

Abstract
Omega-3 and omega-6 long-chain polyunsaturated fatty acids (LC-PUFAs) are essential for the development and function of the human brain. They can be obtained directly from food, e.g., fish, or synthesized from precursor molecules found in vegetable oils. To determine the importance of genetic variability to fatty-acid biosynthesis, we studied FADS1 and FADS2, which encode rate-limiting enzymes for fatty-acid conversion. We performed genome-wide genotyping (n = 5,652 individuals) and targeted resequencing (n = 960 individuals) of the FADS region in five European population cohorts. We also analyzed available genomic data from human populations, archaic hominins, and more distant primates. Our results show that present-day humans have two common FADS haplotypes—defined by 28 closely linked SNPs across 38.9 kb—that differ dramatically in their ability to generate LC-PUFAs. No independent effects on FADS activity were seen for rare SNPs detected by targeted resequencing. The more efficient, evolutionarily derived haplotype appeared after the lineage split leading to modern humans and Neanderthals and shows evidence of positive selection. This human-specific haplotype increases the efficiency of synthesizing essential long-chain fatty acids from precursors and thereby might have provided an advantage in environments with limited access to dietary LC-PUFAs. In the modern world, this haplotype has been associated with lifestyle-related diseases, such as coronary artery disease.
Introduction
The proportionally large human brain relative to body size is unique among primates, and a large portion of the resting-energy budget is allocated to the brain to support it.1 In order to maintain the function of our brain and central nervous system, humans are highly dependent on high amounts of two long-chain polyunsaturated fatty acids (LC-PUFAs), the omega-3 docosahexaenoic acid (DHA) and the omega-6 arachidonic acid (AA).2,3 These fatty acids are essential to humans in the sense that they cannot be synthesized de novo but need to be supplied through dietary intake, either as DHA and AA or as their 18-carbon precursors, alpha-linolenic acid (ALA) and linoleic acid (LA). DHA is mainly found in fish, whereas AA is also present in egg, land-animal fats, and liver. The precursors LA and ALA are found in high quantities in some vegetable oils.4 The conversion of the 18-carbon precursors to LC-PUFAs is done through a series of elongations and desaturations of the fatty-acid molecules. The δ-5 and δ-6 fatty-acid desaturases, which introduce double bonds after the fifth and sixth carbon atoms, respectively, from the carboxyl end of the carbon chain, are rate-limiting enzymes in the biosynthesis of omega-3 and omega-6 LC-PUFAs.5 These two key enzymes are encoded by FADS1 (MIM 606148) and FADS2 (MIM 606149), respectively, located in a head-to-head orientation on chromosome 11.
SNPs in the FADS region have previously been shown to have strong association with levels of blood lipids6–10 and LC-PUFAs.11–14 The variation in the FADS loci has also been shown to modulate breast-feeding effects on intelligence development15 and modify the transfer of DHA and AA from the mother to the child,16,17 and it has been associated with increased risks of inflammation and coronary artery disease.18
Despite the great interest in the FADS region as a key locus for LC-PUFA biosynthesis, there are until now no studies that have investigated the potential role of FADS mutations in human evolution. The greater encephalization of modern humans might have required genetic adaptations of the fatty-acid metabolism to satisfy the high demand of LC-PUFAs needed to sustain the larger brain.19,20 In particular, mutations that increase the efficiency of converting the precursors ALA and LA to longer fatty acids are likely to be favored in environments with limited dietary access to these LC-PUFAs.
In order to elucidate the effect of genetic variants in the FADS region on the production of LC-PUFAs, we combined genotyped and imputed SNPs data with resequencing of the FADS1 and FADS2 loci and estimates of the synthesis of LC-PUFAs in local European populations. The population-sequencing data allowed us to investigate the independent effect of rare variants, an issue that has been the focus of much interest.21 We also analyzed available genomic data from contemporary human populations, archaic hominins, and more distant primates to gain insight into the evolutionary history of the FADS region. Our results show that in humans, two common and very distinct FADS haplotypes are strongly associated with LC-PUFA-synthesis levels. The haplotype associated with the enhanced ability to produce AA and DHA from their precursors is specific to humans and has appeared after the split of the common ancestor of humans and Neanderthals. This haplotype shows evidence of positive selection in African populations, in which it is presently almost fixed; the haplotype is less frequent outside Africa. We propose that the haplotype that provides a more efficient synthesis of LC-PUFAs might act as a thrifty genotype and represents a risk factor for lifestyle-related diseases, such as coronary artery disease.
Material and Methods
Population Sample
The cohorts studied are from populations in Sweden, Italy, Scotland, Croatia, and The Netherlands and are part of the European Special Population Research Network (EUROSPAN). The Northern Swedish Population Health Study (NSPHS) is a cross-sectional study conducted in the community of Karesuando, north of the Arctic Circle in Norrbotten County, Sweden.22 The Orkney Complex Disease Study (ORCADES) is a longitudinal study in the Scottish archipelago of Orkney.23 The VIS study is a cross-sectional study in the villages of Vis and Komiza on the Dalmatian island of Vis, Croatia.24,25 The Microisolates in South Tyrol Study (MICROS) is a cross-sectional study carried out in Venosta Valley, South Tyrol, Italy.26 The Erasmus Rucphen Family Study (ERF) is a longitudinal study of a population that has been living in the Rucphen region, The Netherlands since the 19th century.27 More information on the EUROSPAN populations has been published previously.22 All participants gave their written informed consent,28 and the projects were approved by the institutional review boards in each country.
Lipidomics
As part of the EUROSPAN project, a large number of lipids were measured by mass spectrometry (ESI-MS/MS) in positive-ion mode as described previously.29,30 In particular, phospholipids including phosphatidylcholine (PC) have been quantified in over 4,000 individuals from the five populations.31 PC 36:4 used here is a subgroup of phospholipids containing 36 carbons and four double bonds. Specific fatty acids (e.g., components of the omega-3 and omega-6 biosynthesis) were quantified for 700 individuals from the NSPHS cohort.
SNP Array Genotyping and Imputation
DNA samples were genotyped according to the manufacturer's instructions on Illumina Infinium HumanHap300 or HumanCNV370 SNP bead microarrays as described previously.7 Analysis of the raw data was performed with BeadStudio software according to the recommended parameters for the Infinium assay and with the genotype cluster files provided by Illumina. Samples with a call rate <97%, identical twins, and genetic outliers (identified by classical multidimensional scaling) were excluded from the analysis. Population stratifications were also tested for by multidimensional scaling. In the initial quality control, the primary analysis included most SNPs except for those deviating strongly form the Hardy-Weinberg equilibrium (p value < 10−10). MaCH32 and reference haplotypes from the 1,000 Genomes Database (release 2010-08) were used for performing SNP imputation for a 2.7 Mb region centered over the FADS region. The EUROSPAN genotype data were phased with fastPhase.33 The following are the amounts of individuals with complete genotype and imputation data in each cohort: 2,385 from ERF, 795 from VIS, 1,097 from MICROS, 719 from ORCADES, and 656 from NSPHS.
SNP Association Analysis and Haplotype Reconstruction
Association analyses of the quantitative lipid measurements were performed with the R package GenABEL.34 This tool was developed to enable statistic analyses of genetic data in related individuals with the use of a mixed model.35 Meta-analyses of data from the five populations were carried out with MetABEL.34 We used Haploview36 to examine the association between high and low levels of the lipid PC 36:4 and all SNPs with a call rate above 75%. This resulted in a list of 134 SNPs (see Table S1, available online). The associated haplotypes were constructed by the following procedure. Starting with all 134 SNPs in the region, we iteratively removed SNPs for which (1) the two alleles were found on different haplotypes with the same effect on PC 36:4 and (2) the same allele was found in multiple haplotypes with different effects on PC 36:4. For each round of this analysis, we considered only the haplotypes present in at least 3% of the chromosomes. This resulted in 28 strongly associated SNPs on two common haplotypes accounting for over 95% of the chromosomes. The calculation of p values for individual markers was performed in Haploview with the use of a chi-square test based on allele frequencies between the case and control groups (highest and lowest levels of PC 36:4). The detected haplotype frequencies (for A, D, and mixed) did not deviate from the Hardy-Weinberg equilibrium (chi-square test, p value > 0.9) in any of the five populations or in a combination of the five.
Enrichment and Resequencing of the FADS Region
From each of the five population cohorts, we selected 90–100 unrelated individuals with the lowest levels of PC 36:4 and the same number of individuals with the highest levels of PC 36:4. We pooled equal amounts of genomic DNA from each individual into a high-lipid and a low-lipid pool for each population at a final DNA concentration of 10 ng/μl. The procedure for genomic enrichment and sequencing has been described previously.37 The FADS region (hg18, chr11: 61,311,035–61,390,653) was covered by 26 amplicons. The average amplicon was about 2 kb and had 50–100 nucleotides that overlapped. As a result of repetitive sequences, some parts of the 80 kb region could not be covered by amplicons.
We carried out long-range PCR in the Veriti thermal cycler (Applied Biosystems) by using 50 ng DNA from the pool in a reaction volume of 100 μl containing 5× HF buffer, 200 mM dNTPs, 12 μM of each primer, and 1 unit of Phusion high fidelity polymerase (Finnzymes, Finland). A two-step PCR was performed with an initial denaturation for 30 s at 98°C; this initial denaturation was followed by 30 cycles of denaturation for 10 s at 98°C, an extension for 90 s at 72°C, and a final extension at 72°C for 10 min. Amplicons were purified with the QIAGEN Clean-up kit (QIAquick, QIAGEN Nordic, Sweden), the concentration was determined with NanoDrop (NanoDrop Technologies), and an equal copy number of each amplicon was used in an amplicon pool. We used the amplicon pool from a population to generate a fragment library, and we sequenced this by using 50 bp reads on the SOLiD3 system.
Analysis of Sequence Reads
The SOLiD system-analysis pipeline tool (corona lite) was used for the alignment of the SOLiD reads to a reference consisting of the DNA sequence in the FADS region; up to four mismatches were allowed for each 50 bp read. SNP identification and estimation of allele frequencies in the resequenced pools of individuals were performed as described earlier.37
Analysis and Validation of Rare Variants in the FADS Region
The analysis of rare variants was carried out with the C-alpha statistic as recently described.38 This resulted in 24 rare SNPs that showed significant differences between the individuals with high and low levels of PC 36:4 (Table S3). Fourteen of these 24 SNPs were validated with TaqMan genotyping assays on the individual DNA samples. The assays included 1,308 individuals from four of the populations: 459 from VIS, 192 from ERF, 160 from MICROS, and 497 from NSPHS. For VIS and NSPHS, a cross-section of all individuals was selected and included those that were resequenced in the high and low pools. For ERF and MICROS, only the individuals from the pooled resequencing were included.
Analysis of Effects of Rare Variants with Respect to Haplotypes A and D
In order to elucidate the independent effects of rare variants, we stratified the data on the basis of the haplotypes of the 1,308 genotyped individuals (AA or DD). The allele frequencies for the rare SNPs ranged between 0.04 and 0.50 for the 180 AA individuals. For the 492 individuals in the DD group, the corresponding allele frequencies were considerably lower (0–0.004). Thus, the rare variants are predominantly found on haplotype A. Among individuals homozygous for haplotype A, no statistically significant effect was detected for any of the 14 rare variants (p > 0.1, Wilcoxon-rank sum test, one-sided).
Analysis of the Geographical Distribution of the Two FADS Haplotypes
We used genotype data from all individuals present in CEPH-obtained samples from the Human Genome Diversity Panel39 (HGDP; 2008, final release) to study the frequencies of haplotypes A and D in native populations distributed all over the world. Four of the 28 SNPs that distinguish between the two haplotypes were present in the HGDP data. Each chromosome in the HGDP data was classed as haplotype A, D, or mixed on the basis of the four genotyped SNPs. A similar analysis was performed for individuals of African (ASW [African ancestry in Southwest USA], LWK [Luhya in Webuye, Kenya], and YRI [Yoruba in Ibadan, Nigeria]), European (FIN [Finnish in Finland), GBR [British from England and Scotland, UK], CEU [Utah residents with ancestry from northern and western Europe from the CEPH collection], and TSI [Toscans in Italy]), and Asian ancestry (CHS [Han Chinese South, China], CHB [Han Chinese in Beijing, China], and JPT [Japanese in Tokyo, Japan]) from HapMap40 and the 1,000 Genomes Project.41 For these population samples, the haplotype analyses were based on all of the 28 SNP positions.
Haplotype Evolution Analysis
We used data from primates and archaic hominins to examine the evolutionary history of haplotypes A and D (see Table S2). For chimpanzees, we obtained sequence variants from in-house whole-genome sequence data on the SOLiD 3 system. Two individuals were sequenced at 7× and 13× coverage, respectively, and the reads were aligned to the chimpanzee reference sequence (panTro2). The Denisovan sequence originates from high-coverage Illumina sequencing (30×) of a finger bone from one individual in a recently discovered archaic hominin group.42 The Neanderthal sequence variants were inferred from Illumina sequencing of bones from three different individuals (Vi33.16, Vi33.25, and Vi33.26).43
Age Estimate of Haplotype D
Our estimate of the age of haplotype D is based on a 30 kb region (chr11: 61,324,000–61,354,000) with low heterozygosity in HGDP populations in Africa and America (see Figure S3). As a proof of concept, we first aimed at determining the mutation rate in the FADS region on the basis of a human-chimpanzee comparison. Therefore, we aligned SOLiD mate-pair reads from two chimpanzees to the human reference sequence (hg18) and identified fixed nucleotide differences. The alignment and variant calling were done by two different strategies; the first was with Bioscope and diBayes software, and the second was with Mosaik and SAMtools.44 We required the variants to be detected by both methods. This analysis revealed 154 nucleotide differences within the 30 kb window for one of the chimpanzees and 100 nucleotide differences for the other. The chimpanzee with the higher number of nucleotide differences was sequenced at 13× coverage compared to 7× coverage for the other individual. The correlation between coverage and number of identified variants indicates that some of the nucleotide differences in one of the chimpanzees were not detected because of insufficient coverage. We therefore used the higher number of variants (154) as a measurement of the number of fixed nucleotide differences between humans and chimpanzees. Assuming that 154 mutations have accumulated on the human and chimpanzee lineages since the time of the last common ancestor about 5 million years ago and assuming an average generation time of 20 years, the mutation rate for the region was estimated at 1.03 × 10−8 mutations per base per generation. Our mutation rate is very similar to the estimate from a recent whole-genome sequencing study of a family quartet; in this study, the intergeneration mutation rate was estimated at ∼1.1 × 10−8 per position.45
SNP information in the 30 kb region was extracted for seven human individuals that have been sequenced at high coverage; five of these individuals have a DD genotype, and two have a AA genotype. The five DD individuals are all from Africa; KB1 and ABT are from Southern Africa,46 NA19238 and NA19293 are the parents from a YRI trio sequenced as part of the 1,000 Genomes Project,41 and NA18507 is a HapMap individual also from YRI. The two individuals with the AA genotype are one Paleo-Eskimo (Saqqaq)47 and one Australian Aboriginal.48 We calculated the pair-wise nucleotide differences between all seven human samples, the Denisovan sample, and the 13× chimpanzee sample, and we applied the UPGMA (unweighted pair-group method of analysis) hierarchical clustering as implemented in the phangorn R package.49 The phylogenetic analysis revealed two distinct clusters on the human lineages, one with the individuals homozygous for haplotype A and the other with individuals homozygous for haplotype D. We then counted the number of mutations—within each of the DD and AA clusters—that are not present in other branches in the tree. On average, there were 8.4 nucleotide differences between individuals within the DD group and 20 nucleotide differences between individuals within the AA cluster. From these numbers of mutations, we estimated a divergence time of 255,000 years ago for haplotype D and 606,000 years ago for haplotype A. We also performed similar calculations on the basis of the minimum and maximum number of nucleotide differences within the DD group, and this calculation resulted in a range from 212,000 to 303,000 years ago. In these calculations, we assumed a mutation rate of 1.03 × 10−8 mutations per base per generation (as estimated above) and an average generation time of 20 years.
Furthermore, we used the Bayesean method BEAST50 as an alternative approach to estimate divergence times, and this resulted in an age of 433,000 years ago for haplotype D. One plausible explanation for the discrepancy is that the BEAST program overestimated the age because of input data. BEAST requires the two haploid FADS sequences from each individual as input, and it is impossible to deduce these sequences from the short read data. As a result, the program cannot distinguish between homozygous and heterozygous positions. This probably leads to an overestimate because all variant nucleotides (present in a heterozygous state as is the case for the individuals carrying the haplotype D in the study) are assumed to be present on the same haplotype.
Results
Two Common FADS Haplotypes
To determine the haplotype structure in the FADS region, we used SNP-array genotype data from 5,652 individuals from five European local population cohorts (EUROSPAN) and data from the 1,000 Genomes Project41 to impute additional SNPs, and we phased the chromosomes. The combined data for the five populations showed the presence of two major linkage disequilibrium (LD) blocks, one spanning FADS1 and the first part of FADS2 (block 1) and another spanning the second part of FADS2 (from exon 6 to the 5′UTR) (block 2) (Figure 1). The same LD pattern was found in all five European cohorts.
Figure 1.
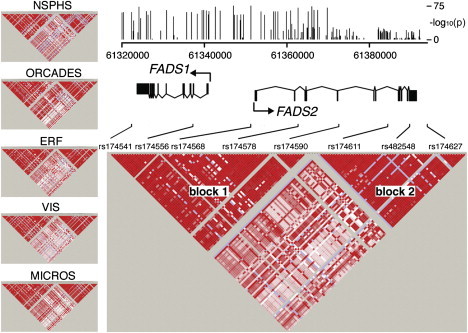
LD Pattern in the FADS Region
LD display of the five population cohorts from Sweden (NSPHS), Scotland (ORCADES), The Netherlands (ERF), Croatia (VIS), and Italy (MICROS) (left) and of all individuals combined (bottom right). Color schemes in all LD maps are based on the standard (D'/LOD) option in the Haploview software. The genomic coordinates on chromosome 11 (hg18) are shown at the top right, and the locations of eight SNPs are drawn out as positional guides. The vertical black bars show p values for each individual SNP; p values range from 1 to 10−75 and represent the association with the lipid PC 36:4. See Table S1 for a complete list of p values for all SNPs in the region.
We studied the association between these blocks and lipid levels by using a lipid (phosphatidylcholine [PC] 36:4) measured in our five European population cohorts. A meta-analysis of the individuals with the highest and lowest levels of this lipid (n = 952) showed that the most significant SNP associations (p < 10−60) were located in block 1 (see Figure 1 and Table S1). We used phased chromosome data to identify in block 1 a set of 28 SNPs (Table S2) that most clearly distinguished the two groups of individuals with high and low lipid levels. A similar analysis for block 2 did not produce a strongly associated set of SNPs (data not shown). Because of this and the fact that the p values were considerably lower in block 1 than in block 2 (see Figure 1), we focused our attention on block 1. The two main haplotypes identified on the basis of the 28 SNPs in block 1 accounted for over 95% of the chromosomes in the five populations; the remaining chromosomes could be accounted for by a set of rare haplotypes that were combinations of the two major ones (Table 1). The most common haplotype, denoted haplotype D, was associated with high lipid levels (p = 1 × 10−65), whereas the less common haplotype (haplotype A) was associated with low levels (p = 1 × 10−52).
Table 1.
Frequency of FADS Haplotypes in European Populationsa
| Haplotype |
SNP |
Frequency (%) | Association (p Value) |
|---|---|---|---|
| 1234567890123456789012345678 | |||
| D | CCCTCGTTAATCTTAAAAACCACCCTAG | 62.1 | 1 × 10−65 |
| A | agtcgaccggctccgggggttgaaaaga | 33.0 | 1 × 10−52 |
| M1 | CgtcCGccggTCTTggAggttgaaaaga | 2.8 | 3 × 10−04 |
| M2 | agtcgaccggctccgggggttgaCaaga | 0.5 | 9 × 10−02 |
| M3 | agtcgaccggTtccgggggttgaaaaga | 0.3 | 2 × 10−01 |
| M4 | CCCTCGTTAATCTTAAAAACCgaaaaga | 0.2 | 4 × 10−02 |
| Total frequency | 98.9 | ||
The haplotypes are defined by 28 SNPs in block 1. The association between FADS haplotypes and lipid levels (PC 36:4) in the five European cohorts is shown in the far right column. Nucleotides on haplotypes D and A are represented by uppercase and lowercase letters, respectively.
From the European Special Population Research Network (EUROSPAN).
To determine whether there could exist in block 1 other strongly associated haplotype structures that were obscured by the presence of haplotypes A and D, we removed the 28 SNPs from the input data and repeated the same analysis procedure for the remaining SNPs in block 1. This resulted in a haplotype consisting of only two SNPs (rs174541 and rs174583). These two SNPs were in very strong LD with the previously identified 28 SNPs. Our analysis thus revealed that in block 1, there were additional significant SNPs that would have been included among the set of 28 if slightly more relaxed filtering criteria had been used. However, in block 1, there are no other independent haplotypes that have strong association with PC 36:4.
To investigate the effect of SNPs not present on the genotyping arrays or in the 1,000 Genomes Database, we designed amplicons for the FADS region and performed deep sequencing by using the SOLiD system in two pools of individuals from each of our five populations (Figure S1). Each pool consisted of a minimum of 90 individuals with either the highest or lowest levels of PC 36:4. From the targeted resequencing, we identified a number of additional variants, but none of these showed as large a difference in frequency between the extreme lipid groups as the 28 SNPs previously found to distinguish haplotypes A and D, described above. However, by applying the C-alpha test38 for analyses of rare variants in high-throughput sequencing data, we detected in the FADS region several low-frequency SNPs that differed significantly between the high- and low-lipid pools (Table S3). Genotyping of 14 of the significant low-frequency SNPs in 1,308 individuals showed that all of them were predominantly located on haplotype A (Table S4). No association was seen for any of the 14 SNPs when the analysis was adjusted for the effects of haplotypes A and D. Our analyses thus show that the association of SNPs with lipid levels can be entirely explained by the two common haplotypes and that there is no independent effect of rare variants.
Effect of FADS Haplotypes on PUFA Metabolism
We studied the functional difference between the two haplotypes by measuring the levels of eight PUFAs in the omega-3 and omega-6 pathways in blood plasma from the NSPHS cohort (Table 2). In both the omega-3 and omega-6 pathways, haplotype D was strongly associated with lower levels of the precursors in fatty-acid synthesis (LA and ALA) and higher levels of EPA, GLA, DHA, and AA (the products), indicating that this haplotype is more efficient in converting the precursors to LC-PUFAs (Figure 2 and Table 2). Individuals homozygous for haplotype D had 24% higher levels of DHA and 43% higher levels of AA than those homozygous for haplotype A. Analysis of the ratios of consecutive products in fatty-acid synthesis (i.e., GLA/LA, ETA/ALA, AA/DGLA, and EPA/ETA) showed that both the δ-5 and δ-6 desaturase steps are affected by the FADS haplotype (Table 2).
Table 2.
Associations between FADS Haplotypes and PUFAs in the NSHPS Cohort
| Common Name | p Value (mmscore)a | Effect of Haplotype D | |
|---|---|---|---|
| Omega-6 | |||
| FA18:2(n-6) | LA | 0.052 | −0.12 |
| FA18:3(n-6) | GLA | 1.3 × 10−18 | 0.56 |
| FA20:3(n-6) | DGLA | 0.26 | 0.07 |
| FA20:4(n-6) | AA | 5.2 × 10−18 | 0.50 |
| Ratio GLA/LAb | 1.6 × 10−27 | 0.68 | |
| Ratio AA/DGLAb | 1.8 × 10−11 | 0.40 | |
| Omega-3 | |||
| FA18:3(n-3) | ALA | 0.024 | −0.14 |
| FA20:4(n-3) | ETA | 2.2 × 10−05 | −0.25 |
| FA20:5(n-3) | EPA | 1.1 × 10−12 | 0.37 |
| FA22:6(n-3) | DHA | 8.3 × 10−05 | 0.20 |
| Ratio ETA/ALAb | 0.042 | −0.12 | |
| Ratio EPA/ETAb | 1.0 × 10−24 | 0.61 | |
The p values represent significance of differences in PUFA levels and have been corrected for relatedness between individuals. The following abbreviations are used: LA, linoleic acid; GLA, gamma-linoleic acid; DGLA, dihomo-gamma-linoleic acid; AA, arachidonic acid; ALA, alpha-linoleic acid; ETA, eicosatetraenoic acid; EPA, eicosapentaenoic acid; and DHA, docosahexaenoic acid.
Mixed model to adjust for relatedness among individuals; Bonferroni-adjusted p values.
Ratios (product/substrate) for the FADS1- or FADS2-catalyzed steps in fatty-acid synthesis.
Figure 2.
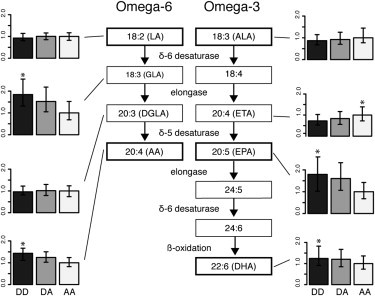
Effect of Haplotype on Synthesis of PUFAs in the Omega-3 and Omega-6 Pathways
Measurements of the omega-3 and 6 fatty-acid levels in the NSPHS population. The three bars in each of the smaller plots (labeled DD, DA, and AA) represent levels of fatty acids in individuals homozygous (AA and DD) and heterozygous (DA) for the A and D haplotypes. Fatty-acid measurements have been scaled so that the average levels for the individuals homozygous for haplotype A are set to 1. The error bars represent the upper and lower quartiles for the PUFA measurements. Asterisks (∗) indicate p values < 10−3.
The conversion of ALA to DHA occurs primarily in the liver,51 and the examination of gene-expression levels in liver samples from 195 individuals52 showed that individuals homozygous for haplotype D have significantly higher expression of FADS1 than those homozygous for haplotype A (see Figure S2). This finding is further supported by results from databases, e.g., the eQTL browser, and other publications,53 in which SNPs within this region have been shown to affect the transcription levels of both FADS1 and FADS2. The increase in expression level by haplotype D in liver agrees with the effect that we estimated for haplotype D on the FADS catalyzed steps in fatty-acid synthesis and the generation of LC-PUFAs (Table 2). Thus, the two FADS haplotypes differ both in transcription levels and in their ability to synthesize AA and DHA from their precursors.
The 28 SNPs defining haplotypes A and D span a 38.9 kb region, including the promoter regions of FADS1 and FADS2. Given that we have performed a comprehensive genetic analysis of the region, the difference in the haplotypes' ability to synthesize LC-PUFAs is probably due to one or several of the 28 SNPs that define the haplotypes or, possibly, some nearby genetic variant(s) in LD. Determining which SNP(s) is causative is difficult because of the complete LD in the region, but the fact that several of the SNPs that define the two haplotypes are located at sites where complexes with regulatory molecules have been shown to bind (Figure 3) advocates for a functional significance of one or several of the SNPs in the haplotype block on the transcription levels.
Figure 3.
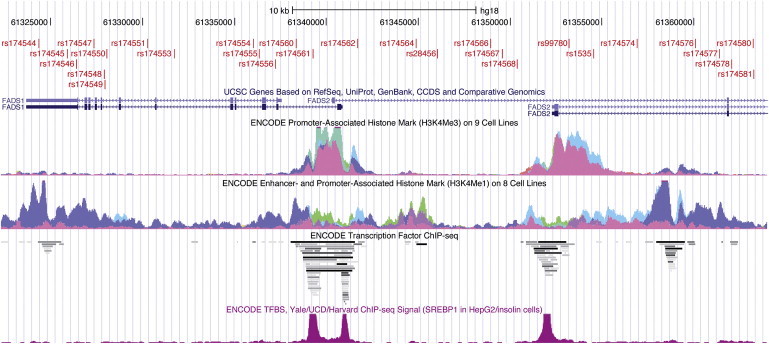
Transcriptional Regulation Elements in the FADS Region
The figure shows a UCSC Genome Browser view of the FADS region; the 28 SNPs define the two haplotypes A and D indicated in red (at the top). The two tracks in the middle show promoter- and enhancer-associated histone marks identified in cell-line studies within the ENCODE project.65 Below is a track displaying transcription-factor binding in the region as identified by ENCODE ChIP-seq experiments. The track at the bottom shows the binding profile for the transcription factor SREBP1 in HepG2 cells; this protein has been shown to affect expression of both FADS1 and FADS2 in mice.66 Many of the SNPs that distinguish haplotype A from D are located inside or in close proximity to the binding sites of regulatory molecules.
Population Distribution and Diversity of FADS Haplotypes
Estimated from the human genome diversity panel (HGDP-CEPH),39 the geographic distributions of haplotypes A and D differ dramatically between continents (Figures 4A and 4B). In African HGDP populations, haplotype A is essentially absent (1% of chromosomes), whereas in Europe, West, South, and East Asia, and Oceania, it occurs at a frequency of 25%–50%. Among the 126 Native Americans included in HGDP, haplotype A accounts for 97% of the chromosomes. A complementary analysis of haplotype frequencies for population samples from HapMap40 and the 1,000 Genomes Project41 confirmed that haplotype A occurs at a very low frequency among individuals of African descent, whereas it is present at moderate to high frequencies in populations of European and Asian ancestry (Figure 4C). Among individuals of African ancestry, 49% carry mixed FADS haplotypes with a higher resemblance to haplotype D than to haplotype A, consistent with a decay of haplotype D by recombination in African populations (Figure 4C).
Figure 4.
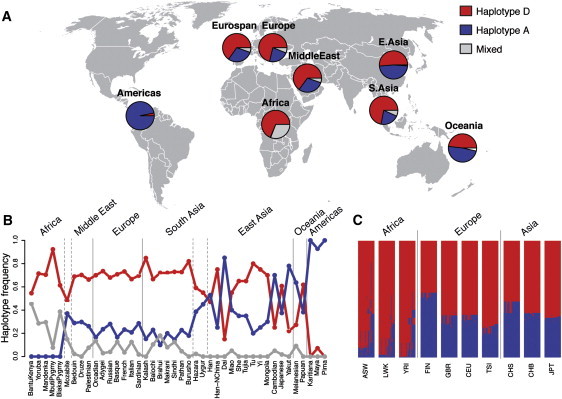
Distribution of FADS Haplotypes in Human Populations
(A) The frequencies of the A (blue) and D (red) haplotypes on different continents are based on four SNPs genotyped in the HGDP populations. The remaining fractions represent mixes (gray) of haplotypes A and D. In Europe, data for the same four SNPs are included for all genotyped individuals in our five local population cohorts (EUROSPAN).
(B) Frequencies of A, D, and mixed haplotypes in HGDP populations in which at least ten individuals have been genotyped.
(C) Frequencies of the 28 SNPs on the FADS haplotypes for ten HapMap and 1,000 Genomes populations of African, European, and Asian ancestry. Phased SNP data for all chromosomes in a population are shown as colored rows. Each row consists of 28 elements, one for each SNP on the two main haplotypes. A SNP is colored blue if it is located on haplotype A and colored red if it is on haplotype D. Mixed haplotypes are represented by horizontal lines that contain both red and blue elements.
Signature of Positive Selection
The very high frequency of haplotype D in Africa and the high LD in the FADS region indicate that this part of the genome has been subjected to positive selection, and this is supported by several lines of evidence from the HGDP data.39 African and American populations show a large reduction in heterozygosity in a 30 kb region encompassing the FADS haplotypes (see Figure S3). The cross-population extended haplotype homozygosity test (XP-EHH), which is designed to detect selective sweeps where the selected allele has approached or achieved fixation in one population but remains polymorphic in the human population as a whole,54 shows a distinct peak in the FADS region in the African Bantu-speaking populations (Figure S3C). Moreover, in a genome-wide analysis that used a composite likelihood ratio (CLR) test of the allele-frequency spectrum,55,56 the FADS region is among the top five candidate-gene clusters that have been under positive selection in African populations.57
Evolutionary History of the FADS Region
Comparative-genomics analyses focusing on the 28 SNPs that distinguish haplotypes D and A showed that rhesus macaques, African apes (chimpanzees and gorillas), and Denisovans42 all have haplotypes that are very similar to haplotype A (Figure 5A and Table S2). The Neanderthal data are based on incomplete sequences from three individuals43 and have nucleotide variants found on both human haplotypes, but overall, they have higher similarity to haplotype A. Our results thus indicate that haplotype D appeared on the lineage leading to modern humans. Intriguingly, the distance between FADS1 and FADS2 has been reduced (though a deletion) from over 75 kb in rhesus macaques and chimpanzees to only 11 kb in humans. This deletion brought the promoters of FADS1 and FADS2 closer to each other, and this might have resulted in coordinated regulation of FADS expression.
Figure 5.
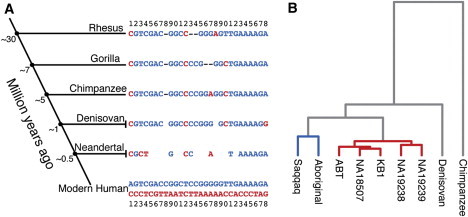
Evolution of FADS Haplotypes
(A) The 28 SNPs distinguishing the two main haplotypes in modern humans are shown at the bottom (haplotype A in red letters, D in blue), and the corresponding nucleotides in primates and archaic hominins are aligned above. The nucleotides for rhesus macaques, gorillas, and chimpanzees are taken from their respective reference genomes (rheMac2, gorGor3, and panTro2). Positions marked by hyphens are missing from the reference assemblies and probably represent deletions. For the archaic hominins, all nucleotides identified by at least ten reads (Denisovan) and two reads (Neanderthal) by Illumina sequencing are shown (see Table S2 for detailed data). Empty cells indicate positions with no sequence-read information as a result of either insufficient coverage or a deletion.
(B) Dendrogram—resulting from a hierarchical clustering via the UPGMA method—of pair-wise nucleotide differences in the FADS region between five DD individuals, two AA individuals, the Denovisan, and one chimpanzee. AA genotypes are depicted with blue branches, and DD genotypes are depicted with red branches.
In order to further understand the evolutionary history of haplotypes A and D, we focused on the 30 kb region of the FADS haplotypes with reduced heterozygosity, and we made use of the data from high-coverage whole-genome sequencing of five African individuals that were all homozygous for haplotype D40,46 and two individuals, one Palaeo-Eskimo47 and one Australian Aboriginal,48 who were both homozygous for haplotype A. For comparison, we also obtained data from a Denisovan and a chimpanzee sequenced at high coverage. For the seven humans, the Denisovan, and the chimpanzee, we then computed all pair-wise nucleotide differences within the 30 kb window and performed a hierarchical clustering by using UPGMA. The results show two clusters in humans, one in which the five African individuals carry the DD genotype and another in which the Eskimo and the Aboriginal carry the AA genotype (Figure 5B). On the basis of the number of SNPs that have accumulated between the DD genotypes, we estimated an age of 255,000 years (range: 212–303 years) for haplotype D diversity. Similarly, based on the variability between AA genotypes, an estimate for haplotype A is an age of 606,000 years. Furthermore, we used the BEAST software50 to analyze the evolutionary history of the haplotypes, and this resulted in an estimate of 433,000 years for the age of haplotype D. Because we were unable to reconstruct the complete haploid sequences that are required as input for BEAST (see Material and Methods), the estimates produced by UPGMA might be more accurate, and the BEAST results should be seen as an upper limit. Our results thus show that haplotype D appeared after the split from Neanderthals (around 500,000 years ago) but prior to the exodus of modern humans from Africa (50,000–100,000 years ago).
Discussion
Single genetic variants in the FADS1 and FADS2 region have previously been associated with lipid-related traits and phenotypes. Our analysis shows the presence of two common human haplotypes with dramatic differences in transcription levels and the ability to synthesize essential omega-3 and omega-6 LC-PUFAs (e.g., AA and DHA). These two haplotypes account for all of the genetic effect seen in FADS activity, and none of the rare SNPs in the region appear to have any additional effect. Haplotype D, which is associated with increased FADS activity, is specific to humans and has appeared on the lineage leading to modern humans well after the split from the common ancestor of humans and chimpanzees. Given that haplotype D has few similarities with the Neanderthal sequences, it does not appear that this human haplotype has been acquired from an archaic species, as proposed for some HLA alleles,58 but that it has evolved on the lineage leading to modern humans.
The age of the diversity seen in haplotype D and the present geographic distribution of this haplotype indicate that both haplotype A and haplotype D were present in Africa at the time of the exodus of modern humans 50,000–100,000 years ago; therefore, both haplotypes were present in European, Asian, and Oceanian populations. The low frequency of haplotype D in the Native American populations included in HGDP indicates that this haplotype might have been lost because of a bottleneck effect in the colonization of the American continent,59,60 possibly in combination with relaxation of the selective pressure as a result of a diet higher in essential LC-PUFAs. It is also possible that haplotype D is more common in other populations on the American continent. In Africa, haplotype D appears to have continued to increase in frequency after the exodus until it reached the present dominating position. Consequently, in the FADS region today, the derived allele (haplotype D) is more frequent in Africa than in other continents; this finding contrasts the more common pattern in which the ancestral allele is more frequent in Africa. Consistent with our results, it has been shown that FADS genetic variants have a stronger effect on PUFA metabolism in African Americans than in Americans of European descent as a result of differences in genotype frequencies.61
We can only speculate about the mechanisms responsible for the increase in frequency of haplotype D. The very rapid increase in the brain size of hominoids probably involved selection on a number of loci and was initiated prior to the appearance of Homo erectus. Such an increase must have resulted in a greater need for LC-PUFAs so that the larger brain volume could be supported. It has been proposed that a shift in diet, characterized by access to food sources that are rich in essential LC-PUFAs, was initiated about 2 million years ago.1,19 This change in the availability of LC-PUFAs might have been important for maintaining the proportionally large hominoid brain relative to body size. Our estimate of the age of haplotype D predates the earliest known anatomically modern humans at about 200,000 years old. The appearance of haplotype D probably did not have any direct effect on brain size per se but might have been selected for because it was highly advantageous under certain circumstances. Humans use a very large portion of dietary fats, predominantly AA and DHA, to feed the brain.1 Consequently, humans' ability to more efficiently synthesize LC-PUFAs from their precursors might have played an important role their ability to survive in periods during which AA- and DHA-rich diets were not available. Haplotype D is likely to have been advantageous to humans living in environments with a limited access to these fatty acids, and this could explain the signature of positive selection seen for this haplotype in African populations.
Regarding the present diet in the Western world, the advantage of having a faster biosynthesis of LC-PUFAs for carriers of haplotype D might have turned into a disadvantage. Because haplotype D increases the biosynthesis of both omega-3 and omega-6 LC-PUFAs, high intake of omega-6 LC-PUFAs augments the amount of AA and thereby the synthesis of arachidonic-acid-derived proinflammatory eicosanoids, which are associated with increased risk of atherosclerotic vascular damage.62 Thus, we hypothesize that the acquisition of a FADS haplotype—which might have been beneficial when food sources rich in the essential LC-PUFAs were in limited supply and when humans had to rely on vegetable oils containing precursors of these fatty acids—would now act as a thrifty genotype and represent a risk factor for lifestyle-related diseases, such as coronary artery disease.18 Consistent with this hypothesis, it has been suggested that differences in the capacity to synthesize LC-PUFA might contribute to health disparities between populations of African and European descent63 and that FADS genotyping should be included as a diagnostic for dietary recommendations.64
Acknowledgments
The SOLiD DNA sequencing and Taqman genotyping were performed by the Uppsala Genome Center, funded by the Knut and Alice Wallenberg foundation (CMS), The Swedish Research Council (SNISS), and Science for Life Laboratory Uppsala. The study was supported by grants from the Swedish Research Council.
Supplemental Data
Web resources
The URLs for data presented herein are as follows:
BioScope and diBayes Software, http://solidsoftwaretools.com
eQTL Browser, http://eqtl.uchicago.edu
Mosaik Alignment Program, http://code.google.com/p/mosaik-aligner/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Leonard W.R., Snodgrass J.J., Robertson M.L. Evolutionary Perspectives on Fat Ingestion and Metabolism in Humans. In: Montmighteur J.P., le Coutre J., editors. Fat Detection: Taste, Texture, and Post Ingestive Effects. CRC Press; Boca Raton, FL: 2010. [PubMed] [Google Scholar]
- 2.Darios F., Davletov B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature. 2006;440:813–817. doi: 10.1038/nature04598. [DOI] [PubMed] [Google Scholar]
- 3.Marszalek J.R., Lodish H.F. Docosahexaenoic acid, fatty acid-interacting proteins, and neuronal function: Breastmilk and fish are good for you. Annu. Rev. Cell Dev. Biol. 2005;21:633–657. doi: 10.1146/annurev.cellbio.21.122303.120624. [DOI] [PubMed] [Google Scholar]
- 4.Ratnayake W.M., Galli C. Fat and fatty acid terminology, methods of analysis and fat digestion and metabolism: A background review paper. Ann. Nutr. Metab. 2009;55:8–43. doi: 10.1159/000228994. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura M.T., Nara T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004;24:345–376. doi: 10.1146/annurev.nutr.24.121803.063211. [DOI] [PubMed] [Google Scholar]
- 6.Aulchenko Y.S., Ripatti S., Lindqvist I., Boomsma D., Heid I.M., Pramstaller P.P., Penninx B.W., Janssens A.C., Wilson J.F., Spector T., ENGAGE Consortium Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009;41:47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hicks A.A., Pramstaller P.P., Johansson A., Vitart V., Rudan I., Ugocsai P., Aulchenko Y., Franklin C.S., Liebisch G., Erdmann J. Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genet. 2009;5:e1000672. doi: 10.1371/journal.pgen.1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kathiresan S., Willer C.J., Peloso G.M., Demissie S., Musunuru K., Schadt E.E., Kaplan L., Bennett D., Li Y., Tanaka T. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatti C., Service S.K., Hartikainen A.L., Pouta A., Ripatti S., Brodsky J., Jones C.G., Zaitlen N.A., Varilo T., Kaakinen M. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka T., Shen J., Abecasis G.R., Kisialiou A., Ordovas J.M., Guralnik J.M., Singleton A., Bandinelli S., Cherubini A., Arnett D. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009;5:e1000338. doi: 10.1371/journal.pgen.1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bokor S., Dumont J., Spinneker A., Gonzalez-Gross M., Nova E., Widhalm K., Moschonis G., Stehle P., Amouyel P., De Henauw S., HELENA Study Group Single nucleotide polymorphisms in the FADS gene cluster are associated with delta-5 and delta-6 desaturase activities estimated by serum fatty acid ratios. J. Lipid Res. 2010;51:2325–2333. doi: 10.1194/jlr.M006205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malerba G., Schaeffer L., Xumerle L., Klopp N., Trabetti E., Biscuola M., Cavallari U., Galavotti R., Martinelli N., Guarini P. SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids. 2008;43:289–299. doi: 10.1007/s11745-008-3158-5. [DOI] [PubMed] [Google Scholar]
- 13.Schaeffer L., Gohlke H., Müller M., Heid I.M., Palmer L.J., Kompauer I., Demmelmair H., Illig T., Koletzko B., Heinrich J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006;15:1745–1756. doi: 10.1093/hmg/ddl117. [DOI] [PubMed] [Google Scholar]
- 14.Zietemann V., Kröger J., Enzenbach C., Jansen E., Fritsche A., Weikert C., Boeing H., Schulze M.B. Genetic variation of the FADS1 FADS2 gene cluster and n-6 PUFA composition in erythrocyte membranes in the European Prospective Investigation into Cancer and Nutrition-Potsdam study. Br. J. Nutr. 2010;104:1748–1759. doi: 10.1017/S0007114510002916. [DOI] [PubMed] [Google Scholar]
- 15.Caspi A., Williams B., Kim-Cohen J., Craig I.W., Milne B.J., Poulton R., Schalkwyk L.C., Taylor A., Werts H., Moffitt T.E. Moderation of breastfeeding effects on the IQ by genetic variation in fatty acid metabolism. Proc. Natl. Acad. Sci. USA. 2007;104:18860–18865. doi: 10.1073/pnas.0704292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lattka E., Rzehak P., Szabó E., Jakobik V., Weck M., Weyermann M., Grallert H., Rothenbacher D., Heinrich J., Brenner H. Genetic variants in the FADS gene cluster are associated with arachidonic acid concentrations of human breast milk at 1.5 and 6 mo postpartum and influence the course of milk dodecanoic, tetracosenoic, and trans-9-octadecenoic acid concentrations over the duration of lactation. Am. J. Clin. Nutr. 2011;93:382–391. doi: 10.3945/ajcn.110.004515. [DOI] [PubMed] [Google Scholar]
- 17.Moltó-Puigmartí C., Plat J., Mensink R.P., Müller A., Jansen E., Zeegers M.P., Thijs C. FADS1 FADS2 gene variants modify the association between fish intake and the docosahexaenoic acid proportions in human milk. Am. J. Clin. Nutr. 2010;91:1368–1376. doi: 10.3945/ajcn.2009.28789. [DOI] [PubMed] [Google Scholar]
- 18.Martinelli N., Girelli D., Malerba G., Guarini P., Illig T., Trabetti E., Sandri M., Friso S., Pizzolo F., Schaeffer L. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 2008;88:941–949. doi: 10.1093/ajcn/88.4.941. [DOI] [PubMed] [Google Scholar]
- 19.Cordain L., Watkins B.A., Mann N.J. Fatty acid composition and energy density of foods available to African hominids. Evolutionary implications for human brain development. World Rev. Nutr. Diet. 2001;90:144–161. doi: 10.1159/000059813. [DOI] [PubMed] [Google Scholar]
- 20.Crawford M.A., Bloom M., Broadhurst C.L., Schmidt W.F., Cunnane S.C., Galli C., Gehbremeskel K., Linseisen F., Lloyd-Smith J., Parkington J. Evidence for the unique function of docosahexaenoic acid during the evolution of the modern hominid brain. Lipids. 1999;34(Suppl):S39–S47. doi: 10.1007/BF02562227. [DOI] [PubMed] [Google Scholar]
- 21.Bansal V., Libiger O., Torkamani A., Schork N.J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 2010;11:773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson A., Marroni F., Hayward C., Franklin C.S., Kirichenko A.V., Jonasson I., Hicks A.A., Vitart V., Isaacs A., Axenovich T., EUROSPAN Consortium Common variants in the JAZF1 gene associated with height identified by linkage and genome-wide association analysis. Hum. Mol. Genet. 2009;18:373–380. doi: 10.1093/hmg/ddn350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McQuillan R., Leutenegger A.L., Abdel-Rahman R., Franklin C.S., Pericic M., Barac-Lauc L., Smolej-Narancic N., Janicijevic B., Polasek O., Tenesa A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudan I., Campbell H., Rudan P. Genetic epidemiological studies of eastern Adriatic Island isolates, Croatia: Objective and strategies. Coll. Antropol. 1999;23:531–546. [PubMed] [Google Scholar]
- 25.Vitart V., Biloglav Z., Hayward C., Janicijevic B., Smolej-Narancic N., Barac L., Pericic M., Klaric I.M., Skaric-Juric T., Barbalic M. 3000 years of solitude: Extreme differentiation in the island isolates of Dalmatia, Croatia. Eur. J. Hum. Genet. 2006;14:478–487. doi: 10.1038/sj.ejhg.5201589. [DOI] [PubMed] [Google Scholar]
- 26.Pattaro C., Marroni F., Riegler A., Mascalzoni D., Pichler I., Volpato C.B., Dal Cero U., De Grandi A., Egger C., Eisendle A. The genetic study of three population microisolates in South Tyrol (MICROS): Study design and epidemiological perspectives. BMC Med. Genet. 2007;8:29. doi: 10.1186/1471-2350-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aulchenko Y.S., Heutink P., Mackay I., Bertoli-Avella A.M., Pullen J., Vaessen N., Rademaker T.A., Sandkuijl L.A., Cardon L., Oostra B., van Duijn C.M. Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 2004;12:527–534. doi: 10.1038/sj.ejhg.5201188. [DOI] [PubMed] [Google Scholar]
- 28.Mascalzoni D., Janssens A.C., Stewart A., Pramstaller P., Gyllensten U., Rudan I., van Duijn C.M., Wilson J.F., Campbell H., Quillan R.M., EUROSPAN consortium Comparison of participant information and informed consent forms of five European studies in genetic isolated populations. Eur. J. Hum. Genet. 2010;18:296–302. doi: 10.1038/ejhg.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liebisch G., Drobnik W., Reil M., Trümbach B., Arnecke R., Olgemöller B., Roscher A., Schmitz G. Quantitative measurement of different ceramide species from crude cellular extracts by electrospray ionization tandem mass spectrometry (ESI-MS/MS) J. Lipid Res. 1999;40:1539–1546. [PubMed] [Google Scholar]
- 30.Liebisch G., Lieser B., Rathenberg J., Drobnik W., Schmitz G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope correction algorithm. Biochim. Biophys. Acta. 2004;1686:108–117. doi: 10.1016/j.bbalip.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Demirkan A., van Duijn C.M., Ugocsai P., Isaacs A., Pramstaller P.P., Liebisch G., Wilson J.F., Johansson A., Rudan I., Aulchenko Y.S., DIAGRAM Consortium. CARDIoGRAM Consortium. CHARGE Consortium. on behalf of the EUROSPAN consortium Genome-Wide Association Study Identifies Novel Loci Associated with Circulating Phospho- and Sphingolipid Concentrations. PLoS Genet. 2012;8:e1002490. doi: 10.1371/journal.pgen.1002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aulchenko Y.S., Ripke S., Isaacs A., van Duijn C.M. GenABEL: An R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 35.Chen W.M., Abecasis G.R. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 37.Zaboli G., Ameur A., Igl W., Johansson A., Hayward C., Vitart V., Campbell S., Zgaga L., Polasek O., Schmitz G., EUROSPAN Consortium Sequencing of high-complexity DNA pools for identification of nucleotide and structural variants in regions associated with complex traits. Eur. J. Hum. Genet. 2012;20:77–83. doi: 10.1038/ejhg.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 40.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reich D., Green R.E., Kircher M., Krause J., Patterson N., Durand E.Y., Viola B., Briggs A.W., Stenzel U., Johnson P.L. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuster S.C., Miller W., Ratan A., Tomsho L.P., Giardine B., Kasson L.R., Harris R.S., Petersen D.C., Zhao F., Qi J. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rasmussen M., Li Y., Lindgreen S., Pedersen J.S., Albrechtsen A., Moltke I., Metspalu M., Metspalu E., Kivisild T., Gupta R. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463:757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasmussen M., Guo X., Wang Y., Lohmueller K.E., Rasmussen S., Albrechtsen A., Skotte L., Lindgreen S., Metspalu M., Jombart T. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schliep K.P. phangorn: Phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott B.L., Bazan N.G. Membrane docosahexaenoate is supplied to the developing brain and retina by the liver. Proc. Natl. Acad. Sci. USA. 1989;86:2903–2907. doi: 10.1073/pnas.86.8.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., International HapMap Consortium Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nielsen R., Williamson S., Kim Y., Hubisz M.J., Clark A.G., Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williamson S.H., Hubisz M.J., Clark A.G., Payseur B.A., Bustamante C.D., Nielsen R. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pickrell J.K., Coop G., Novembre J., Kudaravalli S., Li J.Z., Absher D., Srinivasan B.S., Barsh G.S., Myers R.M., Feldman M.W., Pritchard J.K. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abi-Rached L., Jobin M.J., Kulkarni S., McWhinnie A., Dalva K., Gragert L., Babrzadeh F., Gharizadeh B., Luo M., Plummer F.A. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Titus-Trachtenberg E.A., Rickards O., De Stefano G.F., Erlich H.A. Analysis of HLA class II haplotypes in the Cayapa Indians of Ecuador: A novel DRB1 allele reveals evidence for convergent evolution and balancing selection at position 86. Am. J. Hum. Genet. 1994;55:160–167. [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S., Lewis C.M., Jakobsson M., Ramachandran S., Ray N., Bedoya G., Rojas W., Parra M.V., Molina J.A., Gallo C. Genetic variation and population structure in native Americans. PLoS Genet. 2007;3:e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathias R.A., Sergeant S., Ruczinski I., Torgerson D.G., Hugenschmidt C.E., Kubala M., Vaidya D., Suktitipat B., Ziegler J.T., Ivester P. The impact of FADS genetic variants on ω6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet. 2011;12:50. doi: 10.1186/1471-2156-12-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinelli N., Consoli L., Olivieri O. A ‘desaturase hypothesis’ for atherosclerosis: Janus-faced enzymes in omega-6 and omega-3 polyunsaturated fatty acid metabolism. J Nutrigenet Nutrigenomics. 2009;2:129–139. doi: 10.1159/000238177. [DOI] [PubMed] [Google Scholar]
- 63.Sergeant S., Hugenschmidt C.E., Rudock M.E., Ziegler J.T., Ivester P., Ainsworth H.C., Vaidya D., Case L.D., Langefeld C.D., Freedman B.I. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012;107:547–555. doi: 10.1017/S0007114511003230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koletzko B., Demmelmair H., Schaeffer L., Illig T., Heinrich J. Genetically determined variation in polyunsaturated fatty acid metabolism might result in different dietary requirements. Nestle Nutr. Workshop Ser. Pediatr. Program. 2008;62:35–44. doi: 10.1159/000146246. discussion 44–49. [DOI] [PubMed] [Google Scholar]
- 65.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., ENCODE Project Consortium. NISC Comparative Sequencing Program. Baylor College of Medicine Human Genome Sequencing Center. Washington University Genome Sequencing Center. Broad Institute. Children's Hospital Oakland Research Institute Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsuzaka T., Shimano H., Yahagi N., Amemiya-Kudo M., Yoshikawa T., Hasty A.H., Tamura Y., Osuga J., Okazaki H., Iizuka Y. Dual regulation of mouse Delta(5)- and Delta(6)-desaturase gene expression by SREBP-1 and PPARalpha. J. Lipid Res. 2002;43:107–114. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.