

The kibdelones A-C (1-3) and their isomeric metabolites (cf. isokibdelone C 4) are hexacyclic tetrahydroxanthone natural products recently isolated by Capon and coworkers from the rare Australian actinomycete Kibdelosporangium sp. (Figure 1).[1] An interesting property is the facile equilibration of kibdelones B 2, and C 3 to a mixture of 1-3via keto/enol tautomerizations followed by quinone/hydroquinone redox reactions.[1] Related natural products include simaomicin α 5 which has been shown to sensitize cancer cells to cytotoxic agents including bleomycin at nanomolar concentrations and to exhibit potent antimalarial and anticoccidial activities. [2] Evaluation of the kibdelones in the NCI 60-cell panel of human cancer cell lines revealed that they are active at low nanomolar concentrations against a number of human tumor cell lines. For example, kibdelone A has a GI50 of 1.2 nm against a SR (leukemia) tumor cell line and <1 nm (GI50) against SN12C (renal) cell carcinoma.[1] In addition, the kibdelones were shown to display novel COMPARE analysis profiles for cancer cell growth inhibition.
Figure 1.

Representative Polycyclic Xanthone Natural Products
There has been substantial work towards the synthesis of the polycyclic xanthones including cervinomycin A2 6, but more limited studies on the synthesis of the corresponding tetrahydroxanthones.[3] Our initial strategy to construct the ABCD ring fragment 7 involved Diels-Alder cycloaddition of heterocyclic quinone 8 and hydroxystyrene 9 (Scheme 1). Literature reports have described [4+2] cycloadditions of styrenes and quinones including applications in natural product synthesis.[4] We envisioned that the cycloaddition may proceed through the hydrogen-bonded assembly A (exo cycloaddition geometry shown). There are literature precedents in which intermolecular hydrogen-bonding has been utilized to accelerate and control the stereochemistry of Diels-Alder cycloadditions.[5] In this Communication, we report our efforts directed towards this approach which ultimately led to the serendipitous discovery of a novel Pt(IV)-catalyzed arylation to assemble the ABCD ring fragment of the kibdelones.
Scheme 1.
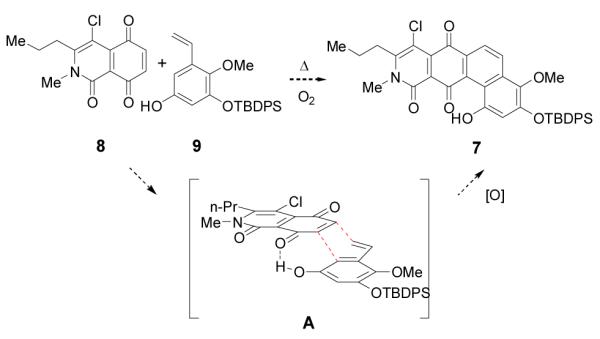
Proposed cycloaddition of heterocyclic quinone 8 and styrene 9
The synthesis of the requisite isoquinoline 8 began with condensation of homophthalic acid 10[6] with butanoic anhydride and pyridine. Treatment of the resulting isocoumarin with methylamine in THF and dehydration afforded isoquinolinone 11.[7] Demethylation of 11 with BBr3 followed by oxidation with Ag2O afforded the heterocyclic quinone 12. To access hydroxystyrene 9, commercially available bromovanillin 13 was demethylated with AlCl3 in pyridine and selectively methylated with Li2CO3 and MeI in DMF [8] which was followed by silyl protection to afford aldehyde 14 (55% yield, 3 steps). Dakin oxidation of 14 with m-CPBA,[9] followed by hydrolysis of the intermediate formate, provided a phenol intermediate which was directly subjected to Stille coupling with vinyl tributylstannane to afford hydroxystyrene 9 (63%, 3 steps).[10]
Unfortunately, attempted thermal and catalyzed Diels-Alder cycloaddition of quinone 12 and hydroxystyrene 9 were unsuccessful, largely due to instability and decomposition of the quinone 12. To improve the stability and handling of the AB quinone reaction partner, we targeted preparation of the corresponding quinone monoketal 16 (Scheme 3). Moreover, we reasoned that 16 may be a candidate for ionic Diels-Alder cycloaddition with styrene 9 under Lewis-acidic conditions.[11] Selective demethylation of isoquinolinone 11 with BCl3, followed by oxidation with iodosobenzene diacetate (PIDA) in methanol, provided quinone monoketal 15 in 90% yield (2 steps). Compound 15 proved to be a suitable substrate for chlorination with catalytic triphenylphosphine (PPh3) and N-chlorosuccinimide [12] enabling access to quinone monoketal 16 in 72% yield.
Scheme 3.
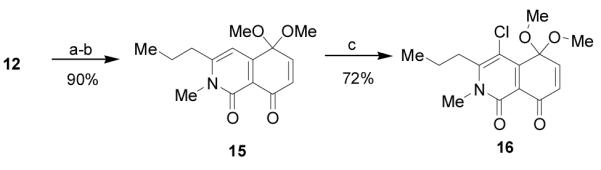
Reagents and conditions: a) BCl3, THF −78 °C-RT b) PIDA, MeOH 0 °C-RT c) NCS, PPh3, DMA. PIDA = Iodobenzene diacetate, NCS = N-chloro-succinimide, DMA =dimethylformamide.
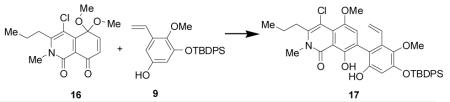
Initial attempts at fragment coupling of 9 and 16 using established ionic Diels-Alder conditions (e.g. TMSOTf in CH3CN) did not lead to observable cycloaddition products. However, under these conditions arylation product 17 was unexpectedly isolated in 15% yield. A report by Sartori and coworkers described use of stoichiometric amounts of Et2AlCl to afford biaryls from quinone monoketals and phenols.[13] Unfortunately latter conditions did not effect conversion of 9 and 16 to biaryl product 17. Since the desired B-D ring connection was established via this unexpected coupling, we conducted an extensive screen of both Brønsted and Lewis acids to improve the yield of the transformation.[6] Following evaluation of reaction conditions, we identified Pt(IV), Au(III), and In(III) catalysts for the arylation (Table 1). AuCl3 and InCl3 provided modest yields (20-25%) of the biaryl product (entries 1-2). Chloroplatinic acid (H2PtCl6-6H2O), bromoplatinic acid (H2PtBr6-9H2O), and PtCl4 (entries 3-5) provided moderate yields of arylation product 17 (25-35%). Ultimately, PtBr4 was found to be optimal providing 2-vinyl biphenyl 17 in yields up to 55% on a multigram scale (Table 1). Interestingly, we also isolated by-product 18, a dimer of the quinone-monoketal, from arylation reactions in 4-6% yield and as the sole product when SnCl4 was employed as a Lewis-acid.[6] The structure of dimer 18 was determined by single X-ray crystal structure analysis [14] (Figure 2).
Table 1.
Evaluation of Conditions for Pt (IV) Arylation
 | |||
|---|---|---|---|
| Entry | Catalyst [a] | time [b] | Yield [c] |
| 1 | AuCl3 (10) | 6 | 25 |
| 2 | InCl3 (10) | 12 | 27 |
| 3 | H2PtCl6-6H2O (5) | 6 | 23 |
| 4 | H2PtBr6-9H2O (5) | 6 | 25 |
| 5 | PtCl4* (5) | 6 | 33 |
| 6 | PtBr4 * (5) | 4 | 55 |
(mol %)
hrs
Isolated yield (%) All reactions run in CH3CN at 65 °C * with 10 mol % water
Figure 2.
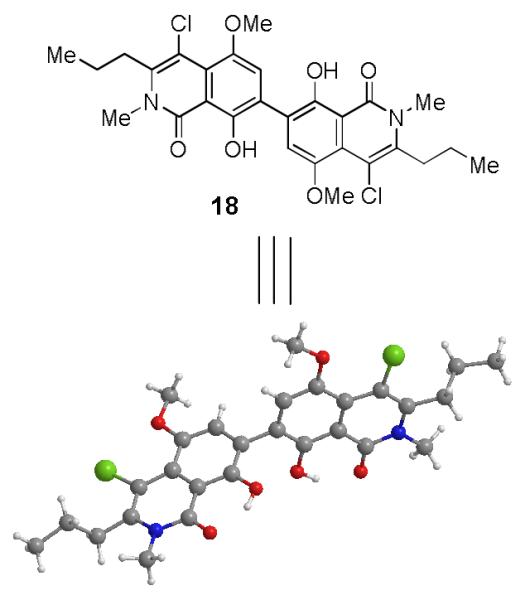
X-Ray Structure of Biaryl Dimer 18
Additional evaluation of the PtBr4 arylation conditions revealed that the presence of water was necessary for catalytic activity. [15] Kobayashi and co-workers have also reported aza-Michael additions of carbamates to enones catalyzed by the platinum aqua complex PtCl4-5H2O.[16] A survey of water stoichiometry using PtBr4 as catalyst and freshly distilled acetonitrile revealed the optimal catalyst:water ratio to be 2:1.[6] A literature report of Pt(IV) carbohydrate complexes indicates that the pKa of water molecules complexed with platinum metal centers can decrease by as much as 13 orders of magnitude.[17b] In addition, a crystal structure of a cis-diaqua-Pt(IV) complex with 18-crown-6 shows that the Pt(IV)-bound water molecules may hydrogen bond to ether oxygens.[17c]
Based on the water requirement for the reaction, we envision two possible activated quinone-monoketal intermediates as shown in Figure 3. In the first case (B), hydrogen bond activation of the ketal may be followed by SN2′ addition to the activated acetal.[18] In the second case (C), dual π-activation of the quinone monoketal by platinum and σ-activation of the quinone monoketal via hydrogen bonding from the platinum-aqua complex may lead to arylation.[19,20] Alternatively, Pt(IV)-aqua complexes may dissociate a water molecule and activate the ketal by direct coordination to Pt(IV).[17a] We believe the regioselectivity of the arylation is influenced by steric bulk of the TBDPS protecting group as formation of the undesired regioisomer is not observed.
Figure 3.
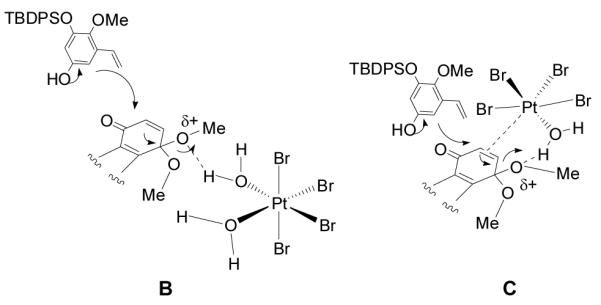
Possible Active Intermediates for Pt(IV)-mediated Arylation
With 2-vinyl biphenyl 17 in hand, we achieved the synthesis of the ABCD ring fragment via photoelectrocyclization[21] in cyclohexane. Silyl-deprotection of the photocyclization product afforded 19 in 61% yield (two steps) (Scheme 3). The structure of dihydrophenanthrene 19 was confirmed by single X-ray crystal structure analysis [22] (Figure 4). We also isolated the corresponding phenanthrene 20 from a gram scale photocyclization in an immersion well in 5% yield. Attempts to conduct the photocyclization of 17 in the presence of oxygen have thus far failed to produce 20 in higher yield. Analysis of molecular models of 17 revealed that the low-energy biaryl conformers are bisected.[6] Optimization of the photocyclization conditions showed higher conversions when the reaction was conducted at room temperature or above. The increased reactivity at elevated temperature may arise from overcoming the rotational barrier of biaryl 17. [21]
Figure 4.
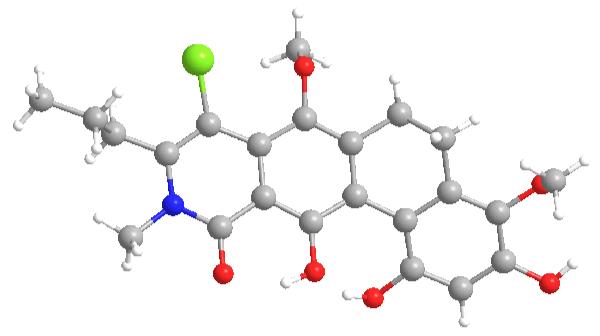
X-ray crystal structure of 19
To elaborate protected dihydrophenanthrene 21 to a B-ring quinone, we first attempted selective demethylation using standard conditions, (e.g. BBr3, BCl3, TMSI, and AlCl3) (Scheme 4). Unfortunately, these conditions were found to be unselective, with mixtures of both B and C ring demethylated products observed. Fortunately compound 21 could be selectively acylated in aprotic solvents due to the strongly hydrogen bonded B-ring phenol. Oxidative cleavage with CAN in acetonitrile/pH 7.0 buffer followed by desilylation afforded the tetracyclic quinone 22. Oxidation of the B vs. D ring was confirmed by HMBC experiments.[6] Further experiments revealed that 23 was sensitive to basic deacylation conditions. Fortunately, treatment of 23 with Otera’s distannoxane catalyst[23] in toluene/methanol afforded tetracycle 23, a compound bearing the ABCD core of kibdelone B in 85% yield. In the presence of aqueous sodium dithionite, the B ring of 23 was cleanly reduced to afford hydroquinone 24 in 90% yield, a compound analogous to the ABCD ring structure of kibdelone C 3.
Scheme 4.
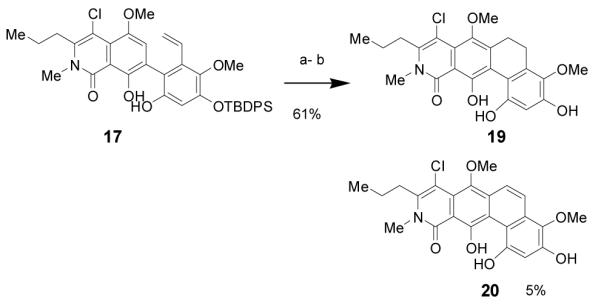
Reagents and conditions: a) hν (Pyrex filter), cyclohexane; b) TBAF, THF 0 °C. TBAF = tetrabutylammonium fluoride.
Compounds 19, 20, 23, and 24 were evaluated in the NCI 60-cell screen.[24] Compounds 19 and 20 were judged to be inactive, with mean cell growth of 82% and 93% of control, respectively, at 10 μM. Compounds 23 and 24 passed the criteria for testing in dose-response format, with mean cell growths of 63% and 54%, respectively. In dose-response format, both 23 and 24 bearing the ABCD functionalities of kibdelones B and C, respectively, showed mean GI50 values of 4.5 μM while having no selectivity. In comparison, kibdelone C (3), the most potent of the natural products in the kibdelone series, had a mean GI50 of 2.4 nM, with 100-fold selectivity between the most and least sensitive cell lines tested. Thus, it is apparent that the contribution of the E and F rings of the kibdelone structure is highly important for cytotoxic activity.
In summary, a proposed Diels-Alder cycloaddition to construct the ABCD ring structure of the kibdelones led to the discovery of a novel Pt(IV)-mediated arylation of quinone monoketals to produce a complex 2-vinyl biphenyl adduct. Photocycloaddition and further oxidation of this compound was used to access ABCD analogs of the kibdelones. Analogs were evaluated for initial biological activity in the NCI 60-cell screen and were found to be ~2000 times less active in comparison to kibdelones B and C, suggesting that the tetrahydroxanthone structure of the kibdelones may be crucial for cytotoxicity. Studies towards the completion of the total synthesis of the kibdelones, as well as mechanistic studies and substrate scope of the Pt(IV) arylation of quinone monoketals, is ongoing and will be reported in due course.
Scheme 2.
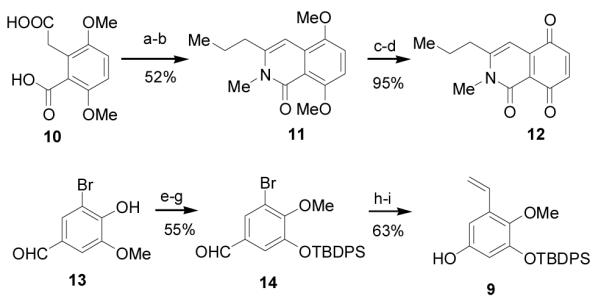
Reagents and conditions: a) 2:1 pyridine: butyric-anhydride reflux; b) 2.0M MeNH2THF RT, CSA, toluene, 85 °C; c) BBr3, THF −78 °C-rt; d) Ag2O, CHCl3; e) AlCl3, pyridine, CH2Cl2 ; f) Li2CO3, MeI, DMF 45 °C; g) TBDPS-Cl, DMAP, TEA, CH2Cl2; h) MCPBA CH2Cl2, NaHCO3, MeOH 0 °C; i) vinyl tributyltin, Pd(PPh3)4, toluene 85 °C. ; THF = tetrahydrofuran, CSA = camphor sulfonic acid, DMAP = 4-dimethylaminopyridine, MCPBA = m-chloroperoxybenzoic acid.
Scheme 5.
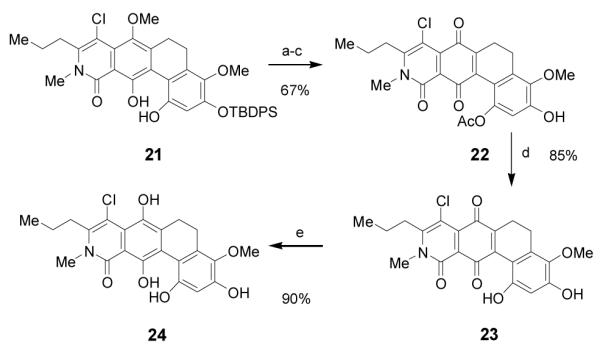
Reagents and conditions: a) acetyl chloride, DIEA CH2Cl20 °C; b) CAN, CH3CN/ pH 7 buffer 0 °C; c) HF-pyrdine, THF 0 °C d) Otera’s catalyst, 2:1 toluene:MeOH 70 °C; e) Na2S2O4 H2O:EtOAc. DIEA = disopropylethylamine, THF = tetrahydrofuran CAN = ceric ammonium nitrate.
Footnotes
Financial support from the National Institutes of Health (R01 CA137270) and Merck Research Laboratories is gratefully acknowledged. We thank Andrew Little and Dr. Stéphane Roche for extremely helpful and stimulating discussions, and Drs. Richard Ball and Jeff Bacon for X-ray crystallographic data.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
David L. Sloman, Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston, MA 02215, USA
Branko Mitasev, Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston, MA 02215, USA.
Stephen S. Scully, Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston, MA 02215, USA
Dr. John A. Beutler, Molecular Targets Laboratory, Bldg. 1052 Room 110, NCI at Frederick Frederick, MD 21702, USA, beutlerj@mail.nih.gov
Prof. John A. Porco, Jr., Department of Chemistry, Center for Chemical Methodology and Library Development (CMLD-BU), Boston University, 590 Commonwealth Avenue, Boston, MA 02215, USA.
References
- [1].a) Ratnayake R, Lacey E, Tennant S, Gill JH, Capon RJ. Org. Lett. 2006;8:5267–5270. doi: 10.1021/ol062113e. [DOI] [PubMed] [Google Scholar]; b) Ratnayake R, Lacey E, Tennant S, Gill JH, Capon RJ. Chemistry--A European Journal. 2007;13:1610–1619. doi: 10.1002/chem.200601236. [DOI] [PubMed] [Google Scholar]; c) Capon RJ, Lacey E, Ratnayake R, Gill JH, Tennant S. PCT Int. Appl. 2007 WO 2007095696.
- [2].a) Lee TM, Carter GT, Borders DB. J. Chem. Soc. Chem. Commun. 1989;22:1771–1772. [Google Scholar]; b) Carter GT, Goodman JJ, Torrey MJ, Borders DB, Gould SJ. J. Org. Chem. 1989;54:4321–4323. [Google Scholar]; c) Maiese WM, Korshalla J, Goodman J, Torrey MJ, Kantor S, Labeda DP, Greenstein M. J. Antibiotics. 1990;43:1059–1063. doi: 10.7164/antibiotics.43.1059. [DOI] [PubMed] [Google Scholar]; d) Arai M, Sato H, Kobayashi H, Suganuma M, Kawabe T, Tomoda H, Omura S. Biochem. Biophys. Res. Comm. 2004;317:817–822. doi: 10.1016/j.bbrc.2004.03.117. [DOI] [PubMed] [Google Scholar]
- [3].cervinomycin A2: Omura S, Nakagawa A, Kushida K, Shimizu H, Lukacs GJ. Antibiot. 1987;40:301–308. doi: 10.7164/antibiotics.40.301. Tanka H, Kawakita K, Suzuki H, Nakagawa PS, Omura SJ. J. Antibiot. 1989;42:431–439. doi: 10.7164/antibiotics.42.431. Mehta G, Venkateswarlu J. Chem Soc., Chem. Commun. 1988:1200–1202. Kelly TR, Jagoe CT, Li Q. J. Am. Chem. Soc. 1989;111:4522–4524. Rao AV, Yadav JS, Reddy K, Upender V. Tetrahedron Lett. 1991;32:5199–5202. Yadav JS. Pure Appl. Chem. 1993;65:1349–1356. Mehta G, Shah SR, Venkateswarlu Y. Tetrahedron. 1994;50:11729–11742. lysolipin. Duthaler RO, Wegmann UH. Helv. Chim. Acta. 1984;67:1755–11766. Walker ER, Leung SY, Barrett AGM. Tetrahedron Lett. 2005;46:6537–6540. Sch 56036.
- [4].Manning WB, Kelly TP, Muschik GM. J. Org. Chem. 1980;45:2535. [Google Scholar]; b) Muschik GM, Kelly TP, Manning WB. J. Org. Chem. 1982;47:4709. [Google Scholar]; c) Tanga MJ, Reist EJ. J. Heterocycl. Chem. 1991:28–29. [Google Scholar]; e) Willmore ND, Hoic DA, Katz TJ. J. Org. Chem. 1994;59:1889. [Google Scholar]; f) Carreño MC, Mahugo J, Urbano A. Tetrahedron Lett. 1997;38:3047. [Google Scholar]; g) Katz TJ, Liu L, Willmore ND, Fox JM, Rheingold AL, Shi S, Nuckolls C, Rickman BH. J. Am. Chem. Soc. 1997;119:10054. [Google Scholar]; h) Krohn K, Loock U, Paavilainen K, Hausen BM, Schmalle WH, Kiesele H. ARKIVOC. 2001;2:88–130. [Google Scholar]
- [5].a) Fisher MJ, Hehre WJ, Kahn SD, Overman LE. J. Am. Chem. Soc. 1988;110:4625. [Google Scholar]; b) Tripathy R, Carroll PJ, Thornton ER. J. Am. Chem. Soc. 1990;112:6743. [Google Scholar]; c) Sorgi KL, McNally JJ, Leo GC. J. Org. Chem. 1994;59:5088. [Google Scholar]; d) Atherton JCC, Jones S. Tetrahedron Lett. 2001;42:8239. [Google Scholar]; e) Huang Y, Rawal VH. J. Am. Chem. Soc. 2002;124:9662. doi: 10.1021/ja0267627. [DOI] [PubMed] [Google Scholar]; f) Domingo LR, Aurell MJ, Arno M, Saez JA. J. Org. Chem. 2007;72:4220–4227. doi: 10.1021/jo070373j. [DOI] [PubMed] [Google Scholar]
- [6].See the Supporting Information for complete experimental details.
- [7].Ahmed H, Rama N, Malana M, Qadeer G. Indian J. Chem, Sec B. 2006;45B:820–822. [Google Scholar]
- [8].a) Wyman WE, Davis R, Patterson JW, Pfister JR. Synthetic Communications. 1988;18:1379–1384. [Google Scholar]; b) Masuno MN, Pessah IN, Olmstead MM, Molinski TF. J. Med. Chem. 2006;49:4497–4511. doi: 10.1021/jm050708u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lanfranchi DA, Hanquet G. J. Org. Chem. 2006;71:4854–4861. doi: 10.1021/jo060486n. [DOI] [PubMed] [Google Scholar]
- [10].Matos M-C, Murphy PV. J. Org. Chem. 2007;72:1803–1806. doi: 10.1021/jo062159l. [DOI] [PubMed] [Google Scholar]
- [11].a) Gassman PG, Singleton DA, Wilwerding JJ, Chavan SP. J. Am. Chem. Soc. 1987;109:2182–2184. [Google Scholar]; b) Schildknegt K, Aube J. J. Chemtracts-Organic Chemistry. 1996;9:237–241. [Google Scholar]; c) Sanghi R, Vankar PS, Vankar YD. J.Indian Chem. Soc. 1998;75:709–715. [Google Scholar]; c) Witulski B, Alayrac C. Science of Synthesis. 2007;25:405–422. [Google Scholar]
- [12].Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- [13].For arylation of quinone monoketals with phenols, see: Sartori G, Maggi R, Bigi F, Grandi M. J. Org. Chem. 1993;58:7271–7273. Sartori G, Maggi R, Bigi F, Giacomelli S, Porta C, Arienti A, Bocelli G. J. Chem. Soc. Perkin Trans.1. 1995:2177–2181.
- [14].CCDC 793693 contains the supplementary crystallographic data for (18). This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- [15].For examples of water as an additive in organic transformations, see: Ribe S, Wipf P. Chem. Commun. 2001:299–307. Yamamoto H, Futatsugi K. Angew. Chem. Int. Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394.
- [16].Kobayashi S, Kakumoto K, Sugiura M. Org. Lett. 2002;4:1319–1322. doi: 10.1021/ol0256163. [DOI] [PubMed] [Google Scholar]
- [17].a) Junicke H, Bruhn C, Strohl D, Kluge R, Steinborn D. Inorg. Chem. 1998;37:4603. doi: 10.1021/ic9801692. [DOI] [PubMed] [Google Scholar]; b) Junicke H, Bruhn C, Kluge R, Serianni AS, Steinborn DJ. Am. Chem. Soc. 1999;121:6232–6241. [Google Scholar]; c) Wagner C, Bruhn C, Gravenhorst O, Steinborn DJ. Z. Kristallogr. 2000;215:61–64. [Google Scholar]
- [18].For SN2′ addition to allylic acetals, see: Ghribi A, Alexakis A, Normant JF. Tetrahedron Lett. 1991;32:5199–5202. Gomez-Bengoa E, Heron NM, Didiuk MT, Luchaco CA, Hoveyda AH. J. Am. Chem. Soc. 1998;120:7649–7650. For a review on carbophilic π activation of platinum see: Fürstner A, Davies PW. Angew. Chem. Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.
- [19].For dual activation by oxophilic/carbophilic catalysts see: Asao N, Kasahara T, Yamamoto Y. Tetrahedron Lett. 2001;42:7903–7905. Kamijo S, Yamamoto Y. J. Org. Chem. 2003;68:4764–4771. doi: 10.1021/jo034254p. Yanada R, Obika S, Kono H, Takemoto Y. Angew. Chem. Int. Ed. 2006;45:3822–3825. doi: 10.1002/anie.200600408. Yamamoto Y. J. Org.Chem. 2007;72:7817–7831. doi: 10.1021/jo070579k.
- [20].For Pt(IV) alkene complexes, see; Parsons EJ, Larsen RD, Jennings PW. J. Am. Chem. Soc. 1985;107:1793–1794.
- [21].For photocyclizations of 2-vinyl biphenyls see: Horgan SW, Morgan DD, Orchin M. J. Org. Chem. 1973;38:3801–3803. Koussini R, Lapouyade R, Fornier de Violet P. J. Am. Chem. Soc. 1978;100:6679–6683. Fornier de Violet P, Bonneau R, Lapouyade R, Koussini R, Ware WR. J. Am. Chem. Soc. 1978;100:6683–6687. Lewis FD, Zuo X. Photochem. Photobiol. Sci. 2003;2:1059–1066. doi: 10.1039/b305831j.
- [22].CCDC 793694 contains the supplementary crystallographic data for (19). This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. See the Supporting Information for complete experimental details.
- [23].Li J, Chen X, Burgett AWG, Harran P. Angew. Chem. Int. Ed. 2001;40:2682–2685. doi: 10.1002/1521-3773(20010716)40:14<2682::AID-ANIE2682>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [24].a) Boyd MR, Paull KD. Drug Dev. Res. 1995;34:91–109. [Google Scholar]; b) Shoemaker RH. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]