] [Google Scholar]
- 28.Soundararajan M., Willard F.S., Kimple A.J., Turnbull A.P., Ball L.J., Schoch G.A., Gileadi C., Fedorov O.Y., Dowler E.F., Higman V.A. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc. Natl. Acad. Sci. USA. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimple A.J., Soundararajan M., Hutsell S.Q., Roos A.K., Urban D.J., Setola V., Temple B.R., Roth B.L., Knapp S., Willard F.S., Siderovski D.P. Structural determinants of G-protein alpha subunit selectivity by regulator of G-protein signaling 2 (RGS2) J. Biol. Chem. 2009;284:19402–19411. doi: 10.1074/jbc.M109.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scrima A., Thomas C., Deaconescu D., Wittinghofer A. The Rap-RapGAP complex: GTP hydrolysis without catalytic glutamine and arginine residues. EMBO J. 2008;27:1145–1153. doi: 10.1038/emboj.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Côté J.F., Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- 33.Miller C.T., Schilling T.F., Lee K., Parker J., Kimmel C.B. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- 34.Kimmel C.B., Ullmann B., Walker M., Miller C.T., Crump J.G. Endothelin 1-mediated regulation of pharyngeal bone development in zebrafish. Development. 2003;130:1339–1351. doi: 10.1242/dev.00338. [DOI] [PubMed] [Google Scholar]
- 35.Reisoli E., De Lucchini S., Nardi I., Ori M. Serotonin 2B receptor signaling is required for craniofacial morphogenesis and jaw joint formation in Xenopus. Development. 2010;137:2927–2937. doi: 10.1242/dev.041079. [DOI] [PubMed] [Google Scholar]
- 36.Jiang H., Lyubarsky A., Dodd R., Vardi N., Pugh E., Baylor D., Simon M.I., Wu D. Phospholipase C beta 4 is involved in modulating the visual response in mice. Proc. Natl. Acad. Sci. USA. 1996;93:14598–14601. doi: 10.1073/pnas.93.25.14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kano M., Hashimoto K., Watanabe M., Kurihara H., Offermanns S., Jiang H., Wu Y., Jun K., Shin H.S., Inoue Y. Phospholipase cbeta4 is specifically involved in climbing fiber synapse elimination in the developing cerebellum. Proc. Natl. Acad. Sci. USA. 1998;95:15724–15729. doi: 10.1073/pnas.95.26.15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagano K., Fukami K., Minagawa T., Watanabe Y., Ozaki C., Takenawa T. A novel phospholipase C delta4 (PLCdelta4) splice variant as a negative regulator of PLC. J. Biol. Chem. 1999;274:2872–2879. doi: 10.1074/jbc.274.5.2872. [DOI] [PubMed] [Google Scholar]
- 39.Ruest L.B., Xiang X., Lim K.C., Levi G., Clouthier D.E. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423. doi: 10.1242/dev.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozeki H., Kurihara Y., Tonami K., Watatani S., Kurihara H. Endothelin-1 regulates the dorsoventral branchial arch patterning in mice. Mech. Dev. 2004;121:387–395. doi: 10.1016/j.mod.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Yanagisawa H., Yanagisawa M., Kapur R.P., Richardson J.A., Williams S.C., Clouthier D.E., de Wit D., Emoto N., Hammer R.E. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 42.Kurihara Y., Kurihara H., Suzuki H., Kodama T., Maemura K., Nagai R., Oda H., Kuwaki T., Cao W.H., Kamada N. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- 43.Clouthier D.E., Hosoda K., Richardson J.A., Williams S.C., Yanagisawa H., Kuwaki T., Kumada M., Hammer R.E., Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- 44.Barron F., Woods C., Kuhn K., Bishop J., Howard M.J., Clouthier D.E. Downregulation of Dlx5 and Dlx6 expression by Hand2 is essential for initiation of tongue morphogenesis. Development. 2011;138:2249–2259. doi: 10.1242/dev.056929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinissen M.J., Gutkind J.S. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.