

Abstract
Mainzer-Saldino syndrome (MSS) is a rare disorder characterized by phalangeal cone-shaped epiphyses, chronic renal failure, and early-onset, severe retinal dystrophy. Through a combination of ciliome resequencing and Sanger sequencing, we identified IFT140 mutations in six MSS families and in a family with the clinically overlapping Jeune syndrome. IFT140 is one of the six currently known components of the intraflagellar transport complex A (IFT-A) that regulates retrograde protein transport in ciliated cells. Ciliary abundance and localization of anterograde IFTs were altered in fibroblasts of affected individuals, a result that supports the pivotal role of IFT140 in proper development and function of ciliated cells.
Main Text
Ciliopathies are an emerging class of genetic disorders caused by altered cilia assembly, maintenance, or function.1,2 They comprise a broad range of phenotypes that result from developmental or functional defects of unique or multiple systems. Syndromic ciliopathies that affect bone development are classified as skeletal ciliopathies.1,2 Mutations in genes that encode components of the intraflagellar transport complex A (IFT-A), which drives retrograde ciliary transport,3 are a major cause of skeletal ciliopathies.4–11 Alterations of all IFT-A components but one (IFT140) have been previously reported to cause Sensenbrenner, Jeune, and/or short-rib polydactyly syndromes (IFT43 [MIM 614068], IFT122 [MIM 606045], IFT139 [MIM 612014], IFT144 [MIM 608151], WDR35 [MIM 613602]).4–11
Mainzer-Saldino syndrome (MSS [MIM 266920]), or conorenal syndrome (CRS), is a rare autosomal recessive disease defined by phalangeal cone-shaped epiphyses (PCSE), chronic renal disease, nearly constant retinal dystrophy, and mild radiographic abnormality of the proximal femur.12 Occasional features include short stature, cerebellar ataxia, and hepatic fibrosis.12 MSS shares retinal dystrophy with Leber congenital amaurosis (LCA [MIM 204000]) and nephronophthisis (NPHP [MIM 256100]), and skeletal features with asphyxiating thoracic dystrophy (ATD [MIM 208500]), or Jeune syndrome, and cranioectodermal dysplasia (CED [MIM 218330]), or Sensenbrenner syndrome, suggesting that it is a ciliopathy.
We collected 15 families presenting three diagnostic criteria of MSS, namely early-onset retinal dystrophy, PCSE, and renal disease, and two families with nonovert renal disease (Tables S1 and S2). Informed consent was obtained for each individual participating in this study, which was approved by the Comité de Protection des Personnes “Ile-de-France II.” To identify the genetic mutations responsible for MSS, we subjected the genomic DNA of an affected individual born to unrelated parents (FI1; Figure 1 and Table S1) to ciliome resequencing, using a 5.3 Mb customized Agilent SureSelect Target Enrichment library to capture 32,146 exons of 1,666 genes. We first focused our analysis on consensus splice-site changes, nonsynonymous variants, and insertion and/or deletion in coding regions. Considering that MSS-causing mutations are rare, we assumed that the affected individual was likely compound heterozygous for variants absent in the dbSNP132, 1000Genome, and in-house databases. This pointed to one candidate gene only, IFT140 (intraflagellar transport protein 140 homolog (chalydomonas); [NM_014714.3]), which encodes the last IFT-A component not yet shown to be related to skeletal ciliopathies (Table S3). We found both a missense and a donor splice-site mutation (Figure 2 and Table S1). The missense mutation altered an acidic residue conserved across species (c.1990G>A [p.Glu664Lys]), whereas we expected the splice-site mutation to result in the skipping of exon 18 and/or the use of a surrounding cryptic splice site (c.2399+1G>T). Sanger sequencing confirmed these results, and segregation analysis excluded allelism of the variants.
Figure 1.
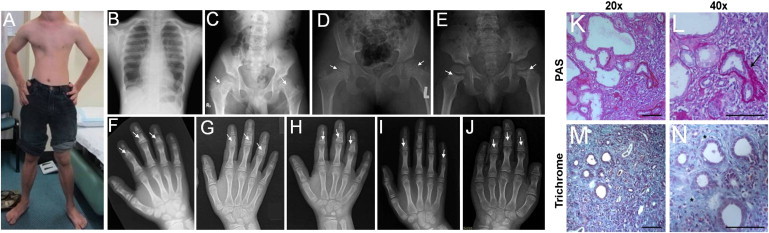
Clinical and Radiological Manifestations of MSS-Affected Individuals
(A and B) Photograph and thorax X-rays of affected individual FI1 at 17 years of age showing narrow chest.
(C–E) Hip X-rays of affected individuals FI1 (17 years), FIII1 (9 years), and FIII2 (5 years) showing flattened femoral epiphyses (C–D) and wide femoral neck with areas of sclerosis in the metaphyseal region (arrows).
(F–J) Hand X-ray of affected individuals FI1, FIII1, FIII2, FII1, and FV1 showing PCSE (arrows).
(K–N) Renal biopsy from affected individual FVI1 at 2 years (periodic acid-Schiff [PAS] in K and L; trichrome staining in M and N) showing severe tubulointerstitial lesions characterized by dedifferentiated and dilated tubules as well as atrophic tubules surrounded by marked thickening of the tubular basement membrane (arrow) and interstitial fibrosis with infiltrates (asterisks). Scale bar: 100 μm.
Figure 2.
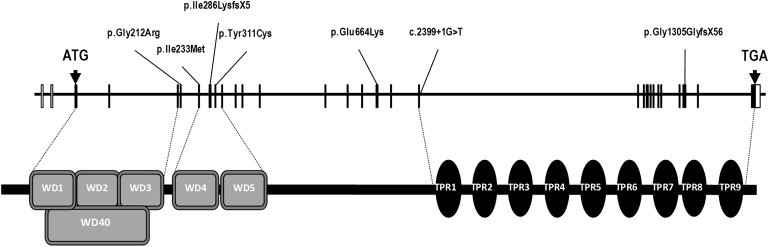
IFT140 Organization, Protein Structure, and Mutations Identified in Homozygous and Compound-Heterozygous Individuals Affected with Skeletal Ciliopathies
Additional ciliome resequencing and/or Sanger sequencing of the 29 IFT140 coding exons (3 to 31, Table S4) and intron-exon boundaries detected homozygous or compound-heterozygous disease-causing mutations in five other MSS families (Figures 1 and 2; Table 1, Tables S1 and S3) and single heterozygote mutations in four additional families (Table 1 and Table S2). All changes were absent from 200 control chromosomes and were predicted to be deleterious with the use of the Alamut Mutation Interpretation Software, a decision support system for mutation interpretation based on Align DGVD, Polyphen-2, SIFT, SpliceSiteFinder-like, MaxEntScan, NNSPLICE and Human Splicing Finder (Table 1). Segregation of compound-heterozygous and homozygous mutations with the disease was confirmed.
Table 1.
IFT140 Mutations Identified in Ten Individuals Affected with Mainzer-Saldino Syndrome and a Child with Jeune Syndrome
| Family | Pedigree | Diagnosis |
Allele 1 |
Allele 2 |
||
|---|---|---|---|---|---|---|
| Mutation | Predicted Effecta | Mutation | Predicted Effecta | |||
| FI | simplex, nonconsanguineous | MSS | c.2399+1G>T | loss of intron 19 donor splice-site | c.1990G>A | p.Glu664Lys (deleterious) |
| FI | simplex, nonconsanguineous | MSS | c.2399+1G>T | loss of intron 19 donor splice-site | c.1990G>A | p.Glu664Lys (deleterious) |
| FII | simplex, nonconsanguineous | MSS | c.932A>G | p.Tyr311Cys (deleterious) | c.857_860del | p.Ile286Lysfs∗6 |
| FIII | multiplex, consanguineous | MSS | c.1990G>A | p.Glu664Lys (deleterious) | c.1990G>A | p.Glu664Lys (deleterious) |
| FIV | multiplex, consanguineous | MSS | c.1990G>A | p.Glu664Lys (deleterious) | c.1990G>A | p.Glu664Lys (deleterious) |
| FV | multiplex, nonconsanguineous | MSS | c.634G>A | p.Gly212Arg (deleterious) and/or alteration exon 6 donor-splice site | c.3916dup | p.Ala1306Glyfs∗56 |
| FVI | simplex, consanguineous | MSS | c.699T>G | p.Ile233Met (deleterious) | c.699T>G | p.Ile233Met (deleterious) |
| FVII | simplex, nonconsanguineous | Jeune syndrome | c.2399+1G>T | loss of intron 19 donor splice-site | c.634G>A | p.Gly212Arg (deleterious) and/or alteration exon 6 donor splice site |
| FVIII | simplex, nonconsanguineous | MSS | c.1565G>A | p.Gly522Glu (deleterious) | no mutation identified | |
| FIX | simplex, nonconsanguineous | MSS | c.874G>A | p.Val292Met (deleterious) | no mutation identified | |
| FX | simplex, nonconsanguineous | MSS | c.1727G>A | p.Arg576Gln (deleterious) | no mutation identified | |
| FXI | simplex, nonconsanguineous | MSS | c.489C>T | p.Gly163Gly and creation of an additional donor splice site 4 bp upstream of intron 5 and predicted to result in c.488_491del (p.Glu164Thrfs∗10) | no mutation identified | |
Variant pathogenicity according to Align DGVD, Polyphen-2, SIFT, SpliceSiteFinder-like, MaxEntScan, NNSPLICE and Human Splicing Finder available through the Alamut Interpretation Software 2.0.
Affected individuals of the nonconsanguineous simplex FII and multiplex FV families were compound heterozygous for missense and truncating mutations, whereas those of the multiplex consanguineous families III and IV from Saudi Arabia were homozygous for missense mutations (Table 1 and Figure 2). Affected cases of these latter two families harbored the c.1990G>A (p.Glu664Lys) change identified in affected individual FI1 (Table 1 and Figure 2). Linkage analysis at the IFT140 locus detected an intragenic IFT140 recombination, precluding the search for linkage disequilibrium in families segregating this latter change (Figure S1). Affected individuals FVIII1, FIX1, and FX1 were single heterozygous for missense mutations. FXI1 harbored a conservative change that we predicted would create 4 bp upstream of the intron 5 donor splice site, a competing donor site, the use of which would result in the appearance of a premature stop codon (c.489C>T [p.Gly163Gly and p.Glu164Thrfs∗10]; Table 1 and Table S2).
The effect of the c.634G>A (p.Gly212Arg), c.699T>G (p.Ile233Met), c.932A>G (p.Tyr311Cys), and c.1990G>A (p.Glu664Lys) changes on the IFT140 localization was assessed in the telomerase-immortalized retinal pigment epithelial cell line (RPE1). Flag-tagged IFT140 mutant proteins showed a partial to nearly complete loss of basal body localization associated with an increase of cytoplasm staining, whereas the wild-type Flag-tagged IFT140 protein predominantly localized to the basal bodies in RPE1 cells (Figure 3A). The c.1990G>A (p.Glu664Lys) change displayed the most severe disorganization (IFT140 mislocalization in 80% of the cells; Figure 3A).
Figure 3.
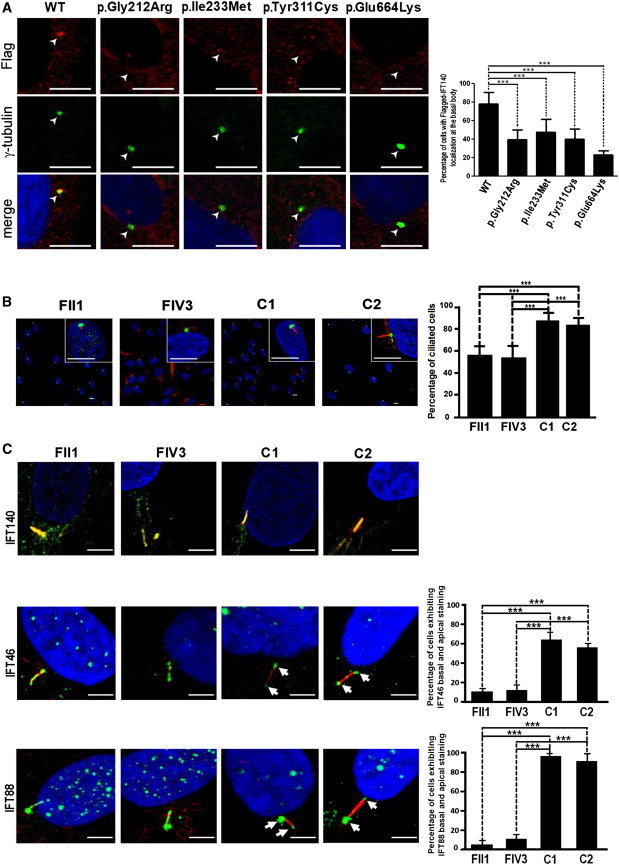
Distribution of Wild-Type and Mutant Endogenous or Flag-Tagged IFT140 in Fibroblasts and RPE1 Cells
(A) Aberrant distribution of Flag-tagged mutant IFT140 proteins in RPE1 cells. Cells were transfected with pCMV-IFT140-WT-Flag, pCMV-IFT140-Glu212Arg-Flag, pCMV-IFT140-Ile233Met-Flag, pCMV-IFT140-Tyr311Cys-Flag, and pCMV-IFT140-Glu664Lys-Flag plasmids, respectively. Flag-tagged proteins were stained with the use of rabbit anti-Flag (1:200, Sigma-Aldrich) and Alexa Fluor 555-conjugated anti-rabbit (1:200, Molecular Probes; red) primary and secondary antibodies, respectively. Basal bodies were stained with the use of mouse monoclonal anti-γ-tubulin (1:1000, Sigma-Aldrich) and Alexa Fluor 488-conjugated anti-mouse (1:200, Molecular Probes) primary and secondary antibodies, respectively. Images were recorded with a Leica SP5 confocal microsocope (Leica). Scale bars 5 μm. Wild-type (WT) IFT140 is clearly visible at the basal body (white arrow), whereas mutant proteins exhibit a significant cytoplasmic staining with decreased basal body labeling. The graph shows the percentage of transfected cells with localization of the Flag-tagged IFT140 protein at the basal body calculated from two independent experiments, and n > 100 cells for each transfection condition (∗∗∗: p < 0.001 calculated via Dunn's Multiple Comparison Test following the analysis of variance [ANOVA] test, GraphPad Prism Software).
(B) Significantly decreased cilia abundance in serum-starved fibroblasts of MSS-affected individuals with IFT140 mutations compared to two controls (mean affected individuals versus mean controls: 55.10% versus 83.61% calculated from two independent experiments and n > 140 cells; ∗∗∗: p < .0001 calculated via the Fisher protected least significant difference [PLSD] test according to the significance of the Student's t test; StatView software version 5.0). Basal bodies were stained with the use of rabbit polyclonal anti-pericentrin (1:500, Abcam) and Alexa Fluor 488-conjugated goat anti-rabbit (1:1000, Sigma-Aldrich; green) primary and secondary antibodies, respectively. Axonemes were stained with the use of mouse monoclonal anti-acetylated alpha-tubulin (1:1000, Sigma-Aldrich) and Alexa Fluor 594 goat anti-mouse (1:1000, Molecular Probes; red) primary and secondary antibodies, respectively. Images were recorded with a Zeiss LSM700 microscope (Carl Zeiss S.A.S.). Scale bars 10 μm.
(C) Distribution of IFT components in fibroblasts of MSS-affected individuals and controls. IFTs were stained with the use of IFT140 (1:100, goat polyclonal), IFT46 (1:200, rabbit polyclonal; gift of F. Mallin-Guerin), and IFT88 (1:100, rabbit polyclonal; gift of C. Desdouets) primary antibodies and Alexa Fluor 488-conjugated goat anti-rabbit or rabbit anti-goat (1:1000; green) secondary antibodies. Axonemes were stained as described previously. Nuclei were labeled with the use of DAPI (Southern Biotech). Images were recorded with a Zeiss LSM700 confocal microscope (Carl Zeiss S.A.S.). Scale bars 5 μm. Acetylated alpha-tubulin and IFT140 were evenly distributed along the axoneme in control, patient FII1, and patient FIV3 cells (Pearson's coefficients calculated via the ImageJ 1.42d JACoP software ≥ .80). Despite apparently normal expression and localization of IFT140, IFT46 and IFT88 were mislocalized in cilia of affected individuals FII1 and FIV1 (even distribution along the axoneme versus predominant staining at the base and the tip [white arrows] of cilia in control cells). The graphs show the percentage of cilia exhibiting both basal and apical staining in controls versus affected individuals (calculated from two independent experiments and n ≥ 40 cells for each cell line and each IFT, ∗∗∗: p < 0.0001 calculated with the Student's t test; StatView software version 5.0).
Error bars represent SD.
To assess the impact of IFT140 mutations on ciliogenesis, abundance and morphology of primary cilia were studied in cultured fibroblasts of affected individuals FII1 and FIV3. By staining the cilia axonemes with acetylated alpha-tubulin, we detected absent cilia in a high proportion of cells of affected cases compared to controls (mean affected cases versus mean controls: 55.10% versus 83.61%, p < .0001), supporting a defect in ciliogenesis and/or cilia maintenance (Figure 3B).
To determine the effect of the IFT140 mutations on retrograde intraflagellar transport, we analyzed the endogenous subcellular localization of IFT140. The fibroblasts of affected individuals FII1 (compound heterozygous for a splice-site mutation and the c.932A>G [p.Tyr311Cys] change) and FIV3 (homozygous for the c.1990G>A [p.Glu664Lys] change) exhibited an unaltered IFT140 localization along the cilia axoneme (Figure 3C). However, two components of the anterograde transport IFT-B complex, IFT88 and IFT46, were evenly distributed along the cilium of both affected individual fibroblasts, whereas they were predominantly detected at the base and the tip of the cilium in control fibroblasts (p < 0.0001), suggesting an alteration in retrograde ciliary transport (Figure 3C).
The defect in ciliogenesis and/or ciliary maintenance and the aberrant distribution of IFT88 and IFT46 in cells of affected individuals is consistent with the report of short cilia and aberrant distribution of IFT88 in mutant reduced mechanoreceptor potential A (rempA), the Drosophila ortholog of IFT140.13 The ternary IFT-B subcomplex, made up of IFT52, IFT88, and IFT46, is crucial for the stabilization of the IFT-B particle.14–16 Therefore, the data we report further support the view that alterations of IFT-A components disorganize assembly and/or maintenance of ciliary structure by impairing retrograde IFT through the redistribution of ciliary proteins (notably IFT-B complex components).5,7,8,17,18
Genetic and clinical heterogeneity are hallmarks of multisystemic ciliopathies.1,2 In addition, several examples have been reported which demonstrate that different mutations in a same gene, e.g., WDR35,6,11 WDR19 [MIM 608151],8 CEP290 [MIM 610142],19 and IQCB1 [MIM 609237],20–22 can give rise to a broad range of phenotypes, from isolated nephronophtisis or LCA to multisystemic and sometimes embryonically lethal conditions. Genotype-phenotype correlations and global mutation load in ciliary genes have been suggested to account for this clinical variability.1,2 Here we report that IFT140 mutations consistently caused PCSE as well as retinal dystrophy and occasional chronic renal failure, hepatic fibrosis, additional skeletal abnormalities, or neurological symptoms.
Interestingly, although retinal dystrophy is an occasional feature of skeletal ciliopathies,1,2 it is very uncommon in patients with IFT-A mutations.5–8,10 Conversely, all affected individuals with IFT140 mutations and full ophthalmological examination (n = 9/10; six families) had LCA or early-onset, severe retinal dystrophy between birth and 4 years of age (Table S1). With regard to individual FVI1, who had no reported retinal disease at the age of 2 years, electrophysiological recordings were not available to assess the function of photoreceptors, the alteration of which typically precedes fundus changes. Retinal dystrophy appears therefore to be a very stringent clinical manifestation of IFT140 alterations. Although there is no clear-cut correlation between tissue expression of IFTs and clinical features, the recent report of the role of IFT140 in photoreceptor cell ciliogenesis24 is consistent with the retinal disease of MSS-affected individuals harboring IFT140 mutations.
Chronic renal failure is an inclusion criterion in MSS. However, age-at-onset and outcome of the dysfunction vary, even within families.12 Therefore, it is possible that renal failure occurs in affected individuals in families III and IV (n = 5, age range 4–17 years) late in the course of the disease. This would be consistent with the recent report of pronounced renal cystic disease in mice resulting from HoxB7-Cre-driven loss of Ift140 in the renal collecting duct.23 Nevertheless, affected individuals from families III and IV were homozygous for a missense mutation (c.1990G>A [p.Glu664Lys]), whereas all affected persons with renal failure but one (FVI1) harbored a severe truncating mutation (in addition to a missense mutation), raising the question of whether some genotype-phenotype correlations may exist. From this point of view, it is worth noting that affected individual FVI1 harbored a homozygous missense mutation (c.699T>G [p.Ile233Met]) and heterozygous mutations in other ciliopathy genes, TMEM67 (MIM 609884; data not shown) and XPNPEP3 (MIM 613553; Table S3B), which could contribute to kidney failure.
Cerebral MRIs were consistently normal, but siblings in two families exhibited neurological symptoms that included mild intellectual impairment or autistic features, seizures, and epilepsy (Families III and IV, respectively; Table S1). Considering the high degree of consanguinity of the two families, it is difficult to determine whether some or all these traits are accounted for by IFT140 mutations, or if these disabilities are independently inherited.
Some of the skeletal abnormalities of individuals with IFT140 mutations overlap with clinical symptoms of Jeune and Sensenbrenner syndromes—namely, short hands, hip and cranial abnormalities, narrow chest, and/or short stature (Table S1). This overlap opens a debate as to whether some affected individuals with IFT140 mutations are affected with complex MSS, or incomplete Jeune or Sensenbrenner syndromes (e.g., FI1, who had a narrow chest but no respiratory deficiency and no trident acetabular roof, or FVI1, who had trident acetabular roof, a narrow chest, and short ribs; Table S1). Additionally, this clinical overlap raises the question of whether other skeletal ciliopathies are accounted for by IFT140 alterations. So far the screening of large cohorts of individuals affected with Sensenbrenner of Jeune syndromes failed to detect IFT140 mutations.8 However, during this study, we had the opportunity to identify IFT140 mutations in a child affected with Jeune syndrome (FVII1, Table S1). The child was compound heterozygous for a splice-site mutation and the c.634G>A (p.Gly212Arg) change identified in Family V. These data suggest that mutations in IFT140 may be a rare cause of Jeune syndrome and that there is no clear correlation between the IFT140 genotype and the severity of the disease.
The clinical presentation of the disease did not differ significantly in MSS-affected individuals with two IFT140 disease alleles compared to single-heterozygous patients (4/17) or affected individuals with no mutation (7/18; Tables S1 and S2). It is possible that some of the latter 10/17 affected individuals may harbor undetected IFT140 mutations lying in unscreened regions, such as untranslated regions or introns. Furthermore, following the example of the deep intronic CEP290 c.1991+1655A>G mutation, which accounts for approximately 60% of CEP290 disease alleles in LCA, it is possible that a common undetected IFT140 mutation may exist.19 However, like most other ciliopathies, MSS may be genetically heterogeneous with some or all 10/17 affected individuals, including single heterozygotes, harboring mutations in other genes.
In summary, we here report on compound heterozygosity or homozygosity for IFT140 mutations in six families affected with MSS and an individual affected with Jeune syndrome. After Sensenbrenner and Jeune syndromes, MSS is the latest skeletal ciliopathy ascribed to IFT disorganization.
Acknowledgments
We are grateful to all affected individuals and their families for their participation in the study. We thank R. Devaux for confocal microscopy and C. Masson and S. Pruvost for technical assistance. This work was supported by grants from Retina France, the Fondation pour la Recherche Médicale (FRM DEQ20071210558 to S.S.), the Agence Nationale de la Recherche (grants R09087KS and RPV11012KK to S.S.), and the Dutch Kidney Foundation (KJPB09.009 and IP11.58 to H.H.A.).
Contributor Information
Josseline Kaplan, Email: josseline.kaplan@inserm.fr.
Jean-Michel Rozet, Email: jean-michel.rozet@inserm.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Alamut Interpretation Software 2.0, http://alamut.interactive-biosoftware.com
UCSC Genome Browser, http://genome.ucsc.edu
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters A.M., Beales P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole D.G., Snell W.J. SnapShot: Intraflagellar transport. Cell. 2009;137:784–784.e1. doi: 10.1016/j.cell.2009.04.053. [DOI] [PubMed] [Google Scholar]
- 4.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 5.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 8.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavalcanti D.P., Huber C., Sang K.H., Baujat G., Collins F., Delezoide A.L., Dagoneau N., Le Merrer M., Martinovic J., Mello M.F. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J. Med. Genet. 2011;48:88–92. doi: 10.1136/jmg.2009.069468. [DOI] [PubMed] [Google Scholar]
- 10.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A., Keighren M., Bahlo M., Bromhead C.J., Budd P. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 2011;88:508–515. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beals R.K., Weleber R.G. Conorenal dysplasia: a syndrome of cone-shaped epiphysis, renal disease in childhood, retinitis pigmentosa and abnormality of the proximal femur. Am. J. Med. Genet. A. 2007;143A:2444–2447. doi: 10.1002/ajmg.a.31948. [DOI] [PubMed] [Google Scholar]
- 13.Lee E., Sivan-Loukianova E., Eberl D.F., Kernan M.J. An IFT-A protein is required to delimit functionally distinct zones in mechanosensory cilia. Curr. Biol. 2008;18:1899–1906. doi: 10.1016/j.cub.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucker B.F., Miller M.S., Dziedzic S.A., Blackmarr P.T., Cole D.G. Direct interactions of intraflagellar transport complex B proteins IFT88, IFT52, and IFT46. J. Biol. Chem. 2010;285:21508–21518. doi: 10.1074/jbc.M110.106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou Y., Qin H., Follit J.A., Pazour G.J., Rosenbaum J.L., Witman G.B. Functional analysis of an individual IFT protein: IFT46 is required for transport of outer dynein arms into flagella. J. Cell Biol. 2007;176:653–665. doi: 10.1083/jcb.200608041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmed N.T., Gao C., Lucker B.F., Cole D.G., Mitchell D.R. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J. Cell Biol. 2008;183:313–322. doi: 10.1083/jcb.200802025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Absalon S., Blisnick T., Kohl L., Toutirais G., Doré G., Julkowska D., Tavenet A., Bastin P. Intraflagellar transport and functional analysis of genes required for flagellum formation in trypanosomes. Mol. Biol. Cell. 2008;19:929–944. doi: 10.1091/mbc.E07-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iomini C., Li L., Esparza J.M., Dutcher S.K. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 2009;183:885–896. doi: 10.1534/genetics.109.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppieters F., Lefever S., Leroy B.P., De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum. Mutat. 2010;31:1097–1108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- 20.Otto E.A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O'Toole J.F., Helou J., Attanasio M. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 21.Estrada-Cuzcano A., Koenekoop R.K., Coppieters F., Kohl S., Lopez I., Collin R.W., De Baere E.B., Roeleveld D., Marek J., Bernd A. IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2011;52:834–839. doi: 10.1167/iovs.10-5221. [DOI] [PubMed] [Google Scholar]
- 22.Stone E.M., Cideciyan A.V., Aleman T.S., Scheetz T.E., Sumaroka A., Ehlinger M.A., Schwartz S.B., Fishman G.A., Traboulsi E.I., Lam B.L. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 2011;129:81–87. doi: 10.1001/archophthalmol.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jonassen J.A., Sanagustin J., Baker S.P., Pazour G.J. Disruption of IFT Complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 2012 doi: 10.1681/ASN.2011080829. in press. Published online January 26, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sedmak T., Wolfrum U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell. 2011;103:449–466. doi: 10.1042/BC20110034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.