

See related article in EMBO Molecular Medicine http://dx.doi.org/10.1002/emmm.201100199
In a rich and comprehensive piece of work, Ferone et al report in this issue of EMBO Molecular Medicine the first mouse model of the AEC (Ankyloblepharon-Ectodermal defects-Cleft lip/palate, OMIM 106260) syndrome (Ferone et al, 2012). The mechanistic insights are of likely general relevance for a number of conditions resulting from deranged keratinocyte growth control, including cancer.
AEC is part of a group of genetic syndromes with compromised skin development that go under the general name of ectodermal dysplasias (EDs). These are caused by mutations of genes with disparate functions, including transcription factors like p63. This close cousin of p53 plays a key role in ectodermal development as first demonstrated by the lack of stratified epithelia and their adnexa in mice with homozygous deletion of the gene. Importantly, no such alterations occur in mice with heterozygous p63 deletion, which are phenotypically normal. In the human population, at least five ED malformation syndromes have been linked to p63 gene mutations (Rinne et al, 2006). These are heterozygous missense mutations likely to cause the disorders by interfering with the function of wild type p63, which controls transcription by binding to DNA in a tetrameric form (Crum & McKeon, 2010).
p63 can be produced in multiple isoforms as the result of transcription from two different promoters and differential splicing. The predominant p63 isoform expressed in stratified epithelia is ΔNp63α, which contains, in its C-terminus region, asterile-alpha-motif (SAM) domain thought to be mediating interactions with other proteins and, possibly, lipid or RNA molecules (Crum & McKeon, 2010). The different ED syndromes result from p63 mutations that tend to cluster at different locations (Rinne et al, 2006). The AEC syndrome is mostly caused by mutations in the p63 SAM domain and differs from the others in the severity of the skin phenotype, the occurrence of ankyloblepharon (eyelid fusion at birth), and absence of ectrodactyly. Cleft palate is also a feature of the AEC syndrome that is shared with the ectrodactyly, ED and cleft lip/palate syndrome (EEC, OMIM 604292) and limb mammary syndrome (LMS, OMIM 603543), but not with the acro-dermato-ungual-lacrimal-tooth syndrome (ADULT, OMIM 103285) syndrome (Rinne et al, 2006). Interestingly, while in EEC syndrome cleft palate occurs only in a fraction of cases (40%) and is almost always associated with cleft lip, in AEC syndrome, cleft palate is a much more frequent feature (80%), and is associated with cleft lip only in half of the cases, suggesting that the cellular/molecular mechanisms involved in the two syndromes are different (Rinne et al, 2006).
A major obstacle to elucidate the molecular basis of the various p63-related syndromes has been the lack of adequate animal models. To this aim, Ferone et al generated mice with a knock-in missense mutation of the p63 SAM domain (p63L514F), which has been found in AEC patients. Mice harbouring this mutation already in a heterozygous form exhibited a phenotype with striking similarities to AEC patients, including hypoplastic and fragile skin, ED (tooth and hair follicle defects) and cleft palate. This is a remarkable achievement in view of previous attempts to develop murine models of p63-dependent ED syndromes (as discussed in Ferone's paper).
To provide mechanistic insights, the authors focused at first on the cleft palate, and found that the palatal shelves of p63+/L514F mutant mice elevated normally at E14.5, but were significantly smaller than the control. They further hypothesized that this reduced size and consequent failure to meet in the midline may be the result of reduced cell proliferation. Indeed, while differentiation of keratinocytes at this and later stage was essentially normal in the mutant mice, their proliferative capability was substantially reduced. Paralleling these observations, they found that the size of the keratinocyte stem cell population is reduced in the mutant mice during development, while the intrinsic self-renewal potential of the stem cells that survive after birth is normal. Molecularly, this is consistent with the fact that expression of various cell cycle regulators previously shown to be under p63 control in keratinocytes, including p21Cip1/Waf1, p16Ink4a, p19Arf and microRNA miR-34a (Antonini et al, 2010; Nguyen et al, 2006; Su et al, 2009), is unaffected in keratinocytes with the p63+/L514F knock-in mutation. This implies that the mutant p63 has a more specific impact on keratinocyte stem cell populations during development and likely uncouples self-renewal from differentiation.
Cleft palate is a relatively common developmental abnormality that can be caused by mutations of a number of genes besides p63 (Dixon et al, 2011). Notable among these are molecules involved in FGF signalling, specifically the fibroblast growth factor receptors (FGFR) 2 or 3 and FGF8 (Dixon et al, 2011). The authors noticed that the reported phenotype of mice lacking the epithelial-specific FGFR2b isoform was remarkably similar to that of their p63 mutant mice, including cleft palate and a strongly hypo-plastic and hypo-proliferative skin (Rice et al, 2004). In a carefully orchestrated series of studies, they established that the FGFR2b gene is a direct p63 target gene and that the p63 gene mutation that they are studying is selectively affecting expression of this gene and of the related FGFR3b gene. A demonstration of the functional importance of the p63-FGFR connection was provided by experiments showing that the hypo-proliferative phenotype of keratinocytes with the p63 mutation could be rescued by increased FGFR expression and signalling. Importantly, this mutant p63–FGFR connection is not limited to the mouse system, as it was also validated in skin from AEC syndrome patients (Fig 1).
Figure 1. Impact of p63-SAM mutations on palate development.
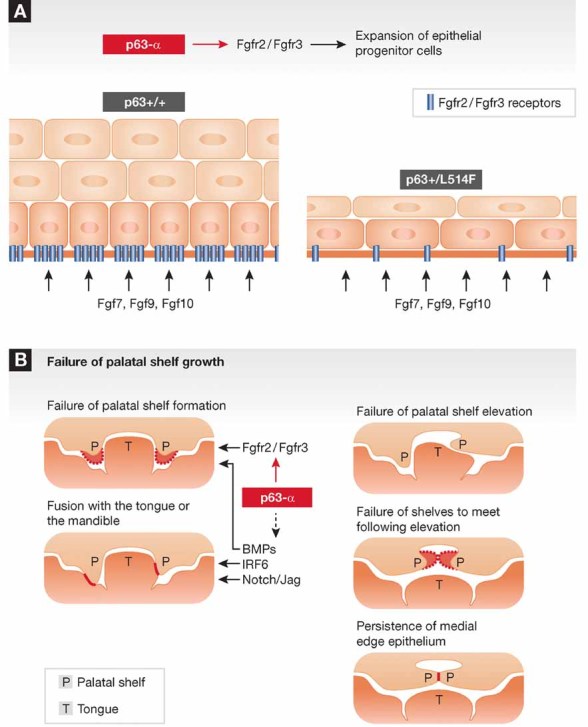
- ΔNp63α functions as a positive determinant of FGFR2 and FGFR3 expression. Mutations found in AEC syndrome are sufficient to suppress, in a heterozygous state, FGFR2 and FGFR3 expression. This causes an impairment of proliferation in embryonic ectodermal cells.
- p63 is interconnected with several pathways that are required for one or more steps of secondary palate development as recently reviewed (Dixon et al, 2011).
As it is always the case, significant advances in a field lead to important new questions. First and foremost is the molecular basis for the selective consequences of the SAM mutation on a selected subset of p63 target genes. Previous in vitro over-expression studies indicated that AEC mutant p63 can suppress the function of wild-type p63α through a dominant-negative mechanism (Koster et al, 2009). However, the basis for the selective role of the SAM domain in control of p63-dependent transcription remains elusive, and a conclusion of the present study is that it may be best understood in vivo, possibly by proteomic approaches to identify functionally relevant, associated partners. In this respect, it has been recently reported that AEC p63 mutations affect the ability of the p63 protein to interact with Special AT-rich Binding Protein-2 (SATB2), which has also been implicated in palate development (Chung et al, 2011).
A second main question is the identity of the additional targets, besides the FGFR genes, that are affected by the AEC-causing p63 mutations. As pointed out by Ferone et al, the phenotypes of the p63 and FGFR2b mutant mice are only partially overlapping, pointing to the existence of additional cross-regulatory targets and effectors. In fact, other genes of relevance to cleft palate development, like IRF6, were also found to be affected in mice with the p63-SAM mutation, even if to a lesser extent than FGFRs. Even for the cleft palate, the authors infer, but do not prove, the existence of a causal link between this abnormality and the keratinocyte hypo-proliferation that they have observed. Besides proliferation, palatogenesis is intimately dependent on other key processes like cell migration, reciprocal epithelial–mesenchymal interactions and differentiation. Cleft palate has been described in more than 300 human syndromes, and a battery of genes has been implicated in the disease by both human and mouse studies (Dixon et al, 2011). Mutations of these genes impinge on pathways already known to functionally and/or biochemically interconnect with p63, such as BMPs/TGF-ßs, IKK1, Jagged/Notch, and, as mentioned, IRF6 (Dixon et al, 2011). Interestingly, recent deep sequencing analysis of a set of oral keratinocyte-derived squamous cell carcinomas has revealed mutations with a possible cancer-driver or -permissive function in both Notch1 and IRF6 genes as well as p63 (Stransky et al, 2011). In contrast to p53, p63 was previously connected with cancer at the mRNA/protein expression rather than gene mutation level (Crum & McKeon, 2010). Interestingly, the recent findings point to a number of p63 missense mutations including one in the SAM domain (Stransky et al, 2011). Given the connection between this region of p63 and FGFR expression and function established by the Ferone's work, it is tempting to speculate that a similar connection may apply to keratinocyte-tumour development.
Acknowledgments
The author declares that there is no conflict of interest.
References
- Antonini D, Russo MT, De Rosa L, Gorrese M, Del Vecchio L, Missero C. Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J Invest Dermatol. 2010;130:1249–1257. doi: 10.1038/jid.2009.438. [DOI] [PubMed] [Google Scholar]
- Chung J, Grant RI, Kaplan DR, Irwin MS. Special AT-rich binding protein-2 (SATB2) differentially affects disease-causing p63 mutant proteins. J Biol Chem. 2011;286:40671–40680. doi: 10.1074/jbc.M111.271189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol. 2010;5:349–371. doi: 10.1146/annurev-pathol-121808-102117. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferone G, Thomason HA, Antonini D, De Rosa L, Hu B, Gemei M, Zhou H, Ambrosio R, Rice DP, Acampora D, et al. Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signaling in AEC syndrome. EMBO Mol Med. 2012 doi: 10.1002/emmm.201100199. DOI: 10.1002/emmm.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MI, Marinari B, Payne AS, Kantaputra PN, Costanzo A, Roop DR. DeltaNp63 knockdown mice: a mouse model for AEC syndrome. Am J Med Genet A. 2009;149A:1942–1947. doi: 10.1002/ajmg.a.32794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, Koster MI, Zhang Z, Wang J, di Vignano GP. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20:1028–1042. doi: 10.1101/gad.1406006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne T, Hamel B, van Bokhoven H, Brunner HG. Pattern of p63 mutations and their phenotypes–update. Am J Med Genet A. 2006;140:1396–1406. doi: 10.1002/ajmg.a.31271. [DOI] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Cho MS, Gi YJ, Ayanga BA, Sherr CJ, Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]