

Abstract
Histone deacetylases (HDACs) and microRNAs (miRs) have pro-survival roles, but the mechanism behind this is unclear. Repression of ceramide synthase 1 (CerS1), altering C18-ceramide generation, was linked to drug resistance and metastasis. Here we report that the CerS1 promoter was repressed by HDAC1-dependent inhibition of Sp1 recruitment to two specific GC-boxes spanning the −177 and −139 region. Moreover, an alternatively spliced variant CerS1 mRNA (CerS1-2) was detected mainly in cancer cells or primary tumour tissues compared to controls, which was targeted by miR-574-5p for degradation. A specific 3′UTR-targeting site, localized within the retained intron between exons 6 and 7, was identified, and its mutation, or miR-574-5p knockdown prevented the degradation of CerS1-2 mRNA. Interference with HDAC1 and miR-574-5p reconstituted CerS1-2 expression and C18-ceramide generation in multiple human cancer cell lines, which subsequently inhibited proliferation and anchorage-independent growth. Accordingly, knockdown of CerS1 partially protected cancer cells from MS-275/miR-574-5p siRNA-mediated growth inhibition. Thus, these data suggest that the HDAC1/miR-574-5p axis might provide a novel therapeutic target to reconstitute tumour suppressor CerS1/ceramide signalling.
Keywords: ceramide synthase 1, lipid signalling, sphingolipids, sphingosine, sphingosine 1-phosphate
INTRODUCTION
HDAC1 belongs to class I histone deacetylases (HDACs), along with HDAC2, HDAC3 and HDAC8. HDAC1 plays important roles in cancer pathogenesis, and several HDAC inhibitors (HDAC-Is), including MS-275, which has some specificity against HDAC1, have been used in clinical trials as an anti-cancer drug (Bolden et al, 2006; Marks, 2010). Also, microRNAs (miRs) are known to target mRNAs at their 3′UTRs for rapid degradation, leading to either tumour proliferation or suppression (Croce, 2009; Garzon et al, 2010). For example, increased miR-574-5p was detected in the serum of lung cancer patients, providing a potential cancer-specific biomarker (Foss et al, 2010). However, mechanisms by which HDAC1 and miR-574-5p influence cancer growth are unknown.
Sphingolipids are structural membrane lipids that also act as bioeffector molecules, and ceramide is the central molecule in sphingolipid metabolism (Ogretmen & Hannun, 2004), which is mainly generated via the hydrolysis of sphingomyelin by sphingomyelinases, or via the de novo pathway involving ceramide synthases 1–6 (CerS1–6; Pewzner-Jung et al, 2006). CerS1–6 regulate the generation of ceramides with distinct fatty acid chain lengths. For example, whereas, CerS1 mainly generates C18-ceramide, CerS6 is generally responsible for C16-ceramide generation. In fact, low C18-ceramide, which was highly associated with decreased CerS1 mRNA, was significantly linked to lymph node metastasis and vascular invasion in head and neck squamous cell carcinoma (HNSCC) patients (Mizutani et al, 2005). Similarly, knockdown of CerS1 resulted in cisplatin resistance (Min et al, 2007). In contrast, reconstitution of CerS1 either by its ectopic expression or by treatment with chemotherapy induces C18-ceramide generation, leading to tumour suppression (Senkal et al, 2007).
Although there have been several important studies which revealed the mechanisms behind regulation of CerS1 protein and/or function (Spassieva et al, 2006; Sridevi et al, 2010), mechanisms that control CerS1 gene expression in human cancer cells have not been described previously. In this study, our data revealed that a novel, two-pronged repressive mechanism involving epigenetic promoter suppression by HDAC1 and post-transcriptional mRNA targeting by miR-574-5p, coordinates the inhibition of alternatively spliced CerS1 isoform 2 (CerS1-2) expression in multiple human cancer cells and primary tumour tissues, providing a novel mechanism by which HDAC1 and miR-574-5p exhibit pro-survival roles. The data indicated that interference with HDAC1 and miR-574-5p using molecular and/or pharmacological tools reconstitutes CerS1-2, and C18-ceramide generation, leading to the inhibition of cancer cell growth and/or proliferation.
RESULTS
Cloning and characterization of the human CerS1 and CerS6 promoters
To determine whether down-regulation of CerS1 mRNA in the majority of HNSCC tumour tissues compared to their adjacent normal head and neck tissues (Karahatay et al, 2007; Koybasi et al, 2004) is also consistent in cell culture conditions, we measured CerS1 and CerS6 mRNA in multiple human cancer cells (UM-SCC-22A, UM-SCC-14A and UM-SCC-1) compared to non-cancerous keratinocytes using quantitative-polymerase chain reaction (Q-PCR). Consistent with HNSCC primary tumours, CerS1 was down-regulated about 10–20-fold in HNSCC cell lines compared to normal human epidermal primary keratinocytes (NHEK) or HPV-E6/E7-immortalized human keratinocytes controls (Fig 1A and B). CerS6 mRNA was similar in HNSCC cell lines and immortalized keratinocytes (Fig 1B). Down-regulation of CerS1 was also associated with lower C18-ceramide, measured with liquid chromatography/mass spectrometry (LC/MS/MS), in UM-SCC-1 and -22A (Fig 1C) compared to immortalized keratinocytes (about 90 and 50%, respectively). Thus, these studies indicate that CerS1/C18-ceramide, but not CerS6/C16-ceramide is down-regulated in HNSCC cancer cell lines compared to non-cancerous keratinocytes, consistent with decreased CerS1/C18-ceramide in HNSCC tumour compared to non-cancerous head and neck tissues (Karahatay et al, 2007; Koybasi et al, 2004).
Figure 1. Regulation of CerS1 versus CerS6 mRNA and promoter activities in HNSCC versus keratinocytes.

A-B. CerS1 mRNA was detected by Q-PCR in primary NHEK (A) or in HPV E6/E7-immortalized skin keratinocytes (B), compared to UM-SCC-1 and UM-SCC-22A cells by Q-PCR.
C. Ceramide measurements were performed in immortalized skin keratinocytes compared to UM-SCC-1 and UM-SCC2A cells using LC/MS/MS. Data were normalized to Pi.
D-E. CerS1 (D) and CerS6 (E) promoter activities were measured using luminometry, normalizing transfection efficiency to β-gal expression using spectrometry. Transcription factor binding sites were predicted using the TFBind software. Samples were run in duplicates at least three-independent times. The error bars represent the standard deviations.
To determine whether the transcriptional down-regulation of CerS1 is due to alterations in its promoter activity, we first cloned the functional promoter using genomic DNA isolated from non-cancerous Wi-38 fibroblasts and UM-SCC-22A cells. A region that contains a promoter was identified to span between +1 (transcriptional start site) and −1556 (Fig S1A of Supporting Information) of the CerS1 gene (http://genome.ucsc.edu/cgi-bin/hgGateway), and it was sub-cloned upstream of luciferase in the pGL3 reporter vector. Similarly, we also cloned the CerS6 promoter as an additional control, spanning +1 to −1625 of the CerS6 gene (Fig S1A of Supporting Information). The sequencing of the CerS1 promoter clones obtained using the genomic DNAs from non-cancerous compared to HNSCC cancer cells did not show any detectable mutations (data not shown). Then, we transiently co-transfected HNSCC cells with the CerS1 or CerS6 promoter-luciferase reporter vectors, and full-length beta-galactosidase (β-gal) expression vector (used to normalize transfection efficiencies) to measure their activities. The promoter activity of CerS1 was approximately 80–90% decreased compared to CerS6 promoter activity in UM-SCC-22A, UM-SCC14A or UM-SCC-1 cells (Fig S1B of Supporting Information), consistent with relative mRNA content in these cells (see Fig 1A).
We next identified the functional core promoter of CerS1 and CerS6 by generating serial deletion mutations, which were then independently sub-cloned upstream of luciferase in the pGL3-luciferase reporter vector for co-transfections in HNSCC cells. The core activity of CerS1 was localized between the −436/200 and −80 regions of the promoter, because the deletion mutants containing −1180, −772 or −80 regions of the CerS1 promoter constructs did not show any significant increase in the promoter activity compared to the full-length promoter (−1556; Fig 1D). However, the promoter mutants spanning between −436/300/200 had around 3-, 7- and 10-fold increased promoter activity, respectively, compared to the full-length promoter (Fig 1D). In addition, deletion of the region between −436 and +1 (−1556–437) completely blunted CerS1 promoter activity, which is consistent with the core promoter activity localized within the −436 and +1 region of the promoter. Using transcription factor binding software (http://tfbind.hgc.jp/), multiple transcription factor recognition sequences, including Sp1/Sp3, AP2, E2A, CREB, HIF-1 or STAT (Fig 1D) were predicted to bind to this region. In addition, these data also suggest that the optimal core promoter activity is localized within the −200 and −81 region of the CerS1 promoter (Fig 1D), which mainly contains Sp1/Sp3 and AP2-recognition sites (Fig 1D). Similar experiments were also performed using serial deletion mutants of the CerS6 promoter. These data showed that unlike the CerS1 promoter, optimal core promoter activity of CerS6 was localized between −1625 and −486 in UM-SCC-22A cells (Fig 1E), which is consistent with the presence of the CAAT box within −998 and −1625 region of the promoter. Thus, these data suggest that the regulation of the CerS1 and CerS6 promoter activities are regulated by distinct mechanisms involving their unique promoter regions.
Regulation of the core CerS1 promoter by Sp1
We then defined the roles of AP2 or Sp1/Sp3 transcription factors, whose recognition sequences are present in the optimal core promoter of CerS1, spanning the −200 and −80 region. To determine whether AP2 and/or Sp1/Sp3 play a role in the regulation of CerS1 promoter activity, their expression was down-regulated using siRNAs, and then effects of their knockdown on the promoter activity of CerS1 (containing the +1 to −200 or +1 to −300 regions) were examined. Knockdown of AP2-α and -γ using siRNAs, which decreased their expression approximately 60–80% compared to scrambled (Scr) non-targeting siRNAs, measured by Q-PCR, did not significantly affect CerS1 promoter activity (data not shown). However, knockdown of Sp1 using siRNAs decreased its expression approximately 70%, as measured by Western blot (Fig 2A), and caused a significant decrease (around 50%, p < 0.05) in CerS1 promoter activity (Fig 2B), which was also consistent with decreased CerS1 mRNA (Fig 2C), measured by Q-PCR, in response to knockdown of Sp1. In reciprocal experiments, overexpression of wt-Sp1-HA, but not wt-Sp3-HA, enhanced the core promoter activity of CerS1 (containing the +1 to −200 or +1 to −300 regions) around 3.5- or 2-fold, respectively, compared to vector-transfected controls (Fig 2D). Expression of an inactive mutant of Sp1-HA (Moorefield et al, 2004), whose transactivation/DNA-binding domain was exchanged with the DNA-binding domain of Sp2 (Sp1/2), did not have any significant effect on the CerS1 promoter activity (Fig 2D). Expression of Sp1, Sp3 and Sp1/2, containing HA-tags at their C-termini, were confirmed by Western blotting using an anti-HA antibody compared to controls (Fig 2E, lanes 2–5 and 1, respectively). Actin served as a loading control (Fig 2E, lower panel). In addition, because ceramide decreased the acetylation of Sp3 at the K551 residue by HDAC1 (Wooten-Blanks et al, 2007), which then down-regulated the promoter of telomerase reverse transcriptase (hTERT), we examined effects of ectopic expression of wt-Sp3 and its acetylation-deficient (K551Q) or acetylation-mimic (K551R) mutants (Wooten-Blanks et al, 2007) on CerS1 promoter activity in UM-SCC-22A cells. Expression of Sp3 or its acetylation mutants (K551Q or K551R) had no significant effect on the CerS1 promoter (Fig S2A of Supporting Information). Expression of wt-, K551Q- or K551R-Sp3 containing an HA-tag on their C termini, was also confirmed by Western blotting (Fig S2B of Supporting Information). Overall, these data suggest that Sp1, but not Sp3, plays an important role in the transactivation of the CerS1 core promoter.
Figure 2. The CerS1 promoter and mRNA activation by Sp1.
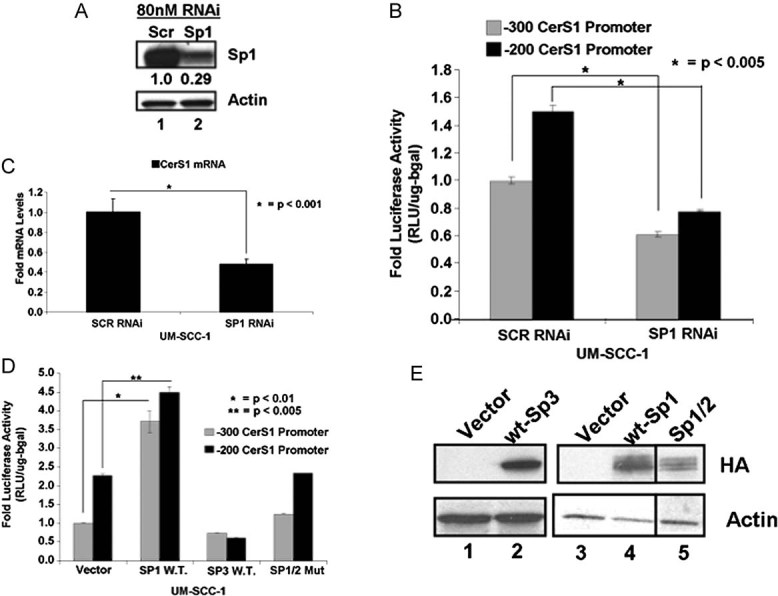
A. Cells were transfected with Sp1 siRNAs and Sp1 protein in UM-SCC-1 was measured by Western Blotting. Beta-actin was used as a control.
B-C. Effects of Sp1 siRNAs on CerS1 promoter activity (B) or mRNA (C) were measured by luciferase activity assay or Q-RT-PCR, respectively. Relative quantities were normalized to ribosomal RNA.
D. CerS1 promoter activity upon over-expression of Sp1, Sp3 or Sp1/2 mutant over-expression (n = 3).
E. HA-tagged proteins were measured upon over-expression of Sp-family proteins in UM-SCC-1 cells with Western blotting. Beta-actin was used as a loading control. Experiments were performed at least in three-independent trials as duplicates. The error bars represent the standard deviations.
Determination of specific Sp1-binding sites involved in the transactivation of the CerS1 core promoter
The optimal core promoter DNA of CerS1 spanning between the −200 and −80 region contains four conserved GC-box Sp1-recognition sequences centred at the −200, −177, −139 and −107 regions, which were referred to as GC-Boxes 1–4, respectively (Fig 3A). To delineate specific roles of these four Sp1-recognition sites in the regulation of the CerS1 promoter, we first generated serial deletion mutants of these GC-box sequences (Fig 3A), and then determined their effects on CerS1 promoter activity in UM-SCC-22A cells using luciferase reporter assays, as described above. Data indicate that deletion of GC-Box 1 had no significant effect on CerS1 core promoter activity (spanning the +1 to −200 region), whereas, deletion of the GC-Boxes 1–2 and 1–2–3 significantly suppressed the CerS1 promoter in an additive manner (about 2.5- and 5-fold, respectively, p < 0.05) compared to the wt (−200) and GC-Box-1-deleted controls (Fig 3B). Deletion of the GC-Box 4 had no additional effect on CerS1 promoter inhibition, compared to the deletions of GC-Boxes 1–2–3 (Fig 3B). Thus, these data suggest that Sp1-recognition sites referred to as GC-Boxes 2 and 3, but not 1 and 4, play important roles in the activation of the core CerS1 promoter, under basal expression conditions. It should also be noted that other transcription factors, such as E2A, CREB, HIF-1 or STAT, whose recognition sites are present on the CerS1 minimal promoter or other factors that associate with the promoter distant regulatory sites, might affect CerS1 transcription in various other cell types and stress conditions.
Figure 3. Determination of specific Sp1-binding/recognition sites for the activation of the CerS1 core promoter.
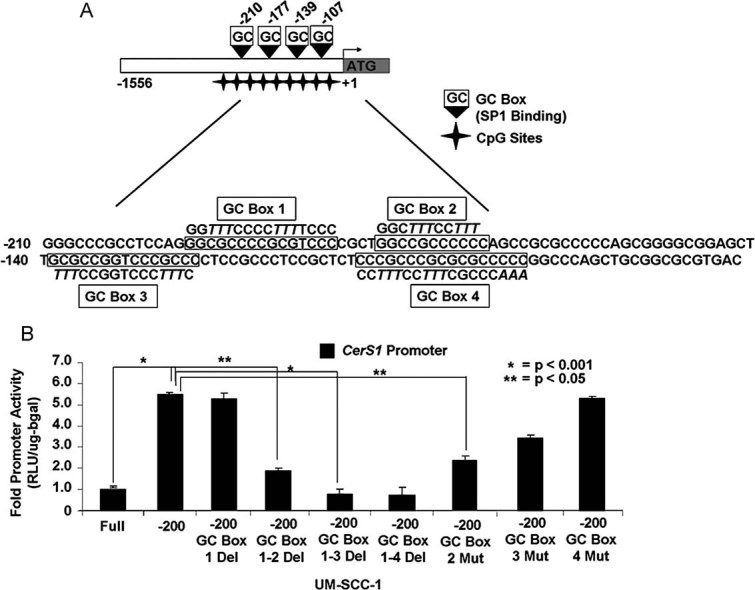
- Transcription factor binding map denoting four potential GC Boxes located within the CerS1 promoter. Sequences in italics are mutated regions of the promoter used in the GC Box mutant constructs.
- Effects of GC Box deletion/mutations on CerS1 promoter activity was measured using luminometry in UM-SCC-1 cells in at least three-independent experiments as triplicates. The error bars represent the standard deviations.
To define specific roles of these Sp1-recognition sequences (GC-boxes) individually in the regulation of the CerS1 promoter, we also generated additional mutants in which Sp1-recognition sequences at the GC-Boxes 1 (data not shown), 2–4 were mutated by replacing their GC-rich sequences by multiple TTT arrays without deletions (Fig 3A). Then, we examined the effects of these individual mutations on the CerS1 promoter. The data showed that mutations of the GC-Boxes 1 (data not shown) or 4 (Fig 3B) had no significant effect on the CerS1 promoter, but mutations of GC-Box 2 or 3 inhibited the promoter approximately 70 or 50%, respectively, compared to controls (Fig 3B). Thus, these data support the specific roles of GC-Boxes 2 and 3 on the regulation of the CerS1 promoter activity.
Then, to assess whether the GC-Boxes 2 and 3 bind Sp1 for the regulation of the CerS1 promoter, we performed EMSA using nuclear extracts isolated from UM-SCC-1 cells, and the [32P]-labelled oligonucleotide (22-mer) containing a conserved Sp1-recognition sequence as a probe with and without unlabelled oligonucleotides containing the Sp1 (cold probe), GC-Boxes 1, 2 or 3 sequences or their mutated variants as competitors. Incubation of the labelled Sp1 probe with the UM-SCC-1 nuclear extracts generated two main shifted bands (complexes 1 and 2), which were competed off when unlabelled wt-probe, but not its mutant, were used as competitors (Fig 4A, lanes 2–4, respectively). As expected, co-incubation of the labelled Sp1 probe with the wt- or mutant GC-Box-1 oligonucleotides had no effect on probe binding with C1 or C2 compared to probe-only control (Fig 4A, lanes 5–6 and 2, respectively). Conversely, while unlabelled oligonucleotides containing wt-GC-Boxes 2 and 3 almost completely prevented the Sp1-C1/C2 binding, their mutated isoforms had no effect compared to probe-only control (Fig 4A, lanes 7–10, compared to lane 2, respectively). We also confirmed that C1/C2 complexes indeed associate with the Sp1 transcription factor using a super-shift assay in the presence of an anti-Sp1 or anti-IgG antibodies in EMSA assays. As seen in Fig 4B, pre-incubation of nuclear extracts with the anti-Sp1 antibody, but not the anti-IgG antibody, caused a super-shift on the mobility of the probe-protein complex (C1), indicating that the C1 contains Sp1-probe complex (Fig 4B). Taken together, both functional (promoter mutations) and biochemical (EMSA) studies suggest that Sp1 induces CerS1 core promoter with the specific involvement of GC-rich sequences within the −177 and −139 regions (GC-Boxes 2 and 3) of the promoter.
Figure 4. Endogenous Sp1 specifically binds CerS1 promoter at the GC Boxes 2 and 3, whose recruitment to the promoter is modulated by HDAC-dependent histone deacetylation.

- UM-SCC-1 nuclear extracts were isolated as previously described (Ogretmen and Safa, 2000) and pre-incubated with competitor unlabelled oligonucleotides before incubation with P32-Sp1 consensus oligonucleotides.
- Pre-incubation of nuclear extracts with a-Sp1 antibody, but not non-specific a-IgG antibody, caused a super-shift in the protein/DNA complex, indicating Sp1-specific binding to oligonucleotides creates C1 and C2.
- Sp1 association with the endogenous CerS1 core promoter region was assessed using Q-ChIP. Relative quantities normalized to 1% input DNA before pull-down with Sp1 or IgG antibody.
- Cells plated in a 6-well plates (4 × 105 cells/well) were treated with TSA (200 ng/ml) for 24 h, and CerS1 promoter activity was measured by luciferase assay.
- Sp1 and acetylated histone-H3 (Ac-Histone-H3) association with the core promoter was assed after treatment with 200 ng/ml TSA for 24 h in UM-SCC-1 via Q-ChIP. Sp1 and Ac-Histone-H3 relative quantities normalized to 1% input DNA and IgG controls. These data represent at least two-independent experiments. The error bars represent the standard deviations.
Identification of the binding of endogenous Sp1 with the CerS1 promoter in HNSCC cells versus keratinocytes to assess possible epigenetic regulation
Then, we determined whether down-regulation of CerS1 transcription is linked to alteration of endogenous Sp1 recruitment to the promoter in UM-SCC-1 cells compared to keratinocytes using quantitative-chromatin immuno-precipitation (Q-ChIP). The data showed that, whereas Sp1 was highly associated with the CerS1 promoter in keratinocytes, Sp1 was undetectable on the CerS1 promoter in UM-SCC-1 cells (Fig 4C). There were no differences in the Sp1 protein content, its phosphorylation and/or acetylation status or it's nuclear versus cytoplasmic localization in UM-SCC-1 compared to keratinocytes (data not shown). These data suggested to us that epigenetic mechanisms, such as histone deacetylation by HDACs, might be involved in the inhibition of Sp1-dependent CerS1 promoter activity in cancer cells. Accordingly, treatment of UM-SCC-1 cells with the pan HDAC inhibitor, trichostatin (TSA), induced the CerS1 promoter (Fig 4D) compared to untreated controls. Moreover, TSA treatment also increased the association of Sp1 and acetylated histone H3 with the CerS1 promoter DNA in UM-SCC-1 cells, determined by Q-ChIP (Fig 4E). Thus, these data indicate that although Sp1 is an activator of the core CerS1 promoter, its recruitment to the endogenous CerS1 promoter is significantly reduced in UM-SCC-1 cells compared to keratinocytes possibly due to HDAC-dependent histone deacetylation and repression of the CerS1 promoter.
Moreover, we determined which HDAC was involved in the repression of the CerS1 promoter in HNSCC cells. First, UM-SCC-1 cells were treated with the known HDAC1-specific inhibitor MS-275, a pan class I-HDAC inhibitor BML-210 or a class II-HDAC inhibitor dPAHA (Yang & Seto, 2008), and then their effects on CerS1 promoter activity were examined. Whereas, the class II-HDAC inhibitor dPAHA had no significant effect, inhibition of class-I HDACs using BML-210 or HDAC1 using MS-275 enhanced the CerS1 promoter by about 3- or 4-fold, respectively (Fig 5A). Accordingly, MS-275 treatment increased Sp1 recruitment to the CerS1 promoter DNA compared to controls (Fig 5B), as determined by Q-ChIP. Thus, these data suggest that a class I HDAC, possibly HDAC1 might be involved in the repression of the CerS1 promoter in HNSCC cells. Indeed, knockdown of HDAC1, but not other class-I HDACs (HDAC2, HDAC3 or HDAC8) using siRNAs (decreasing their target gene mRNA expression by about 50–90%, as determined by Q-PCR, Fig S3A of Supporting Information) increased the activity of the CerS1 core promoter (Fig 5C). Thus, these data suggest that the CerS1 promoter is repressed mainly by an HDAC1-dependent mechanism, and that inhibition of HDAC1 using MS-275 or HDAC1-siRNAs activates the CerS1 promoter.
Figure 5. Regulation of CerS1 expression via decreased mRNA stability in HNSCC versus keratinocytes.

A. UM-SCC-1 cells in 6-well plates (4 × 105 cells/well) were treated with 10 µM BML-210, 10 µM dPAHA or 5–10 µM MS-275 for 24 h. CerS1 promoter activity was measured by luciferase assay.
B. Effects of MS-275 (10 µM for 24 h) on the association of Sp1 with the endogenous CerS1 promoter DNA were determined using Q-ChIP. Relative quantities of Sp1 were normalized to 1% input DNA and IgG controls.
C. Effects of knockdown of HDAC1, HDAC2, HDAC3 and HDAC8 using siRNAs on CerS1 promoter activity were measured in UM-SCC-22A cells.
D-E. The half-life of CerS1 mRNA in immortalized keratinocytes versus UM-SCC-1 or UM-SCC-22A cells, or in primary NHEK versus UM-SCC-22A cells were measured using Q-PCR in the absence/presence of DMSO or Act D (25 µg/ml) for 0.5–6 h. GAPDH mRNA was used as a control. These data represent at least two-independent trials performed in duplicates. The error bars represent the standard deviations.
Post-transcriptional regulation of CerS1 mRNA in HNSCC versus keratinocytes
Effects of HDAC inhibitors, which increased the CerS1 promoter activity by approximately 2- to 7-fold, respectively, on CerS1 mRNA were examined using Q-PCR in UM-SCC-1 cells. Interestingly, TSA, the class I HDAC inhibitor BML-210, or the HDAC1 specific inhibitor MS-275 treatment did not significant increase CerS1 mRNA compared to untreated controls (Fig S3B of Supporting Information). Similar data were also obtained using HDAC1 siRNAs, or overexpression of wt-Sp1, which did not increase CerS1 mRNA compared to controls (data not shown). Thus, these data suggest that in addition to epigenetic HDAC1-dependent repression of the CerS1 promoter, CerS1 transcription might also be modulated by post-transcriptional mechanisms. To test this hypothesis, we determined the half-life of CerS1 mRNA in keratinocytes versus UM-SCC-1 and -22A cells using Actinomycin D (Act D) at various time points. The data showed that the half-life of CerS1 was around 1.5 h in UM-SCC-22A or UM-SCC-1 cells, respectively, whereas its half-life was >2 h in immortalized keratinocytes (Fig 5D), or NHEK (Fig 5E), which was around 6 h (data not shown). Thus, these data suggest that the half-life of CerS1 mRNA is decreased in HNSCC cells compared to non-cancerous keratinocytes.
We then determined the mechanisms involved in the decreased stability of CerS1 mRNA in HNSCC cells compared to keratinocytes. It is known that CerS1 has two possible alternatively spliced variants. CerS1-1 is a bi-cistronic mRNA containing both CerS1 and GDF1 (Wang et al, 2007), growth and differentiation factor 1, coding sequences (Fig 6A). CerS1/GDF1 bi-cistronic mRNA is controlled by a common promoter element at the upstream of the 5′-end of the CerS1 gene (exons1–7), and by a 3′UTR poly-adenylation (polyA) sequence (AATAAA), present at the 3′-end of the GDF1 gene. According to mirBase, there are no known miR-target sequences at the 3′UTR of the CerS1/GDF1 bicistronic pre-mRNA (CerS1-1). The alternatively spliced variant of CerS1-2 contains exons 1–6, but not exon 7 and the intronic sequence between exons 6 and 7 contains six putative poly(A)-signals (A1–A6), indicating that this intronic sequence might be utilized as a 3′UTR of the CerS1-2 mRNA (Fig 6A). In fact, analysis of the CerS1-2 3′UTR sequence revealed that there is a hsa-miRNA-574-5p-target sequence upstream of the putative A5–A6 poly-A sequences (Fig 6A and Fig S4A of Supporting Information). This finding suggested that the expression of CerS1-2 mRNA might be regulated by miR-574-5p.
Figure 6. CerS1-2 mRNA containing a miR-574-5p target site within its 3′UTR region is predominantly expressed in HNSCC versus keratinocytes.

A. CerS1/GDF1 bicistronic mRNA transcript with six potential polyA signal sequences within the intronic region between exons 6 and 7 of the CerS1 mRNA transcript is shown. The miR-574-5p target site is located upstream of the polyA sequence 5.
B. Expression of CerS1-1 spanning exons 2–7, or CerS1-exon 7/GDF1-exon 1 juncture were detected using RT-PCR in keratinocytes, UM-SCC-1 and/or UM-SCC-22A cells (left and right panels, lanes 1–3 or 1 and 2, respectively).
C. Expression of CerS1-2 containing 3′UTR sequences down-stream of polyA signal sequences without (3′UTR #1, upper panel), or without a potential miR-574-5p binding site (3′UTR #5, middle panel) were measured using RT-PCR in keratinocytes versus UM-SCC-1 and UM-SCC-22A cells.
D. Relative expression of CerS1-1, CerS1-2 containing 3′UTR #1 and CerS1-2 containing 3′UTR #5 mRNAs were measured using RT-PCR in UM-SCC-1 and UM-SCC-22A cells compared to immortalized keratinocytes.
E-F. Levels of CerS1 (E) and miR-574-5p (F) mRNAs were measured with Q-PCR and RT-PCR in primary HNSCC tumour tissues compared to the paired and adjacent pathologically non-cancerous head and neck tissues, respectively. These data represent at least two-independent trials performed as duplicates. The error bars represent the standard deviations.
To define the expression of CerS1-1 and CerS1-2 in non-cancerous versus cancerous cells, we first examined their mRNAs by PCR (after reverse transcription; RT-PCR) in keratinocytes versus UM-SCC1 and UM-SCC-14A cells. Two different primer-pair sets were designed for CerS1-1: one set spans CerS1 exon 7 and GDF1 exon 1 junction, while the second set spans exons 2–7 of CerS1 (Fig 6B, right and left panels, respectively). These data showed that CerS1-1 mRNA was down-regulated around 80–90% in UM-SCC-1 and UM-SCC-14A cells compared to keratinocytes (Fig 6B, left and right panels, lanes 2–3 and 1, or 2–1, respectively), consistent with the Q-PCR data (Fig 1A), which detected both CerS1-1 and CerS1-2 mRNAs at exon 2. Next, to characterize the expression of CerS1-2 mRNA with specific polyA sites, six primer-pair sets were designed spanning exon 2-and A1–A6, putative poly(A) sequences within the retained intronic sequence between exons 6 and 7. The data showed that, whereas, keratinocytes mainly express CerS1-1 mRNA (Fig 6B), or CerS1-2 containing the A1-polyA sequence (Fig 6C, upper panel, lanes 1 and 2–3, respectively), HNSCC cells express CerS1-2 mRNA spanning the A5-polyA sequence (Fig 6C, middle panel, lanes 2–3 and 1, respectively), which contains the miR-574-5p-target sequence. We did not detect any CerS1-2 transcripts spanning exon 2 and A2–A4 or A6 in keratinocytes or cancer cells (data not shown). The relative expression of CerS1-1, CerS1-2(A1) and CerS1-2(A5) between in UM-SCC-1 or UM-SCC-22A compared to keratinocytes was 0.2, 0.1 and 0.8, respectively (Fig 6D). These data suggest that while keratinocytes mainly express CerS1-1 and CerS1-2 with the A1 polyA signal, cancer cell lines mainly express CerS1-2 with the A5 polyA sequence, which contains a putative target sequence for miR-574-5p.
To assess the in vivo (clinical) relevance of CerS1-1 versus miR-574-5p-targeted CerS1-2 mRNA, we measured mRNA in 10 primary tumour tissues obtained from HNSCC patients compared to their adjacent pathologically non-cancerous head and neck tissue pairs using Q-PCR and RT-PCR. As expected, CerS1 mRNA was down-regulated in the majority (9/10) of HNSCC tumour tissues compared to controls (Fig 6E). Interestingly, while CerS1-1 (without the miR-574-5p target sequence) was mainly expressed in normal tissues, miR574-5p-targeted CerS1-2 (with the A5-polyA) was expressed in all of the primary tumour tissues (Fig S4B of Supporting Information). In fact, when miR-574-5p was examined using Q-PCR, it was observed that miR-574-5p was detected in 10/10 of the HNSCC tumours, and its levels were higher in 5/10 of HNSCC tumours compared to their paired controls (Fig 6F). Interestingly, in patient 6, decreased miR-574-5p correlated with increased CerS1-2 mRNA (Fig 6E and F). These data were also consistent in primary keratinocytes, in which the miR-574-5p levels were lower compared to UM-SCC-14, or UM-SCC-1 cells (Fig S4C of Supporting Information). Thus, these data suggest that miR-574-5p-targeting of CerS1-2 might also be detected in some human primary tumour tissues, which decreased CerS1-2 is detected in the majority of HNSCC tumours compared to their non-cancerous head and neck tissues.
Defining the miR-574-5p-dependent regulation of the CerS1-2 3′UTR
To define the involvement of the putative miRNA-574-5p target sequence for mRNA degradation, the 3′UTR sequences of CerS1-2 with or without the miRNA-574-5p target sequence were sub-cloned down-stream of the full-length luciferase cDNA, whose expression was constitutively activated by the CMV promoter (Fig 7A–C). We used UM-SCC-14A cells to examine the effects of CerS1-2 3′UTR elements on luciferase stability, with/without the miRNA-574-5p target sequence in the absence/presence of the miRNA-574-5p-siRNA (which reduced its expression approximately 75% compared to Scr siRNA controls, as measured by Q-PCR, Fig S5A–D of Supporting Information). The data showed that the stability of luciferase mRNA containing the 3′UTR of CerS1-2 without the miRNA-574-5p target sequence did not change significantly in the presence of Act D at 2 h, and was not affected by miRNA-574-5p siRNA (Fig 7A). However, the stability of the luciferase mRNA was decreased around 60% (at 2 h) in the presence of miRNA-574-5p target sequence in the 3′UTR of the CerS1-2, compared to controls in UM-SCC-14A (Fig 7B). Remarkably, knockdown of miRNA-574-5p expression using siRNA prevented the degradation of luciferase mRNA in the presence of Act D at 2 h compared to scr-siRNA-transfected controls (Fig 7B). Previous studies indicate that for miR to bind its target site, it must recognize a ‘seed sequence’ which is localized within first 2–8 bases of the target sequence in the 3′UTR of its target mRNA (Bartel, 2009; Carthew & Sontheimer, 2009; Huntzinger & Izaurralde, 2011). Thus, we generated a mutation with deletion of second and third bases of the miR-574-5p-target/seed sequence of the CerS1-2 3′UTR (Fig S4A of Supporting Information), and determined its effects on luciferase mRNA stability. The data showed that when the mutant miR-574-5p-target sequence was introduced, the luciferase mRNA did not decrease in the presence of Act D at 2 h (Fig 7C). Overall, these data suggest that an alternatively spliced variant CerS1-2, a major CerS1 isoform expressed in HNSCC cells or primary tumour tissues, is targeted for degradation by a specific miRNA-574-5p, whose target sequence is localized within the 3′UTR (in the intronic sequence between exons 6 and 7 of CerS1-2 pre-mRNA) containing the TACACACA/(CACA)6-seed/recognition sequence (Fig S4A of Supporting Information). These data suggest also that knockdown of miRNA-574-5p expression using a siRNA reduces the degradation of CerS1-2 mRNA in these cancer cells.
Figure 7. MiR-574-5p targets CerS1-2 3′UTR, decreasing mRNA stability in HNSCC.

A-C. Cells plated in 6-well plates (2 × 105 cells/well), and transfected with luciferase expression vector containing CerS1-2 3′UTR1-1000 bp (A), CerS1-2 3′UTR-574-5p (B) and CerS1-2-3′UTR-mutant 574-5p (C) sequences in the absence/presence of siRNA against miR-574-5p, with/without Act D (25 µg/ml, 2 h) or DMSO. Effects of 3′UTR sequences on luciferase mRNA were determined by Q-PCR. Actin was used as a loading control. These data represent at least two-independent trials performed as duplicates. The error bars represent the standard deviations. Two vertical lines on the map of the construct (C) represent the deletion mutations.
Reconstitution of CerS1 expression by HDAC1 inhibition and knockdown of miRNA574-5p expression inhibits HNSCC growth
These data suggested that CerS1 transcription is regulated by both epigenetic repression and post-transcriptional mRNA degradation via HDAC1 and miR-574-5p, respectively, and to optimally increase CerS1 expression, both of these negative regulators might require simultaneous inhibition. Indeed, knockdown of miR-574-5p and HDAC1, but not HDAC3, using siRNAs increased CerS1 mRNA approximately 4-fold compared to controls (Fig 8A) in UM-SCC-1 cells. Accordingly, treatment of multiple human cancer cells (UM-SCC-1 or UM-SCC-22A-HNSCC, K562-chronic myeloid leukaemia or Daoy-medulloblastoma) cells with HDAC1 inhibitor MS-275 in the presence of the siRNA against miR-574-5p, which effectively decreased miR-574-5p by approximately 70% in these cell lines (Fig S5A–D of Supporting Information), increased CerS1 mRNA approximately 2.0-, 1.7-, 22- and 2.5-fold, respectively (Fig 8B–E). Indeed, combination treatment with MS-275 and knockdown of miRNA-574-5p using siRNA increased CerS1-2 mRNA around 2- and 4-fold in UM-SCC-22A and Daoy cells compared to controls (Fig 8F). These data were also consistent with increased CerS1-2 protein expression around 1.9- or 3.0-fold compared to controls in UM-SCC-22A or UM-SCC-1 cells, respectively (Fig 9A). Importantly, treatment of UM-SCC-22A cells with MS-275 in combination with knockdown of miRNA-574-5p using siRNA increased (2.5-fold) C17–C18-ceramide (Spassieva et al, 2007) generation (Fig 9B). There were no significant changes in C17–C16-ceramide, which is generated by CerS5 and CerS6 (Fig S5E of Supporting Information). Treatment with MS-275 or siRNA against miRNA-574-5p alone had no significant effect on ceramide generation (Fig 9B). Accordingly, ectopic expression of CerS1-2-FLAG was as effective as CerS1-1-FLAG (their equal expression was determined by Western blotting using anti-FLAG antibody, see Fig S6A of Supporting Information) in the generation of C18- and C18:1-ceramides, but not other ceramide species (Fig S6B and C of Supporting Information), confirming that CerS1-2 plays a role in the selective generation of C18-ceramide as CerS1-1. Overall, these data suggest that interference with HDAC1 and miR-574-5p in combination reconstitutes CerS1-2, which increases C18-ceramide generation.
Figure 8. Combination treatment with MS-275 or HDAC1 RNAi and miR-574-5p siRNA reconstitutes CerS1 expression in multiple human cancer cell lines.

A. Effects of knockdown of HDAC1 or HDAC3 using siRNAs on CerS1 mRNA were detected using Q-PCR in UM-SCC-1 cells.
B-F. Cells were plated in 6-well plates (4 × 105 cells/well) and transfected with 100 nM miR-574-5p siRNA for 24 h before treatment with MS-275. CerS1 mRNA or miR-574-5p were measured with Q-PCR, and normalized to ribosomal RNA in UM-SCC-1 (B), UM-SCC-22A (C), K562 (D) and Daoy (E) cells. (F) Effects of treatment with MS-275 with/without siRNA-mediated miR-574-5p knockdown on CerS1-2 mRNA were determined using semi-quantitative RT-PCR in UM-SCC-22A or Daoy medulloblastoma cells compared to controls (upper and lower panels, lanes 2 and 4 and 1 and 3, respectively). These data represent at least two-independent trials performed as duplicates.
Figure 9. Reconstitution of CerS1-2 expression by inhibition of HDAC1 and miR-574-5p.

A. CerS1 protein in response to MS-275 (lanes 2 and 4) in the absence/presence of siRNA against miR-574-5p (lanes 3 and 4), compared to controls (lanes 1 and 3) was detected by Western blotting, and normalized to actin in UM-SCC-22A or UM-SCC-1 cells (left and right panels, respectively).
B. Levels of C17–C18-ceramide in response to MS-275 in the absence/presence of siRNA against miR-574-5p were detected in UM-SCC cells using RT-PCR and LC/MS/MS, respectively.
C-D. Effects of MS-275 and knockdown of miR-574-5p using siRNA on the growth of UM-SCC-22A cells were measured by trypan blue exclusion (C), or in UM-SCC-1 cells by Cyquant (D) in the absence/presence of Scr or CerS1 siRNAs.
E. Regulation of anchorage-independent growth of UM-SCC-22A cells by combination of MS-275 and miR-574-5p siRNA treatment was assessed by measurement of growth on soft agar. These data represent at least two-independent trials performed as duplicates. The error bars represent the standard deviations.
F. Suggested novel mechanism for the regulation of CerS1 expression and C18-ceramide generation by the coordinated functions of HDAC1 and miR-574-5p in human cancer cells is shown.
Reconstitution of CerS1-2 inhibits the growth of cancer cells
We determined whether reconstitution of CerS1 expression, and subsequent increase in C18-ceramide generation using the HDAC1 inhibition and knockdown of miRNA-574-5p alone or in combination inhibit HNSCC proliferation, as detected by trypan blue exclusion and modulated anchorage-independent growth in soft agar. Treatment of UM-SCC-22A cells with MS-275 and miR-574-5p siRNA inhibited the growth by around 80%, whereas, knockdown of endogenous CerS1 using siRNAs slightly but significantly protected (approximately 40%, p < 0.05) growth inhibition in response to this combination treatment (Fig 9C). Similarly, suppression of growth mediated by MS-275 and knockdown of miR-574-5p by siRNA in combination, as measured by detection of DNA replication using the CyQUANT cell proliferation assay kit (Invitrogen), was completely prevented by siRNA-mediated down-regulation of CerS1 (Fig 9D). Moreover, results also showed that dual treatment with MS-275 and miR-574-5p siRNA significantly decreased the growth of UM-SCC-22A cells approximately 90% compared to controls in soft agar (Fig 9E). Overall, these data indicate that prevention of epigenetic repression of CerS1 promoter by HDAC1, and inhibition of post-transcriptional targeting of CerS1-2 mRNA by miR-574-5p reconstitutes CerS1 expression, resulting in increased C18-ceramide generation, which then leads to cancer cell growth inhibition.
DISCUSSION
Our data suggest that down-regulation of alternatively spliced CerS1-2 is coordinated by two independent mechanisms, involving the epigenetic repression of CerS1 transcription via inhibition of Sp1 recruitment to the promoter by HDAC1-dependent histone deacetylation, and miR-574-5p-dependent targeting of CerS1-2 mRNA, which contains a specific miR-574-5p targeting sequence at its 3′UTR localized within the intronic sequence between exons 6 and 7. Importantly, inhibition of HDAC1 and knockdown of miR-574-5p expression using pharmacologic and molecular tools reconstituted CerS1-2 mRNA and protein, resulting in increased C18-ceramide generation, and growth inhibition in multiple cancer cells (Fig 9F).
CerS function was associated first with longevity in yeast, and its deletion resulted in increased life span, and thus it was named ‘longevity associated gene 1’ (LAG1; D'Mello et al, 1994). CerS1 gene encodes a polycistronic mRNA including exons 1–7, which is referred to as CerS1-1, and exons 1–2 of GDF1. There is also an alternatively spliced variant of CerS1, CerS1-2, which encodes only exons 1–6. It was reported previously that both CerS1-1 and CerS1-2 are catalytically active for the generation of ceramide (Sridevi et al, 2010). Mechanisms that regulate CerS1 expression have been unknown. In this study, we cloned the promoter sequences of both CerS1 and CerS6. Recruitment of Sp1 to specific GC-Boxes localized between −176 to −166 and −139 to −125 regions was shown to play key roles for the activation of the CerS1 promoter. Interestingly, Sp1 was previously shown to regulate glucosylceramide synthase (Uchida et al, 2004), and acid ceramidase (Lucki & Sewer, 2011). Our data also showed that recruitment of Sp1 without alterations to its acetylation and/or phosphorylation, which has been shown to control its transactivation function (Spengler et al, 2008), is significantly decreased in HNSCC cells via HDAC1-dependent histone deacetylation.
HDAC1 has been identified as a valuable drug target against many cancers, including HNSCC, and various inhibitors of HDAC have been developed as potential anti-cancer agents (Bolden et al, 2006; Marks, 2010). Ceramide signalling is important for HDACI-mediated tumour suppression (Maggio et al, 2004; Park et al, 2010). However, whether HDAC-dependent epigenetic alterations regulate ceramide metabolism is unclear. In this study, we defined a clear role for HDAC1 in the repression of CerS1 transcription, and interference of HDAC1 using siRNAs or pharmacologic inhibitor MS-275 enhanced the promoter activity of CerS1. These data are in agreement with a previous study, in which anti-cancer activity of MS-275 against Jurkat lymphoblastic leukaemia cancer cells was linked to increased ceramide generation (Maggio et al, 2004). Although our results suggest a specific role for HDAC1 in the regulation of CerS1 expression, it should be noted that involvement of other class I HDACs might also be possible in this process, due to known large complex formation between HDAC1 with other HDACs and accessory proteins (Yang & Seto, 2008; You et al, 2001). Interestingly, recent data showed that sphingosine kinase-2-generated nuclear sphingosine 1-phosphate is a direct regulator of HDAC1/2, which then up-regulates the expression of p21 or c-Fos (Hait et al, 2009). It is not clear if SK-2/S1P plays a role in the regulation of CerS1 expression via HDAC regulation.
In addition to HDAC1-dependent epigenetic repression, CerS1-2 expression was also regulated at the post-transcriptional level, which significantly reduced the stability and half-life of CerS1 mRNA. We then determined that, whereas, keratinocytes mainly express CerS1-1, HNSCC cells mainly express CerS1-2 mRNAs. Further analysis of the 3′UTR of CerS1-2 showed that there is a specific miR-574-5p target sequence, which is involved in targeting CerS1-2 mRNA for miR-574-5p-dependent degradation. To this end, increased serum miR-574-5p was implicated in poor-survival and negative therapeutic response of patients with lung cancers (Foss et al, 2010). Our data also showed that miR-574-5p levels of patients are higher in only 5 of the 10 tumour samples compared to normal adjacent tissues. These data suggest that other factors might also contribute to the repression of CerS1 mRNA in tumour tissues. Interestingly, these data also revealed that all of 10 tumours express miR-574-5p, and since tumour tissues preferentially express CerS1-2, which is a target for miR574-5p, the overexpression of miR-574-5p might not be needed, and its steady state expression should be sufficient for targeting CerS1-2 in tumour tissues. However, in normal tissues, CerS1-1 is preferentially expressed, which is not targeted by miR-574-5p.
In summary, this study suggests a novel mechanism by which HDAC1/miR-574-5p axis coordinates the repression of alternatively spliced CerS1-2, which is mainly detected in cancer cells and primary tumour tissues compared to controls, modulating the generation of tumour suppressor ceramide. Data also indicate here that interference with HDAC1 and miR-574-5p reconstitutes CerS1 expression, and results in ceramide-mediated growth inhibition in multiple human cancer cells, providing a mechanism-based strategy for the treatment of human cancers.
MATERIALS AND METHODS
Identification of CerS1 and CerS6 promoter
CerS1 and CerS6 promoter DNA sequences were determined by using UCSC Genome Browser ‘Get Genomic Sequence Near Gene’ tool (http://genome.ucsc.edu/cgi-bin/hgGateway). The promoter was analysed for the presence of TATA-, CAAT- and GC-boxes via The European Bioinformatics Institute (EBI) ClustalW sequence analysis tool (http://www.ebi.ac.uk/Tools/clustalw2/index.html). TFBIND was used to determine transcription factor-recognition sequences. CpG Island Searcher software was used to determine the presence of CpG islands in the CerS1 promoter region as previously described (Takai & Jones, 2002). Sequence alignment was performed using Genomatix Software (www.genomatix.de). MiR binding sites within the intronic region of CerS1 exons 6 and 7 were scanned using miRBase (http://www.mirbase.org/search.shtml), and potential human miRNA's were selected if a score of >90 was assigned to a given sequence.
Luciferase reporter constructs
Promoter constructs were generated using a pGL3-Promoter (pGL3) luciferase construct reporter vector (Promega, Madison, WI), as described previously. The CerS1 and CerS6 promoter regions in non-cancerous WI-38 foetal lung fibroblasts were amplified using the PCR using the GC-Rich PCR Kit (Roche). Serial deletions and GC-Box mutations on the promoter DNA were obtained by two-step PCR as described previously (Mukhopadhyay et al, 2009). Sequences of primer sets used for cloning or generating mutants of the CerS1 and CerS6 promoters are shown in Table S1 of Supporting Information.
Transfections and promoter activity measurements
Promoter constructs (0.5 µg) were co-transfected with 0.4 µg of β-gal normalization vector (Promega), using Effectene as described by the manufacturer (Qiagen, Valencia, CA). The transfection efficiencies were normalized to β-gal activity (Promega), as described (Wooten-Blanks et al, 2007).
Detection of Sp1/CerS1 promoter DNA interactions by EMSA
Nuclear extracts (5–8 µg of protein) were isolated, and EMSA was performed as described previously (Flamigni et al, 1997; Wooten-Blanks et al, 2007). Double-stranded (ds) oligonucleotides containing wt or mutant Sp1-recognition sequences (Santa Cruz Biotechnology, Santa Cruz, CA) were used as specific and non-specific competitors, respectively. Ds-CerS1 promoter GC-Boxes 1–4 wild-type or mutant sequences (as shown in Table S2 of Supporting Information; Integrated DNA Technologies, Coralville, IA) were also used as competitors. Super-shift assays were performed using 5 µg anti-Sp1 antibody (sc-59) or IgG (Santa Cruz Biotechnology). Three to five nanograms of 5′-end-labelled Sp1 consensus ds-oligonucleotide was used as a probe (Wooten & Ogretmen, 2005).
Quantitative-PCR-based chromatin immuno-precipitation (Q-ChIP)
Q-ChIP was performed using the ChIP Assay Kit (Millipore, Massachusetts) as described by the manufacturer. Q-PCR primer/probes were designed for use with TaqMan gene expression assays, specific for the CerS1 -300 promoter region using IDT DNA PrimerQuest software. The probe sequence was: 5′-FAM-AGGGGACTGCCTGCT-AGTGT-IowaBlack-3′. Q-PCR reactions were performed using Applied Biosystems StepOnePlus cycler under normal (slow) cycling conditions as described by the manufacturer. Samples were normalized to 1% of input material. Primer sequences are presented in Table S3 of Supporting Information.
Small interfering RNAs (siRNA)
All siRNA information can be found in Table S4 of Supporting Information. Cells were transfected by siRNAs (80 nM for 48 h) using the DharmaFECT™ transfection kit (Dharmacon, ThermoFisher Scientific).
Detection of alternatively spliced CerS1-1 and CerS1-2 mRNAs using RT-PCR
CerS1-1 expression was determined by Q-PCR using the primers spanning sequences of exons 2–7, as described. To determine the predominant alternatively spliced variants of CerS1-2, reverse primers were designed for potential poly(A) signals with AAUAAA, or AAGAAA, UAUAAA, or TATAAA as previously described (Tian et al, 2005). The primer sequences are shown in Table S3 of Supporting Information.
Detection of the role of miRNA-574-5p in targeting the 3′UTR of CerS1-2
Three luciferase reporter constructs containing the 3′UTR of CerS1-2 were designed by GeneCopoeia (Rockville, MD). Construct ‘3′UTR-no miRNA sequence’ consisted of the first 1051 bp after the translational stop site, located at the end of exon 6, which does not contain the proposed miR-574-5p binding sequence. Construct ‘3′UTR-wt-miRNA’ was designed to encompass a 200 bp 3′UTR sequence containing the putative miR-574-5p recognition sequence, located within the intronic region of exon 6 and 7 of the CerS1 mRNA. Construct ‘3′UTR-mutant-miRNA’ contains a deletion mutation within the second and third ‘seed’ bases of the miR-574-5p-recognition/targeting sequence. Firefly luciferase mRNA (161 bp amplicon) was detected via RT-PCR with forward primer: 5′-TCTGGGTCTACCGGCCTGCC-3′ and reverse primer: 5′-ACATGCCGAAGCCGTG GTGG-3′, and normalized to beta-actin (sequences were presented in Table S1 of Supporting Information). PCR reactions were carried out using BioRad SsoFast Master Mix.
The paper explained
PROBLEM
Histone deacetylases (HDACs) and micro-RNAs (miRs) have pro-survival roles but the mechanism behind this is unclear. Repression of ceramide synthase 1 (CerS1), altering C18-ceramide generation was linked to resistance to chemotherapy-induced cell death and nodal metastasis. However, how CerS1 mRNA is down-regulated, and whether HDACs and/or miRs are involved in its regulation in cancerous versus non-cancerous cells and/or tissues remained unknown. Therefore, we have investigated the mechanisms involved in the regulation of CerS1 transcription via HDAC/miR in various human cancer cell lines and tumour tissues, compared to non-cancerous control counterparts.
RESULTS
We determined that decreased CerS1 mRNA, which leads to lower C18-ceramide generation, is associated with repression of its promoter activity by HDAC1-dependent inhibition of Sp1 recruitment to the promoter DNA in various human HNSCC cells compared to non-cancerous primary and immortalized skin keratinocytes. Interestingly, our data revealed that an alternatively spliced variant CerS1 mRNA (CerS1-2) was detected mainly in cancer cells or primary tumour tissues compared to controls, which was targeted selectively by miR-574-5p for degradation, decreasing its mRNA stability. Inhibition of HDAC1 and siRNA-mediated knock-down of miR-574-5p reconstituted CerS1-2 expression, and C18-ceramide generation, which subsequently inhibited cancer cell proliferation.
IMPACT
These data suggest that concerted functions of HDAC1 and miR-574-5p play key roles in the repression of CerS1 expression, leading to alterations of the generation of tumour suppressor sphingolipid C18-ceramide in cancer cells. Therefore, we propose that the role of HDAC1/miR-574-5p in pro-survival might be, at least in part, related to repression of CerS1/C18-ceramide signalling in some cancer models, including HNSCC. Our data also suggest that the HDAC1/miR-574-5p axis might provide a novel therapeutic target to reconstitute tumour suppressor CerS1/C18-ceramide signalling.
Measurement of ceramide by LC/MS/MS
Endogenous ceramides were measured using high performance LC/MS/MS as described previously (Koybasi et al, 2004; Senkal et al, 2007). In short, after cells were collected by centrifugation, lipids were extracted directly from cell pellets, and normalized to Pi, measured in the same extracts as described (Koybasi et al, 2004).
Statistical analysis
Data are represented as mean ± SD, unless otherwise indicated. An unpaired Student's t-test was performed using Prism/GraphPad software; p < 0.05 was considered significant (Mukhopadhyay et al, 2009).
See Supporting Information for details of cell lines, primary tumour tissues and their controls obtained from HNSCC patients, isolation of DNA, RNA and q-PCR, Western blotting, measurement of cancer cell growth and/or proliferation using trypan blue, Cyquant and soft agar growth assays, and image quantification in EMBO Mol Med online.
Acknowledgments
Sp1, Sp3 and Sp1/2 mutant plasmids were obtained from Dr. Jonathan Horowitz at North Carolina State University. We thank Dr Viswanathan Palanisamy (MUSC) for helpful discussions. We also thank Dr J. Schnellmann for her editorial review. This work was supported by research grants from the National Institutes of Health (CA088932, DE016572 and CA097132 to B. O.). M. M.-N. was supported by the GAANN, and T32 training grants. Lipid measurements were conducted in facilities constructed with support from NIH (C06 RR015455).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that there is no conflict of interest.
Author contributions
MMN and BO designed experiments; MMN, SG and SP performed experiments and analysed data; WJ measured the half-life of CerS1 mRNA in NHEK cells; RJT and CES measured the ceramide synthase activity for C18-ceramide generation of CerS1-1 and CerS1-2 in HNSCC cells.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello NP, Childress AM, Franklin DS, Kale SP, Pinswasdi C, Jazwinski SM. Cloning and characterization of LAG1, a longevity-assurance gene in yeast. J Biol Chem. 1994;269:15451–15459. [PubMed] [Google Scholar]
- Flamigni F, Faenza I, Marmiroli S, Stanic I, Giaccari A, Muscari C, Stefanelli C, Rossoni C. Inhibition of the expression of ornithine decarboxylase and c-Myc by cell-permeant ceramide in difluoromethylornithine-resistant leukaemia cells. Biochem J. 1997;324:783–789. doi: 10.1042/bj3240783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss KM, Sima C, Ugolini D, Neri M, Allen KE, Weiss GJ. miR-1254 and miR-574-5p: serum-based microRNA biomarkers for early-stage non-small cell lung cancer. J Thorac Oncol. 2010;6:482–488. doi: 10.1097/JTO.0b013e318208c785. [DOI] [PubMed] [Google Scholar]
- Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Karahatay S, Thomas K, Koybasi S, Senkal CE, Elojeimy S, Liu X, Bielawski J, Day TA, Gillespie MB, Sinha D, et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007;256:101–111. doi: 10.1016/j.canlet.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koybasi S, Senkal CE, Sundararaj K, Spassieva S, Bielawski J, Osta W, Day TA, Jiang JC, Jazwinski SM, Hannun YA, et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem. 2004;279:44311–44319. doi: 10.1074/jbc.M406920200. [DOI] [PubMed] [Google Scholar]
- Lucki NC, Sewer MB. Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. J Biol Chem. 2011;286:19399–19409. doi: 10.1074/jbc.M110.195826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio SC, Rosato RR, Kramer LB, Dai Y, Rahmani M, Paik DS, Czarnik AC, Payne SG, Spiegel S, Grant S. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004;64:2590–2600. doi: 10.1158/0008-5472.can-03-2631. [DOI] [PubMed] [Google Scholar]
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19:1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Mesika A, Sivaguru M, Van Veldhoven PP, Alexander H, Futerman AH, Alexander S. (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol Cancer Res. 2007;5:801–812. doi: 10.1158/1541-7786.MCR-07-0100. [DOI] [PubMed] [Google Scholar]
- Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. 2005;390:263–271. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorefield KS, Fry SJ, Horowitz JM. Sp2 DNA binding activity and trans-activation are negatively regulated in mammalian cells. J Biol Chem. 2004;279:13911–13924. doi: 10.1074/jbc.M313589200. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- Ogretmen B, Safa AR. Identification and characterization of the MDR1 promoter-enhancing factor 1 (MEF1) in the multidrug resistant HL60/VCR human acute myeloid leukemia cell line. Biochemistry. 2000;39:194–204. doi: 10.1021/bi991943f. [DOI] [PubMed] [Google Scholar]
- Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Haussinger D, Reinehr R, Larner A, Spiegel S, Fisher PB, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res. 2010;70:6313–6324. doi: 10.1158/0008-5472.CAN-10-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- Senkal CE, Ponnusamy S, Rossi MJ, Bialewski J, Sinha D, Jiang JC, Jazwinski SM, Hannun YA, Ogretmen B. Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol Cancer Ther. 2007;6:712–722. doi: 10.1158/1535-7163.MCT-06-0558. [DOI] [PubMed] [Google Scholar]
- Spassieva S, Seo JG, Jiang JC, Bielawski J, Alvarez-Vasquez F, Jazwinski SM, Hannun YA, Obeid LM. Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J Biol Chem. 2006;281:33931–33938. doi: 10.1074/jbc.M608092200. [DOI] [PubMed] [Google Scholar]
- Spassieva S, Bielawski J, Anelli V, Obeid LM. Combination of C(17) sphingoid base homologues and mass spectrometry analysis as a new approach to study sphingolipid metabolism. Methods Enzymol. 2007;434:233–241. doi: 10.1016/S0076-6879(07)34012-3. [DOI] [PubMed] [Google Scholar]
- Spengler ML, Guo LW, Brattain MG. Phosphorylation mediates Sp1 coupled activities of proteolytic processing, desumoylation and degradation. Cell Cycle. 2008;7:623–630. doi: 10.4161/cc.7.5.5402. [DOI] [PubMed] [Google Scholar]
- Sridevi P, Alexander H, Laviad EL, Min J, Mesika A, Hannink M, Futerman AH, Alexander S. Stress-induced ER to Golgi translocation of ceramide synthase 1 is dependent on proteasomal processing. Exp Cell Res. 2010;316:78–91. doi: 10.1016/j.yexcr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Itoh M, Taguchi Y, Yamaoka S, Umehara H, Ichikawa S, Hirabayashi Y, Holleran WM, Okazaki T. Ceramide reduction and transcriptional up-regulation of glucosylceramide synthase through doxorubicin-activated Sp1 in drug-resistant HL-60/ADR cells. Cancer Res. 2004;64:6271–6279. doi: 10.1158/0008-5472.CAN-03-1476. [DOI] [PubMed] [Google Scholar]
- Wang B, Shi G, Fu Y, Xu X. Cloning and characterization of a LASS1-GDF1 transcript in rat cerebral cortex: conservation of a bicistronic structure. DNA Seq. 2007;18:92–103. doi: 10.1080/10425170601060947. [DOI] [PubMed] [Google Scholar]
- Wooten LG, Ogretmen B. Sp1/Sp3-dependent regulation of human telomerase reverse transcriptase promoter activity by the bioactive sphingolipid ceramide. J Biol Chem. 2005;280:28867–28876. doi: 10.1074/jbc.M413444200. [DOI] [PubMed] [Google Scholar]
- Wooten-Blanks LG, Song P, Senkal CE, Ogretmen B. Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 2007;21:3386–3397. doi: 10.1096/fj.07-8621com. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.