

Abstract
Psoriasis is a common inflammatory disorder of the skin and other organs. We have determined that mutations in CARD14, encoding a nuclear factor of kappa light chain enhancer in B cells (NF-kB) activator within skin epidermis, account for PSORS2. Here, we describe fifteen additional rare missense variants in CARD14, their distribution in seven psoriasis cohorts (>6,000 cases and >4,000 controls), and their effects on NF-kB activation and the transcriptome of keratinocytes. There were more CARD14 rare variants in cases than in controls (burden test p value = 0.0015). Some variants were only seen in a single case, and these included putative pathogenic mutations (c.424G>A [p.Glu142Lys] and c.425A>G [p.Glu142Gly]) and the generalized-pustular-psoriasis mutation, c.413A>C (p.Glu138Ala); these three mutations lie within the coiled-coil domain of CARD14. The c.349G>A (p.Gly117Ser) familial-psoriasis mutation was present at a frequency of 0.0005 in cases of European ancestry. CARD14 variants led to a range of NF-kB activities; in particular, putative pathogenic variants led to levels >2.5× higher than did wild-type CARD14. Two variants (c.511C>A [p.His171Asn] and c.536G>A [p.Arg179His]) required stimulation with tumor necrosis factor alpha (TNF-α) to achieve significant increases in NF-kB levels. Transcriptome profiling of wild-type and variant CARD14 transfectants in keratinocytes differentiated probably pathogenic mutations from neutral variants such as polymorphisms. Over 20 CARD14 polymorphisms were also genotyped, and meta-analysis revealed an association between psoriasis and rs11652075 (c.2458C>T [p.Arg820Trp]; p value = 2.1 × 10−6). In the two largest psoriasis cohorts, evidence for association increased when rs11652075 was conditioned on HLA-Cw∗0602 (PSORS1). These studies contribute to our understanding of the genetic basis of psoriasis and illustrate the challenges faced in identifying pathogenic variants in common disease.
Introduction
Psoriasis is a chronic, inflammatory disease of the skin and other organs. It affects approximately 2% of individuals of European descent,1 and in up to 30% of cases, it is associated with chronic inflammatory psoriatic arthritis.2 Genome-wide association studies (GWASs) have identified over 20 susceptibility loci for psoriasis.3–11 However, with the exception of psoriasis susceptibility locus 1 (PSORS1 [MIM 177900]), for which the odds ratio (OR) is approximately 3.0,12,13 risk conferred by these loci is generally small (ORs ≤ 1.5). Moreover, less than 20% of disease variance has been explained.14,15 This implies that additional low-risk loci, genetic interactions, or rare variants of large effect account for the remaining variance.
In our accompanying paper, we identified rare, gain-of-function mutations in caspase recruitment domain family, member 14 (CARD14 [MIM 607211])16 in two large multiplex families affected by Mendelian forms of psoriasis and psoriatic arthritis (see the accompanying paper17 in this issue of AJHG). We also identified a de novo mutation in CARD14 in a child with early-onset, severe pustular psoriasis (PSORP [MIM 614204]). These mutations are responsible for the elusive psoriasis susceptibility locus 2 (PSORS2 [MIM 602723]) in chromosomal region 17q25. These results led us to hypothesize that additional rare and common variants in CARD14 might contribute to psoriasis and/or psoriatic arthritis in the general population.
Here, we identify and characterize 15 additional rare missense variants within CARD14 and determine their frequencies in a large cohort of approximately 6,000 psoriasis cases and 4,000 controls. Statistical analyses revealed an excess of rare variants in psoriasis cases relative to controls. The potential pathogenicity of variants was demonstrated by their ability to increase transcriptional activation by nuclear factor of kappa light chain enhancer in B cells (NF-kB) and to enhance production of a subset of psoriasis-associated transcripts. A common missense variant within CARD14 was also associated with psoriasis, and that evidence for association increased when this locus was conditioned on the presence of PSORS1. Our findings indicate that a range of NF-kB responses in the skin are mediated by CARD14 and that a subset of rare CARD14 variants leads to psoriasis and psoriatic arthritis.
Subjects and Methods
Subjects
Cases and controls for sequencing and genotyping were recruited from multiple institutions. Samples were organized into cohorts as shown in Table 1. There were six cohorts of European ancestry and one of Asian ancestry. Referring to Table 1, European cases in cohort A were recruited from either Washington University in St. Louis or the Department of Dermatology at the University of California, San Francisco (UCSF). Controls in cohort A were unaffected individuals who were over 20 years of age and who had no family history of psoriasis; they were recruited from the Texas Scottish Rite Hospital for Children or from the Cardiovascular Research Institute and Center for Human Genetics at the University of California, San Francisco or they were CEU (Utah residents with Northern and Western European ancestry from the CEPH collection) grandparents. Cohort B samples were from the National Psoriasis Foundation Victor Henschel Tissue Repository (NPF). Cases and controls in cohort C were recruited from the Department of Dermatology at the University of Utah. For cohorts A–C, psoriasis was diagnosed by a dermatologist.
Table 1.
Psoriasis Cases and Controls Included in this Study
| Cohort | Cohort Description | Cases | Controls |
|---|---|---|---|
| A | St. Louis/Dallas/UCSF | 676 | 570 |
| B | NPF (European) | 486 | 154 |
| C | Utah | 931 | 236 |
| D | Michigan | 2,768 | 2,749 |
| E | Toronto | 981 | 483 |
| F | Newfoundland | 340 | 379 |
| G | Asians | 194 | 193 |
| H | Combined European (rows A–F) | 6,182 | 4,571 |
| I | NPF (all samples) | 976 | 758 |
Individuals were recruited from multiple institutions and organized into cohorts, as shown in this table. Cohorts A–F are independent case/control cohorts of Northern European ancestry. Cohort G includes samples of Asian ancestry from the St. Louis/Dallas/UCSF (University of California, San Francisco) and the NPF (National Psoriasis Foundation Victor Henschel Tissue Repository) cohorts (cohorts A and B, respectively). Cohorts A–G include samples of known ancestry. Cohort H lists all cases and controls contributed by the NPF, including those of unknown ancestry. Demographic data were available only for approximately half of the NPF samples.
Cohort D samples were recruited from the Department of Dermatology at the University of Michigan. Cases had at least two psoriatic plaques or a single plaque occupying at least 1% of the total body surface outside the scalp. Individuals presenting with only palmoplantar psoriasis, inverse psoriasis, or sebopsoriasis were excluded. Controls were at least 18 years of age and had no personal or family history of psoriasis.
Cohort E samples were gathered from the University of Toronto and Toronto Western Hospital. Cohort F samples were gathered from the Department of Medicine, Division of Rheumatology, Memorial University of Newfoundland. Psoriasis was diagnosed by a dermatologist. When psoriatic arthritis was suspected, cases were evaluated according to clinical history and rheumatologic and radiologic evaluation. Control individuals showed no evidence of psoriasis, psoriatic arthritis, or any other autoimmune disease.
Asian samples in Cohort G were recruited from the Cardiovascular Research Institute and Center for Human Genetics at the University of California, San Francisco, from the University of Toronto and Toronto Western Hospital and the Department of Medicine, or from the NPF.
DNA was isolated from whole blood or saliva by standard methods. Protocols were approved by local institutional review boards. All subjects or their parents (if the subjects were minors) provided informed consent.
Sanger Resequencing and Genotyping
As a first pass, all coding exons of CARD14 (full-length, CARD14fl) were resequenced in 192 psoriasis cases and 96 controls of European ancestry. Exons in which rare missense mutations were identified were resequenced in 95 more controls of European ancestry. Exon 4 of CARD14 was resequenced in an additional 1,856 cases and 882 controls of European ancestry. Primers are available upon request. Samples were genotyped with the Sequenom MassARRAY at Washington University and by TaqMan at the University of Michigan (Table S2, available online). All samples from the NPF were genotyped, but only samples of known ethnicity were included in statistical analyses.
Expression Plasmids
Full-length CARD14sh (GenBank BC018142, coding for 740 amino acids) and CARD14cl (RefSeq NM_052819, coding for 434 amino acids) were cloned into pReceiver-M11 (Capital Biosciences). The CARD14sh construct was subjected to in vitro mutagenesis with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The numbering of all CARD14 mutations in this manuscript is based on RefSeq NM_024110.3. For rare, nonannotated missense variants, constructs were generated with the mutant allele. For polymorphisms (rare and common), constructs were generated as follows (the allele and amino acid used are listed in parentheses): rs115582620 (c.185A [p.Gln185]), p.Ser200Asn (c.599A [p.Asn200]), rs146214639 (c.449G [p.Arg150]), rs144475004 (c.526C [p.His176]), rs2066964 (c.930C [p.Ser547]), rs34367357 (c.1042A [p.Ile585]), rs117918077 (c.2044T [p.Trp682]), and rs151150961 (c.2140A [p.Ser714]). Full-length CARD14fl was not available for subcloning. As a result, constructs could not be created for rs144285237 (c.2919C>G [p.Asp973Gln]) or rs11652075 (c.2458C>T [p.Arg820Trp]).
NF-kB Luciferase Reporter Assay
The NF-kB luciferase reporter assay was performed with the pNFkB-luc system (Clontech) as described in our accompanying manuscript.17 The CARD14cl clone was used as a negative control in this assay because it lacks the sequence encoding the CARD domain, which is necessary for CARD14-induced NF-kB activation.
Expression Profiling and qRT-PCR
HEK 001 cells (human-papillomavirus-16-transformed keratinocytes) were transfected with wild-type CARD14sh or expression constructs encoding CARD14sh substitutions. Cells were cultured for 24 hr, and then RNA was isolated with the miRNeasy kit (QIAGEN). Global expression profiling of RNA from cells was performed with the HumanHT-12 v4 Expression BeadChip (Illumina). Experiments were conducted in compliance with MIAME (minimum information about a microarray experiment) guidelines. Raw and normalized expression data are deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) as series GSE36381. 100 ng to 1 μg of randomly primed total RNA was used for quantitative RT-PCR (qRT-PCR) according to standard procedures. Expression levels were normalized to 18S rRNA. Relative expression levels were calculated as follows: 2 × 106((Ct 18S)−(Ct CARD14)). We also normalized expression levels in transfected cells to levels of FLAG to correct for differences in transfection efficiency.
Clustering of Variants on the Basis of Expression Levels in Keratinocytes
On the basis of the NF-kB reporter assays, a subset of CARD14 variants were classified as (1) leading to enhanced basal NF-kB activation when they were compared to the effects of wild-type CARD14sh (these are c.349G>A [p.Gly117Ser], c.413A>C [p.Glu138Ala], c.424G>A [p.Glu142Lys], and c.425A>G [p.Glu142Gly]), (2) leading to downregulation of NF-kB activation (c.112C>T [p.Arg38Cys]), or (3) having no effect on NF-kB activation (these are c.185G>A [p.Arg62Gln] [rs115582620], c.930G>C [p.Arg547Ser] [rs2066964], c.449T>G [p.Leu150Arg] [rs146214639], c.854A>G [p.Asp285Gly], c.1778T>A [p.Ile593Asn], c.2044C>T [p.Arg682Trp] [rs117918077], and c.2140G>A [p.Gly714Ser] [rs151150961]). After performing mRNA transcriptome analysis on cells that were transfected with either wild-type CARD14sh or CARD14 variants, we found that 1,531 transcripts had at least a 2-fold change (up or down) in expression as well as a significant p value in transfectants with the CARD14 variants causing enhanced NF-kB activation relative to transfectants with wild-type CARD14sh. The variants were then clustered with the use of the reduced probe set and the R (v2.10.1) randomForest package (v4.6-2). Random forests were generated with 500 trees and unweighted classes, and importance was calculated for a total of 10,000,000 forests. The probes were then ranked from highest to lowest mean decrease in Gini. A heat map of the clustering of the full set of variants was generated with the top 30 probes.
Pathway Analysis of the “CARD14 Pathogenic Keratinocyte Signature”
A list was generated of genes differentially expressed as a consequence of the introduction of CARD14 psoriasis-specific alterations into HEK 001 cells as described above. We obtained this list, termed the “CARD14 pathogenic keratinocyte signature,” by comparing the global transcriptomes of keratinocyte transfectants with overtly pathogenic CARD14 substitutions (p.Gly117Ser, p.Glu138Ala, p.Gly142Lys, and p.Glu142Gly) to those with nonpathogenic substitutions (p.Leu150Arg [rs146214639], p.Val191Leu, p.Asp285Gly, and wild-type CARD14sh). Expression data from each sample were quantile normalized, log2 transformed, and fitted to linear models in R (R v2.13.1; Biobase v2.12.2; BeadArray v2.2.0; and limma v3.8.3). The contrast was defined as “pathogenic versus nonpathogenic,” and the t tests, fold changes, and false-discovery-rate-corrected p values were calculated with ImFit and eBayes. Given the overexpression of the constructs, larger sample sizes would be required for the detection of significant group-wise effects. However, by taking the genes with a nominal group-wise p value of 0.05 and ranking them by fold change, we generated a list of the top 200 upregulated and top 200 downregulated genes. This list was analyzed with Ingenuity pathway analysis (IPA).
Statistical Analysis
Analysis of variants was performed with PLINK.18 The variable threshold test19 and a straightforward burden test for association of rare variants with disease were performed with PLINK/SEQ version 0.05. For both tests, we included CARD14 variants with minor allele frequencies of less than or equal to 0.01 among our controls of northern European ancestry. This included the following 16 variants: c.112C>T (p.Arg38Cys), c.185G>A (p.Arg62Gln) (rs115582620), c.349G>A (p.Gly117Ser) (altered in family PS117), c.413A>C (p.Glu138Ala) (altered in generalized pustular psoriasis17), c.424G>A (p.Glu142Lys), c.425A>G (p.Glu142Gly), c.449T>G (p.Leu150Arg) (rs146214639), c.511C>A (p.His171Asn), c.526G>C (p.Asp176His) (rs144475004), c.536G>A (p.Arg179His), c.571G>T (p.Val191Leu), c.599G>A (p.Ser200Asn), c.854A>G (p.Asp285Gly), c.1778T>A (p.Ile593Asn), c.2140G>A (p.Gly714Ser) (rs151150961), and c.2919C>G (p.Asp973Gly) (rs144285237). Meta-analysis of polymorphisms and generation of forest plots were performed with R version 2.12.220 with the rmeta package.
Results
Rare-Variant Screening
In our accompanying manuscript, we identified rare gain-of-function CARD14 mutations that lead to psoriasis.17 These included the familial c.349G>A (p.Gly117Ser) mutation in family PS1, affected by multiple cases of psoriasis and psoriatic arthritis,21 and the de novo c.413A>C (p.Glu138Ala) germline mutation in a case of childhood generalized pustular psoriasis. To determine whether there were additional rare variants predisposing to psoriasis in CARD14, we resequenced all coding exons of CARD14 (full-length, CARD14fl) in over 192 psoriasis cases and 96 controls (see Subjects and Methods). This revealed ten rare missense mutations in CARD14 (Figure 1, Table 2, and Table S1): c.112C>T (p.Arg38Cys), c.185G>A (p.Arg62Gln) (rs115582620), c.425A>G (p.Glu142Gly), c.449T>G (p.Leu150Arg) (rs146214639), c.599G>A (p.Ser200Asn), c.854A>G (p.Asp285Gly), c.1778T>A (p.Ile593Asn), c.2044C>T (p.Arg682Trp) (rs117918077), c.2140G>A (p.Gly714Ser) (rs151150961), and c.2919C>G (p.Asp973Gln) (rs144285237). On the basis of the observation that several rare mutations were clustered in exon 4, which encodes part of the critical coiled-coil domain, we reasoned that it might be a mutation hotspot and resequenced an additional 1,800 cases and 900 controls for that exon. This revealed five additional rare variants within CARD14: c.424G>A (p.Glu142Lys), c.511C>A (p.His171Asn), c.526G>C (p.Asp176His) (rs144475004), c.536G>A (p.Arg179His), and c.571G>T (p.Val191Leu) (Figure 1, Table 2, and Table S1). A search in dbSNP135 revealed that 8 of the 15 identified rare variants have not been previously annotated; these eight are c.112C>T (p.Arg38Cys), c.424G>A (p.Glu142Lys), c.425A>G (p.Glu142Gly), c.511C>A (p.His171Asn), c.536G>A (p.Arg179His), c.571G>T (p.Val191Leu), c.599G>A (p.Ser200Asn), c.854A>G (p.Asp285Gly), and c.1778T>A (p.Ile593Asn). Furthermore, this search revealed that the c.349G>A (p.Gly117Ser) and c.413A>C (p.Glu138Ala) mutations have also not been previously annotated.
Figure 1.
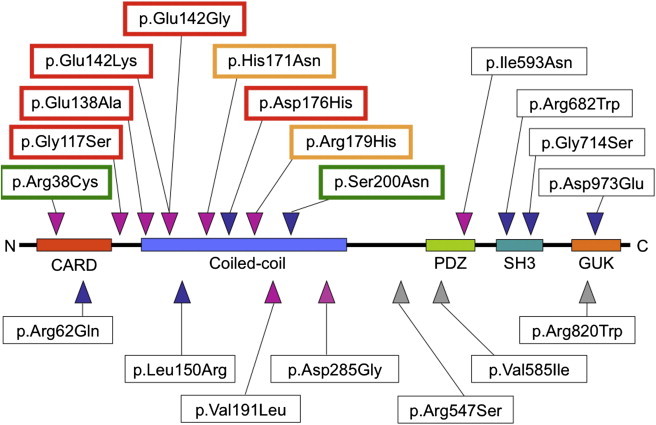
CARD14 Protein Domains and Locations of Amino Acid Substitutions
Missense variants identified in CARD14 by resequencing are shown relative to key protein domains. Red outlining indicates increased NF-kB activation by a variant (relative to wild-type); green outlining indicates reduced activation. Gold outlining indicates variants that showed increased NF-kB activation only in response to TNF-α stimulation. SNPs and variants also identified in dbSNP135, which includes data from the 1,000 Genomes Project and the National Heart, Lung, and Blood Institute (NHLBI) and National Human Genome Research Institute (NHGRI) Exome Project, are indicated with blue triangles. A polymorphism, rs114688446, was observed at the same site as p.Ser200Asn, but a different substitution, p.Ser200Ile, was detected. SNPs used for meta-analysis are indicated with gray triangles.
Table 2.
Characteristics and Frequencies of CARD14 Coding Variants
| CARD14 Exon | cDNA Mutation and Corresponding Protein Change | Protein Domain | PolyPhen227-Predicted Effect on Protein Function | Effect on NF-kB Activation (FC versus Wild-Type CARD14sh) | Allele Frequency in Cases (Number Sampled) | Allele Frequency in Controls (Number Sampled) |
|---|---|---|---|---|---|---|
| 2 | c.112C>T (p.Arg38Cys) | CARD | probably damaging | 0.11 | 0.00019 (2,691) | 0 (1,271) |
| 2 | c.185G>A (p.Arg62Gln) (rs115582620) | CARD | benign | 1.06 | 0.0014 (3,284) | 0.00084 (1,797) |
| 3 | c.349G>A (p.Gly117Ser) | none | possibly damaging | 3.71 | 0.00023 (6,630) | 0 (4,731) |
| 3 | c.349+5G>A | none | NA | ND | 0 (2,871) | 0 (1,339) |
| 4 | c.413A>C (p.Glu138Ala) | coiled-coil | probably damaging | 8.95 | 0.00015 (3,488) | 0 (1,902) |
| 4 | c.424G>A (p.Glu142Lys) | coiled-coil | probably damaging | 4.03 | 0.00012 (4,107) | 0 (1,874) |
| 4 | c.425A>G (p.Glu142Gly) | coiled-coil | probably damaging | 5.00 | 0.00019 (2,848) | 0 (1,451) |
| 4 | c.449T>G (p.Leu150Arg) (rs146214639) | coiled-coil | probably damaging | 1.79 | 0.0025 (6,140) | 0.0016 (4,614) |
| 4 | c.511C>A (p.His171Asn) | coiled-coil | benign | 0.68 (5.95 with TNF-α stimulation) | 0.00025 (4,077) | 0 (1,858) |
| 4 | c.526G>C (p.Asp176His) (rs144475004) | coiled-coil | probably damaging | 2.78 | 0.00056 (3,575) | 0.00062 (1,609) |
| 4 | c.536G>A (p.Arg179His) | coiled-coil | probably damaging | 1.38 (2.19 with TNF-α stimulation) | 0.00025 (4,061) | 0.00027 (1,848) |
| 4 | c.571G>T (p.Val191Leu) | coiled-coil | benign | 1.02 | 0.00014 (3,575) | 0 (1,613) |
| 4 | c.599G>A (p.Ser200Asn) | coiled-coila | benign | 0.67 | 0.011 (6,163) | 0.0084 (4,624) |
| 6 | c.854A>G (p.Asp285Gly) | none | possibly damaging | 1.14 | 0.00019 (2,673) | 0 (1,467) |
| 13 | c.1778T>A (p.Ile593Asn) | PDZ | probably damaging | 1.30 | 0.00024 (2,049) | 0.00048 (1,039) |
| 15 | c.2044C>T (p.Arg682Trp) (rs117918077) | SH3 | probably damaging | 0.95 | 0.013 (2,169) | 0.012 (1,042) |
| 15 | c.2140G>A (p.Gly714Ser) (rs151150961) | SH3 | benign | 1.02 | 0.0021 (2,105) | 0.0014 (1,038) |
| 21 | c.2919C>G (p.Asp973Glu) (rs144285237) | GUK | benign | NDb | 0.0024 (5,177) | 0.0015 (4,099) |
CARD14 missense variants are listed with details on their locations in critical CARD14 protein domains, their predicted effect on protein function from PolyPhen2,27 their effect on NF-kB activation (fold change compared to unstimulated wild-type CARD14sh; see also Figure 3), and frequencies in unrelated cases and controls of European ancestry. The number of individuals screened is in parenthesis. The following abbreviations are used: FC, fold change; and ND, not done.
The p.Ser200 residue lies within a 6 bp sequence separating two predicted coiled-coil regions and thus could be considered part of an overarching coiled-coil domain. Also, variant rs114688446 was identified at this location in dbSNP, but the amino acid change (p.Ser200Ile) was different.
The impact of p.Asp973Glu on NF-kB activation could not be tested because it is exclusive to CARD14fl, for which a full-length cDNA clone was unavailable. Additional data on these variants are presented in Table S1.
Population-Based Frequency Estimates of Rare Variants
We determined the frequencies of all rare CARD14 variants, including those encoding the familial p.Gly117Ser alteration (causing psoriasis and/or psoriatic arthritis) and the p.Glu138Ala alteration (causing generalized pustular psoriasis), by high-throughput genotyping seven independent case/control cohorts (>10,000 individuals; Tables 1 and 2 and Tables S1 and S2). This revealed two other unrelated psoriasis cases with the CARD14 c.349G>A (p.Gly117Ser) mutation; one woman was from the NPF repository and was diagnosed with psoriasis at 10 years old and psoriatic arthritis at 20 years old, and another woman was from Utah and had a history of psoriasis, which was diagnosed at 65 years old. This latter woman transmitted the mutation to her daughter, who developed psoriasis at age 32. This mutation was also detected in a male NPF control for whom additional data (e.g., ethnicity, age, and family history of psoriasis) were not available. None of these individuals harbored the SLC26A1122 (solute carrier family 26, member 11, [SLC26A11 (MIM 610117)]) c.365A>G (p.Tyr122Cys) mutation, which cosegregated with c.349G>A (p.Gly117Ser) in family PS1,17 providing evidence that these variants can arise independently. Numbering of the SLC26A11 mutation is based on RefSeq NM_001166347.1. In total, the frequency of the CARD14 c.349G>A (p.Gly117Ser) mutation in cases of European ancestry was 0.0005. The CARD14 c.349+5G>A mutation, which segregated with psoriasis in a large multiply affected Taiwanese family,17 was not detected in any other cases or controls. However, given the relatively small number of Asian samples screened, we were not well powered to detect variants at low frequencies in that population. The mutation encoding p.Glu138Ala seen in the single pustular psoriasis case was also not seen in any other cases or controls.
Five rare CARD14 variants (encoding p.Arg38Cys, p.Glu142Gly, p.Glu142Lys, p.Val191Leu, and p.Asp285Gly; Figure 1 and Tables S1 and S2) were seen in only one case and in no controls. The variant encoding p.His171Asn was only seen in two psoriasis- and psoriatic-arthritis-affected cases from Newfoundland and in no controls. We performed a simple burden test and a variable threshold test19 to compare the distribution of rare variants in cases and controls. These tests provided evidence of an excess of rare CARD14 variants in cases versus controls (burden test p value = 0.0015; variable threshold test p value = 0.0053).
Common-Variant Association Tests
Resequencing validated several common missense polymorphisms described in dbSNP: rs2066964, rs34367357, and rs11652075. We genotyped these and 20 other previously described SNPs in our seven psoriasis case/control cohorts (Table S2) and looked for association with psoriasis. Most variants were present at similar frequencies in cases and controls. However, three missense SNPs (rs2066964, rs34367357, and rs11562075) within CARD14 and rare variant c.599G>A (p.Ser200Asn) provided evidence of association with psoriasis in some of the cohorts (Table S2). A meta-analysis of these four missense variants was performed for the six cohorts of European ancestry (Figure 2 and Figure S1). This revealed further evidence of association between psoriasis and rs11652075 (c.2458C>T [p.Arg820Trp]) (fixed effects p value = 2.1 × 10−6, OR = 0.87 [0.83–0.92]; random effects p value = 0.031, OR = 0.86 [0.75–0.99]); c.2458C was the risk allele (Figures 2A and 2B). This SNP, as well as the same risk allele, was also associated with psoriasis in Asians (p value = 0.0029, OR = 0.64 [0.48–0.86]). The meta-analysis also revealed evidence of association with p.Ser200Asn (fixed and random effects p values = 0.05, OR = 1 [1.3–1.82]); the rare c.599A allele increased psoriasis risk (Figure 2C). However, this would not be significant if it were adjusted for multiple testing.
Figure 2.
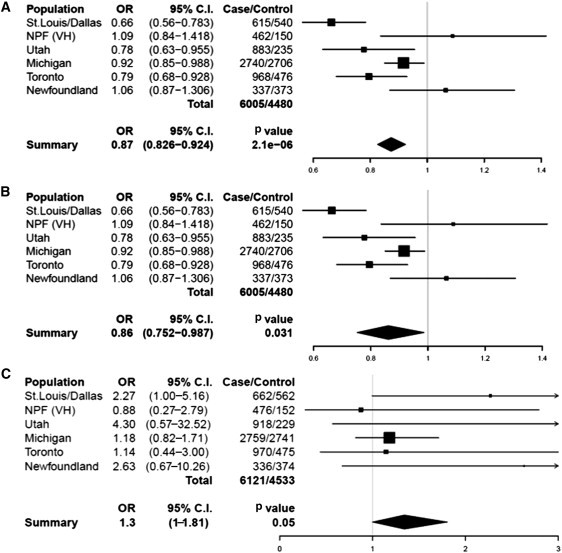
Meta-Analyses of rs11652075 and p.Ser200Asn across Six Case/Control Cohorts of European Ancestry
The results of the fixed- (A) and random-effects (B) meta-analyses for rs11652075 (c.2458C>T [p.Arg820Trp]) and the fixed-effects meta-analysis for p.Ser200Asn (c.599G>A) (C) are shown. The random-effects meta-analysis for p.Ser200Asn was identical to that of the fixed-effects meta-analysis and is therefore not pictured. Forest plots indicate the direction of effect, relative weight, and confidence interval for the odds ratio of this SNP in each cohort. The number of cases and controls successfully genotyped in each cohort is shown, and the meta-analysis OR and p value are listed below each plot. The following abbreviations are used: OR, odds ratio; and C.I., confidence interval.
Because of the large effect of HLA-Cw∗0602 from the major histocompatibility complex (MHC) class I region (PSORS1),12,13 we investigated its connection with the four CARD14 variants described above. In the Michigan and Utah psoriasis cohorts, rs11652075 was found to have a higher association with psoriasis when it was conditioned on HLA-Cw∗0602 (p value of rs11652075 alone = 0.023 (Michigan), 0.017 (Utah); stratified on HLA-Cw∗0602, p value = 0.0021 (Michigan), 0.0086 (Utah); Table S3). No such evidence was observed with the other variants.
Effect of Variants on CARD14 Function In Vitro
CARD14 encodes a 1,004 amino acid protein that activates NF-kB.16 In our companion paper, we observed that compared to wild-type CARD14, the familial p.Gly117Ser and de novo p.Glu138Ala substitutions lead to enhanced NF-kB activation (3.71- and 8.95-fold enhancement, respectively). To test the effect of rare variants described here on this activity, we again used an NF-kB luciferase reporter assay. Several rare variants (p.Glu142Lys, p.Glu142Gly, and p.Asp176His [rs144475004]) in the coiled-coiled domain increased NF-kB activation two to five times more than did wild-type CARD14sh (Table 2, Figure 3A). The p.Arg38Cys, p.His171Asn, and p.Ser200Asn substitutions led to less NF-kB activation than did CARD14sh, but compared to wild-type CARD14sh, other rare variants and the CARD14 missense polymorphisms (p.Arg62Gln [rs115582620], p.Arg547Ser [rs2066964], p.Arg682Trp [rs117918077]) did not significantly alter NF-kB activation levels (Table 2, Figure 3A). As discussed below, variants that increased NF-kB activation at least 2.5× more than that seen with CARD14sh also induced greater upregulation of psoriasis-associated genes. Relative to wild-type CARD14sh, variants that resulted in a more modest increase in NF-kB activation, those that reduced NF-kB activation, and those that did not change it did not induce upregulation of those genes to the same degree. Therefore, when compared with the level of NF-kB activation caused by wild-type CARD14, a 2.5× or greater increase in NF-kB activation is predictive of putative pathogenic CARD14 amino acid substitutions.
Figure 3.
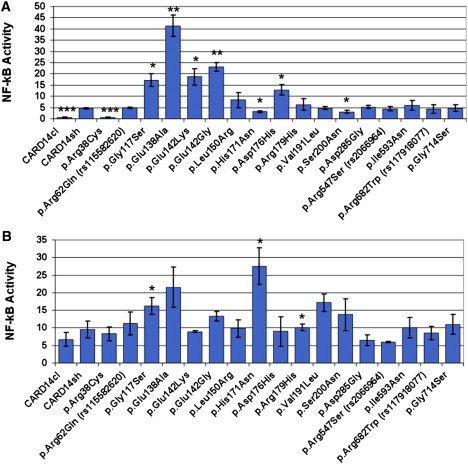
Effect of Wild-Type and Altered CARD14 on NF-kB Activation
HEK 293 cells were transfected with the construct that codes for CARD14sh, the same construct harboring one of the rare variants shown, or a construct that codes for CARD14cl and lacks the CARD domain. NF-kB activity was determined by measuring relative luciferase activity. Transfection efficiency was controlled for by first normalizing all values to Renilla expression, and activity of the empty background vector, pTAL-luc, was controlled for by adjusting the values. Change in NF-kB activity relative to the background vector was determined for each variant (y axis, NF-kB activity). Every data point represents the average value of three replicates. Error bars represent the standard deviation of replicates. For experiments involving TNF-α stimulation, treated cells were exposed to 20 ng/ml TNF-α in culture media for 24 hr. Asterisks show results from two-tailed, unpaired student's t tests comparing NF-kB activation induced by the indicated variant to either that of unstimulated cells with CARD14sh (A) or that of TNF-α-stimulated cells with CARD14sh (B). ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
Onset of psoriatic lesions is thought to be triggered by an inflammatory stimulus. We therefore examined the effects of wild-type and variant CARD14 on NF-kB activation after stimulation with tumor necrosis factor alpha (TNF-α). Compared with unstimulated CARD14sh, TNF-α-stimulated p.His171Asn and p.Arg179His resulted in a 5.95- and a 2.95-fold increase in NF-kB activation, respectively; compared with TNF-α-stimulated CARD14sh, TNF-α-stimulated p.His171Asn and p.Arg179His resulted in a 2.87- and a 1.06-fold increase in NF-kB activation, respectively (Table 2 and Figure 3B, discussed further below).
Effect of CARD14 Substitutions on Keratinocyte Gene Expression
We have shown that CARD14 is localized in keratinocytes and that the familial and pustular-psoriasis variants (p.Gly117Ser and p.Glu138Ala, respectively) lead to enhanced production of some chemokines and other transcripts that are upregulated in psoriatic skin.17 To evaluate the effect of the additional rare variants on transcription in keratinocytes, we transfected all altered constructs into the keratinocyte cell line HEK 001. The transcriptome of each transfectant was then evaluated after 24 hr by interrogation with Illumina bead arrays. A heat map with 30 probes (see Subjects and Methods, random forest classification) revealed clustering of the p.Glu142Lys and p.Glu142Gly transfectants with those with the p.Gly117Ser and p.Glu138Ala alterations (Figure 4). The p.Glu138Ala substitution clustered at the extreme end of other pathogenic variants and on its own branch of the tree. This might be expected given the severity of the disease in the child (who has generalized pustular psoriasis) in whom it was found.
Figure 4.
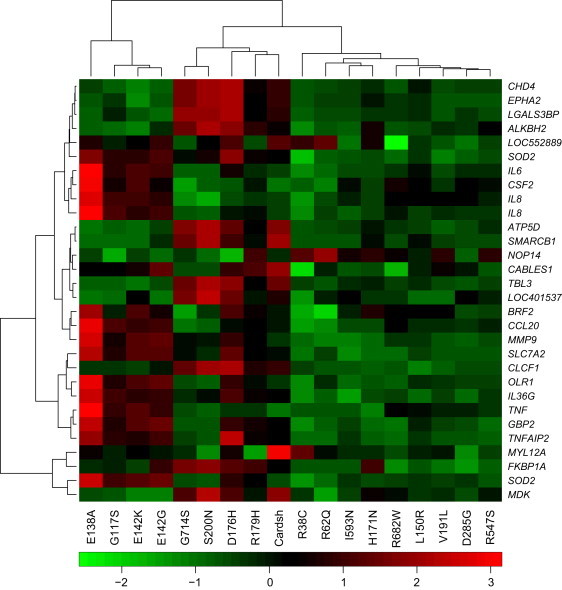
Heat Map Generated after Transcriptome Analysis of Wild-Type and Mutant CARD14 Transfectants
Cells were transfected with CARD14cl, wild-type CARD14sh, or variant CARD14 expression constructs. After 24 hr, RNA was extracted and used for the interrogation of Illumina bead arrays. A subset of differentially expressed genes was used for clustering of CARD14 variants (see Subjects and Methods).
Within this heat map, the following 13 genes were upregulated in keratinocytes transfected with pathogenic substitutions: superoxide dismutase 2, mitochondrial (SOD2 [MIM 147460]); interleukin 6 (interferon, beta 2 [IL6 (MIM 147620)]); colony stimulating factor 2 (granulocyte-macrophage [CSF2 (MIM 138960)]); interleukin 8 (IL8 [MIM 146930]); matrix metallopeptidase 9 (gelatinase B, 92kDa gelatinase, 92kDa type IV collagenase [MMP9 (MIM 120361)]); BRF2, subunit of RNA polymerase III transcription initiation factor, BRF1-like (BRF2 [MIM 607013]); chemokine (C-C motif) ligand 20 (CCL20 [MIM 601960]); solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 (SLC7A2 [MIM 601872]); oxidized low density lipoprotein (lectin-like) receptor 1 (OLR1 [MIM 602601]); interleukin 36, gamma (IL36G [MIM 605542]); guanylate binding protein 2, interferon-inducible (GBP2 [MIM 600412]); tumor necrosis factor, alpha-induced protein 2 (TNFAIP2 [MIM 603300]); and tumor necrosis factor (TNF [MIM 191160]). We used g:Profiler23 to look for specific functions of these genes. The most significant Gene Ontology24 terms associated with this group of genes included: immune-system process/development, hematopoietic- or lymphoid-organ development, and response to lipopolysaccharide/bacterium/molecule of bacterial origin. IL6, MMP9, and BRF2 have also been implicated in cell migration and could mediate immune cell infiltration into the skin. CSF2, IL8, and SLC7A2 have been specifically implicated in myeloid leukocyte activation. Psoriasis is a disease of keratinocyte and immune cell proliferation, and genes involved in cell proliferation include SOD2, IL6, CSF2, and IL8. Other terms included response to wounding (SOD2, IL6, IL8, CCL20, SLC7A2, and OLR1), nitric oxide biosynthetic process (SOD2, IL6, and SLC7A2), and antiapoptosis (SOD2, IL6, and CSF2). IL6 and CSF2 are implicated in the regulation of the JAK-STAT pathway, and IL6, IL8, and MMP9 are implicated in angiogenesis. Genes involved in the response to biotic stimuli include SOD2, IL6, IL8, MMP9, and CCL20. Psoriasis increases risk of cardiovascular disease,25,26 and five of these genes have been implicated in the development of the cardiovascular system (SOD2, IL6, IL6, MMP9, and TNFAIP2). Upregulation of these thirteen genes thus constitutes a pathogenic psoriatic signature.
Two variants clustered together: c.526G>C (p.Asp176His) (rs144475004) and c.536G>A (p.Arg179His). Both amino acid substitutions were predicted to be damaging by PolyPhen2,27 and both lead to upregulation of NF-kB activation (2.78- and 1.38-fold increases, respectively). In the case of p.Arg179His, TNF-α was required for NF-kB activation to achieve pathogenic levels; compared with unstimulated CARD14sh and TNF-α-stimulated CARD14sh, TNF-α-stimulated p.Arg179His produced a 2.95- and a 1.06-fold increase, respectively. Both variants were seen in a small number of cases and controls (4:2 and 2:1, respectively).
Three other constructs clustered in the same branch with p.Asp176His (rs144475004) and p.Arg179His. These included wild-type CARD14sh, c.2140G>A (p.Gly714Ser) (rs151150961), and c.599G>A (p.Ser200Asn). However, the latter three did not induce expression of the pathogenic psoriatic signature described above to the same degree as the overtly pathogenic variants. One noteworthy example of this is that the IL8 expression produced by these three constructs was much lower than that seen with the pathogenic variants. Moreover, clustering of these constructs was due to other transcripts that are not considered part of the pathogenic signature. Other variants exhibited reduced levels of genes in this psoriasis signature, even when compared to wild-type CARD14sh.
We confirmed altered expression of all thirteen transcripts (SOD2, IL6, CSF2, BRF2, MMP9, IL8, CCL20, SLC7A2, OLR1, IL36G, GBP2, TNFAIP1, and TNF) by qRT-PCR (Figure S2).
We also performed a group-wise comparison of the global expression profiles of the overtly pathogenic substitutions (p.Gly117Ser, p.Glu138Ala, p.Glu142Lys, and p.Glu142Gly) and several nonpathogenic variants (p.Leu150Arg [rs146214639], p.Val191Leu, p.Asp285Gly, and wild-type CARD14sh). The top 200 upregulated and top 200 downregulated genes were identified (see Subjects and Methods), and pathway analysis was performed with IPA (Table S4). A number of cytokine signaling pathways were significant (including IL-17 signaling, IL-6 signaling, and TNFR2 signaling). Also significant were communication between innate and adaptive immune cells, dendritic cell maturation, mTOR signaling, notch signaling, and atherosclerosis signaling pathways. This latter pathway is interesting given the association between psoriasis and other systemic comorbidities, including cardiovascular disease.25,26
A comparison of these results with a published psoriasis transcriptome28 revealed that a number of these pathways are significantly represented in both groups. For example, the atherosclerosis signaling pathway, the NF-kB signaling pathway, and many of the cytokine signaling pathways were significant in both the published psoriasis transcriptome and the CARD14 pathogenic keratinocyte signature (Table S4). These results indicate that some of the pathways upregulated in keratinocytes in which CARD14 harbors a pathogenic substitution are also upregulated in classic psoriatic skin. This suggests that altered keratinocyte activation might significantly contribute to the transcriptome signatures in classic psoriasis lesions.
Discussion
Here, we describe a spectrum of rare and common variation within CARD14, an activator of NF-kB16 in skin epidermis, and we demonstrate enrichment of rare variants in cases by using two independent statistical tests. The burden test, which performs a straightforward comparison of the number, or “burden,” of rare variants in cases and controls, provided a p value of 0.0015. The variable threshold test,19 which compares rare variants subject to a variable allele-frequency threshold in cases and controls, gave a p value of 0.0053. We also demonstrate that pathogenic alterations were enriched in the coiled-coil domain of CARD14. This domain is predicted to be involved in the oligomerization of CARD14 with other proteins and the formation of its active conformation.29,30 Interestingly, although some of the rare variants we identified have been annotated in dbSNP135, none of the putative pathogenic alterations are annotated. A coding polymorphism in CARD14, rs11652075 (p.Arg820Trp), and c.599G>A (p.Ser200Asn) were also associated with psoriasis in several large cohorts. In the two largest psoriasis cohorts, evidence of association between psoriasis and rs11652075 increased when rs11652075 was conditioned on PSORS1.
Two rare variants, c.424G>A (p.Glu142Lys) and c.425A>G (p.Glu142Gly), were identified in cases but not in controls and manifested as overtly causing of disease. Compared with wild-type CARD14sh, they significantly enhanced NF-kB activation (4.03- and 5.00-fold enhancement, respectively), and they clustered with p.G117Ser and p.Glu138Ala after transfection into the keratinocyte line HEK 001 and global expression profiling. The c.424G>A (p.Glu142Lys) variant was identified in a Caucasian male who was diagnosed with psoriasis at 42 years of age and who responded well to treatment with UV light and a topical mixture of corticosteroid and a vitamin D analog. The c.425A>G (p.Glu142Gly) variant was found in a Caucasian male who was diagnosed with psoriasis in infancy and whose father also had psoriasis. He experienced a partial remission of psoriasis with methotrexate treatment. It is noteworthy that after these variants were stimulated with TNF-α, levels of NF-kB activation induced by these variants and the pustular-psoriasis substitution, p.Glu138Ala, decreased at the 24 hr mark. This suggests that at this time, downregulation of the NF-kB response might have been initiated in our cell-culture system, which merits further study. Both of these variants lie in the coiled-coil domain of CARD14, as does the de novo pustular-psoriasis substitution, p.Glu138Ala.
Compared with wild-type CARD14sh, a third variant, p.Asp176His (rs144475004), leads to enhanced NF-kB activation. However, its frequency was similar in cases and controls, and it didn't increase NF-kB activity in vitro as much as other variants did. Hence, it might lie below the NF-kB-activation threshold required for disease. Alternatively, disease might require a specific stimulus or interaction with a second genetic factor. Other variants such as p.Arg38Cys and p.Ser200Asn exhibited significantly less NF-kB activation than did wild-type CARD14sh. Previous studies have shown that decreased activation of NF-kB, much like increased activation, can induce inflammation and epidermal hyperplasia.31,32 It might be interesting to examine clinical features, such as inflammation after skin wounding, of individuals with these variants. However, it should be noted that the p.Arg38Cys and p.Ser200Asn substitutions did not induce expression of the pathogenic psoriasis signature when transfected into keratinocytes. Thus, although we cannot completely rule out a role for these variants in some aspects of disease, they are neither overtly pathogenic nor likely to be causative for initiation of psoriasis.
Two variants, p.His171Asn and p.Arg179His, required stimulation with TNF-α to achieve maximal levels of NF-kB activation. The p.Arg179His substitution was observed in two unrelated cases from Toronto and one control. The cases included a female who was diagnosed with psoriasis at 40 years of age and a male who was diagnosed with psoriasis at 64 years of age and who had a family history of psoriasis. The female responded well to oral and topical steroids, but the male was not treated.
The p.His171Asn alteration was seen in two unrelated psoriasis- and psoriatic-arthritis-affected individuals from Newfoundland and was not seen in controls. One individual was diagnosed with psoriasis at 40 years of age and psoriatic arthritis at 41 years of age and had a family history of psoriasis. The second individual was diagnosed with psoriasis at 55 years of age after being diagnosed with psoriatic arthritis at 53 years of age. The identification of the c.511C>A (p.His171Asn) variant in only the Newfoundland population suggests that it arose as a result of a founder effect in this population, but confirming this will require further studies. Inspection of the genotypes of 13 polymorphisms in a 75 kb region harboring CARD14 revealed a shared haplotype that is common in cases and controls from the Newfoundland cohort and our other case/control cohorts from the United States (e.g., St. Louis/Dallas/UCSF, Michigan, and Utah). Therefore, further evidence of a founder effect for the p.His171Asn substitution seen in these two cases was not possible.
The altered transcriptome signature with pathologic substitutions included upregulation of psoriasis-specific transcripts SOD2, IL6, CSF2, IL8, MMP9, BRF2, CCL20, SLC7A2, OLR1, IL36G, GBP2, TNFAIP2, and TNF. Expression of these molecules is expected to be an early event in the pathogenesis of psoriasis. Many of these genes have been implicated in immune-system development. However, BRF2 is implicated in the development of squamous cell carcinoma of the lung.33 This suggests that it might have global effects on the transcriptional profile of squamous cell epithelia in general and might help elicit a wound-healing or regenerative response in psoriatic keratinocytes.
Despite the dramatic effects of some CARD14 variants as keratinocyte transfectants, there was a wide range of phenotypes, even among individuals who carried the same substitution. This suggests that in many instances, the variable phenotypes are likely to be due to genetic background and/or environmental factors. For example, affected members of family PS1 all harbor the c.349G>A (p.Gly117Ser) mutation, but they have variable ages of onset (ranging from infancy to 83 years of age) and variable levels of disease severity, including the presence of psoriatic arthritis. Similarly, age of onset and response to treatment differed among individuals with the putative pathogenic variants from the coiled-coil domain (p.Glu142Lys, p.Glu142Gly, and p.Glu138Ala).
However, there might be some genotype-phenotype correlations because the pustular-psoriasis substitution, p.Glu138Ala, led to the most severe phenotype (in terms of both clinical presentation and increased NF-kB activation) relative to that produced by wild-type CARD14sh. The child with this alteration presented with a spectrum of plaque-type lesions, but she mostly presented with pustular lesions. This implies that some forms of plaque psoriasis might be pathogenetically linked to pustular psoriasis at the severe end of the disease spectrum. The child's lesions also exhibited a pronounced infiltration of neutrophils. Interestingly, in keratinocyte transfectants with this CARD14 substitution, there was a higher level of IL8 than there was with other variants, which could lead to higher levels of neutrophil infiltration. The observation that the p.Glu138Ala alteration led to the most severe clinical phenotype and induced the greatest increase in NF-kB activation and upregulation of psoriasis-associated transcripts suggests that the phenotype of psoriasis could, in some cases, be predicted by the detection of pathogenically increased levels of NF-kB activation and signaling. How the CARD14 substitutions translate to variable levels of chemokine activation and how genetic background and environment trigger variable phenotypes are important areas for further study.
CARD14 missense variants rs11652075 (c.2458C>T [p.Arg820Trp]) and c.599G>A (p.Ser200Asn) were associated with psoriasis in cohorts of European ancestry. In the Asian cohort, the c.2458T>C polymorphism was also associated with psoriasis, but c.599G was monomorphic. CARD14 was not shown to be associated with psoriasis in a previous GWAS.8 However, none of the polymorphisms with evidence of association with psoriasis in this study were included in that GWAS and would not have been likely to exceed genome-wide significance if they had been. From the PSORS2 region of linkage, the SNP with the most significant p value in the GWAS was rs7216577 (p value = 0.00261).8 That SNP is located within an intron of SLC26A11 and might regulate levels of CARD14 mRNAs in the skin. However, rs11652075, although not on the Perlegen microarray in the CASP-GWAS,8 was part of the HapMap-based imputed dataset that was used for the association analyses in that study. Its association p value was of nominal significance (p = 0.039, OR = 1.10 for the C risk allele reported here) and would not have been reported because it was considerably below the threshold for genome-wide significant association.
PSORS2 was originally identified by linkage,21,34 and our studies with a common missense SNP (rs11652075) also indicate that it can be associated with psoriasis. Linkage and association have also been seen in some genes for other common diseases, e.g., NOD2 (nucleotide-binding oligomerization domain containing 2, [MIM 605956] in inflammatory bowel disease (IBD1 [MIM 266600])35–37 and CFH (complement factor H, [CFH (MIM 134370)]) in age-related macular degeneration (ARMD1 [MIM 603075]).38,39
In two cohorts of European ancestry, evidence of association between psoriasis and rs11652075 increased when the PSORS1 variant HLA-Cw∗0602 was included as a covariate, suggesting a genetic connection between PSORS1 and PSORS2. This variant resides in the CARD14fl C-terminal GUK domain, which is predicted to relay external signals to the cellular milieu.16 This could explain the possible genetic connection because antigen stimulation via PSORS1 might increase the risk of psoriasis by upregulating signaling through the CARD14 pathway. This is consistent with the fact that CARD14 affects signaling downstream of antigen stimulation. However, it is important to note that the PSORS1 risk variant, HLA-Cw∗060240, was not present in the affected members of the 17q-linked multiplex families, the pustular-psoriasis case described previously,17 or the cases with the p.Glu142Lys or p.Glu142Gly variants, indicating that rare CARD14 variants can be sufficient to lead to disease.
Our study illustrates some of the difficulties in searching for rare variants associated with common disease, even with an established gene. For example, it is not always easy to differentiate pathogenic rare variants from others when their numbers are very small. This also has an impact on the detection of gene-gene or gene-environment interactions. Moreover, it is sometimes necessary to recreate the cellular milieu (e.g., an inflammatory stimulus in the case of CARD14) when one attempts to differentiate disease-causing variants from neutral variants through functional studies. Nevertheless, our findings provide evidence that some rare CARD14 variants predispose to psoriasis and, possibly, to psoriatic arthritis, and they suggest that common variants in this region might also predispose to disease. They illustrate the challenges faced in identifying truly pathogenic rare variants in common disease and contribute to our understanding of the genetic basis of psoriasis.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH): AR050266 and 5RC1AR058681 (A.M.B.), T32AR007279 (E.D.O.R.), T32HL083822 and T32 GM07200 (C.T.J), AR060222 (M.A.L), T32HG000045 (C.E.J), and K08AR057763 (W.L.). J.T.E., R.N., and P.S. were supported by NIH grants R01 AR042742, R01 AR050511, and AR054966 and by the Babcock Memorial Trust. J.T.E. is supported by the Ann Arbor Veterans Affairs Hospital. R.G.M. is supported by the Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Additional funding came from the Arthritis Society of Canada, the Atlantic Innovation Fund, the Canadian Institute of Health Research, the Arthritis Society, the Dana Foundation, and the Academia Sinica and National Science Council (National Clinical Core, National Genotyping Core) of Taiwan. The authors thank the many individuals with psoriasis and the controls who participated in this study. We thank Linus Schwantes-An, Weimin Duan, and Nancy Saccone for advice on statistical analyses. We also thank Mayte Suarez-Farinas for biostatistical assistance, Haoyan Chen, Olivia Lai, Fawnda Pellet, and M.J. Malloy for assembling the control population, Pui-Yan Kwok for sample procurement, Trilokraj Tejasvi for clinical evaluation of study subjects, Nikki Plass for patient scheduling and study logistics, Damaris Garcia for the management of samples, and Debbie Stone and Dawn Chapelle for patient care. Mike Lovett provided helpful comments on the manuscript. The authors are indebted to the National Psoriasis Foundation for continuing support during the course of this study.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1,000 Genomes Project, http://www.1000genomes.org
g:Profiler, http://biit.cs.ut.ee/gprofiler/index.cgi
Gene Ontology, http://www.geneontology.com
Ingenuity Pathway Analysis (IPA), http://www.ingenuity.com/
Microarray Gene Expression Data Society (MIAME), http://www.mged.org/Workgroups/MIAME/miame_checklist.html
NCBI Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
NCBI Reference Sequence (RefSeq), http://www.ncbi.nlm.nih.gov/RefSeq/
NHLBI/NHGRI Exome Project, http://exome.gs.washington.edu/
Online Mendelian Inheritance in Man (OMIM), http://omim.org
PLINK/SEQ version 0.05, http://atgu.mgh.harvard.edu/plinkseq/
PolyPhen-2.0, http://genetics.bwh.harvard.edu/pph2/
rmeta, http://cran.r-project.org/web/packages/rmeta/index.html
SequenomARRAY, http://hg.wustl.edu/info/Sequenom_description.html
SIFT, http://sift.jcvi.org/
References
- 1.Lowes M.A., Bowcock A.M., Krueger J.G. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 2.Nograles K.E., Brasington R.D., Bowcock A.M. New insights into the pathogenesis and genetics of psoriatic arthritis. Nat. Clin. Pract. Rheumatol. 2009;5:83–91. doi: 10.1038/ncprheum0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capon F., Bijlmakers M.J., Wolf N., Quaranta M., Huffmeier U., Allen M., Timms K., Abkevich V., Gutin A., Smith R. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum. Mol. Genet. 2008;17:1938–1945. doi: 10.1093/hmg/ddn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cargill M., Schrodi S.J., Chang M., Garcia V.E., Brandon R., Callis K.P., Matsunami N., Ardlie K.G., Civello D., Catanese J.J. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellinghaus E., Ellinghaus D., Stuart P.E., Nair R.P., Debrus S., Raelson J.V., Belouchi M., Fournier H., Reinhard C., Ding J. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hüffmeier U., Uebe S., Ekici A.B., Bowes J., Giardina E., Korendowych E., Juneblad K., Apel M., McManus R., Ho P. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y., Helms C., Liao W., Zaba L.C., Duan S., Gardner J., Wise C., Miner A., Malloy M.J., Pullinger C.R. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuart P.E., Nair R.P., Ellinghaus E., Ding J., Tejasvi T., Gudjonsson J.E., Li Y., Weidinger S., Eberlein B., Gieger C. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010;42:1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L.D., Cheng H., Wang Z.X., Zhang A.P., Wang P.G., Xu J.H., Zhu Q.X., Zhou H.S., Ellinghaus E., Zhang F.R. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat. Genet. 2010;42:1005–1009. doi: 10.1038/ng.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elder J.T. PSORS1: Linking genetics and immunology. J. Invest. Dermatol. 2006;126:1205–1206. doi: 10.1038/sj.jid.5700357. [DOI] [PubMed] [Google Scholar]
- 13.Nair R.P., Stuart P.E., Nistor I., Hiremagalore R., Chia N.V., Jenisch S., Weichenthal M., Abecasis G.R., Lim H.W., Christophers E. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006;78:827–851. doi: 10.1086/503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vineis P., Pearce N.E. Genome-wide association studies may be misinterpreted: Genes versus heritability. Carcinogenesis. 2011;32:1295–1298. doi: 10.1093/carcin/bgr087. [DOI] [PubMed] [Google Scholar]
- 15.Chen H., Poon A., Yeung C., Helms C., Pons J., Bowcock A.M., Kwok P.Y., Liao W. A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS ONE. 2011;6:e19454. doi: 10.1371/journal.pone.0019454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertin J., Wang L., Guo Y., Jacobson M.D., Poyet J.L., Srinivasula S.M., Merriam S., DiStefano P.S., Alnemri E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J. Biol. Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 17.Jordan C.T., Cao L., Roberson E.D.O., Pierson K.C., Yang C.-F., Joyce C.E., Ryan C., Duan S., Helms C.A., Liu Y. PSORS2 is Due to Mutations in CARD14. Am. J. Hum. Genet. 2012;90 doi: 10.1016/j.ajhg.2012.03.012. in press. Published online April 19, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price A.L., Kryukov G.V., de Bakker P.I., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R Development Core Team. (2011). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org.
- 21.Tomfohrde J., Silverman A., Barnes R., Fernandez-Vina M.A., Young M., Lory D., Morris L., Wuepper K.D., Stastny P., Menter A. Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science. 1994;264:1141–1145. doi: 10.1126/science.8178173. [DOI] [PubMed] [Google Scholar]
- 22.Vincourt J.B., Jullien D., Amalric F., Girard J.P. Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J. 2003;17:890–892. doi: 10.1096/fj.02-0787fje. [DOI] [PubMed] [Google Scholar]
- 23.Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(Web Server issue) doi: 10.1093/nar/gkm226. W193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: Tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta N.N., Yu Y., Pinnelas R., Krishnamoorthy P., Shin D.B., Troxel A.B., Gelfand J.M. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med. 2011;124:775e1–775e6. doi: 10.1016/j.amjmed.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidovici B.B., Sattar N., Prinz J.C., Puig L., Emery P., Barker J.N., van de Kerkhof P., Ståhle M., Nestle F.O., Girolomoni G., Krueger J.G. Psoriasis and systemic inflammatory diseases: Potential mechanistic links between skin disease and co-morbid conditions. J. Invest. Dermatol. 2010;130:1785–1796. doi: 10.1038/jid.2010.103. [DOI] [PubMed] [Google Scholar]
- 27.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suárez-Fariñas M., Lowes M.A., Zaba L.C., Krueger J.G. Evaluation of the psoriasis transcriptome across different studies by gene set enrichment analysis (GSEA) PLoS ONE. 2010;5:e10247. doi: 10.1371/journal.pone.0010247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno-García M.E., Sommer K., Shinohara H., Bandaranayake A.D., Kurosaki T., Rawlings D.J. MAGUK-controlled ubiquitination of CARMA1 modulates lymphocyte NF-kappaB activity. Mol. Cell. Biol. 2010;30:922–934. doi: 10.1128/MCB.01129-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thome M., Charton J.E., Pelzer C., Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2010;2:a003004. doi: 10.1101/cshperspect.a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009;9:778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 32.Wullaert A., Bonnet M.C., Pasparakis M. NF-κB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–158. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockwood W.W., Chari R., Coe B.P., Thu K.L., Garnis C., Malloff C.A., Campbell J., Williams A.C., Hwang D., Zhu C.Q. Integrative genomic analyses identify BRF2 as a novel lineage-specific oncogene in lung squamous cell carcinoma. PLoS Med. 2010;7:e1000315. doi: 10.1371/journal.pmed.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwu W.L., Yang C.F., Fann C.S., Chen C.L., Tsai T.F., Chien Y.H., Chiang S.C., Chen C.H., Hung S.I., Wu J.Y., Chen Y.T. Mapping of psoriasis to 17q terminus. J. Med. Genet. 2005;42:152–158. doi: 10.1136/jmg.2004.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 36.Hugot J.P., Laurent-Puig P., Gower-Rousseau C., Olson J.M., Lee J.C., Beaugerie L., Naom I., Dupas J.L., Van Gossum A., Orholm M. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 37.van Heel D.A., Fisher S.A., Kirby A., Daly M.J., Rioux J.D., Lewis C.M., Genome Scan Meta-Analysis Group of the IBD International Genetics Consortium Inflammatory bowel disease susceptibility loci defined by genome scan meta-analysis of 1952 affected relative pairs. Hum. Mol. Genet. 2004;13:763–770. doi: 10.1093/hmg/ddh090. [DOI] [PubMed] [Google Scholar]
- 38.Fisher S.A., Abecasis G.R., Yashar B.M., Zareparsi S., Swaroop A., Iyengar S.K., Klein B.E., Klein R., Lee K.E., Majewski J. Meta-analysis of genome scans of age-related macular degeneration. Hum. Mol. Genet. 2005;14:2257–2264. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 39.Raychaudhuri S., Iartchouk O., Chin K., Tan P.L., Tai A.K., Ripke S., Gowrisankar S., Vemuri S., Montgomery K., Yu Y. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011;43:1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elder J.T., Cluster 17 Collaboration Fine mapping of the psoriasis susceptibility gene PSORS1: A reassessment of risk associated with a putative risk haplotype lacking HLA-Cw6. J. Invest. Dermatol. 2005;124:921–930. doi: 10.1111/j.0022-202X.2005.23729.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.