

Abstract
Coordinated release of calcium (Ca2+) from the sarcoplasmic reticulum (SR) through cardiac ryanodine receptor (RYR2) channels is essential for cardiomyocyte function. In catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited disease characterized by stress-induced ventricular arrhythmias in young patients with structurally normal hearts, autosomal dominant mutations in RYR2 or recessive mutations in calsequestrin lead to aberrant diastolic Ca2+ release from the SR causing arrhythmogenic delayed after depolarizations (DADs). Here, we report the generation of induced pluripotent stem cells (iPSCs) from a CPVT patient carrying a novel RYR2 S406L mutation. In patient iPSC-derived cardiomyocytes, catecholaminergic stress led to elevated diastolic Ca2+ concentrations, a reduced SR Ca2+ content and an increased susceptibility to DADs and arrhythmia as compared to control myocytes. This was due to increased frequency and duration of elementary Ca2+ release events (Ca2+ sparks). Dantrolene, a drug effective on malignant hyperthermia, restored normal Ca2+ spark properties and rescued the arrhythmogenic phenotype. This suggests defective inter-domain interactions within the RYR2 channel as the pathomechanism of the S406L mutation. Our work provides a new in vitro model to study the pathogenesis of human cardiac arrhythmias and develop novel therapies for CPVT.
Keywords: CPVT, dantrolene, disease modelling, induced pluripotent stem cells, ryanodine receptor 2
INTRODUCTION
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited life-threatening arrhythmia leading to syncope and sudden cardiac death at a young age. CPVT patients, who usually do not show any detectable cardiac disease, manifest ventricular premature beats and bidirectional or polymorphic ventricular tachycardia in response to emotional or physical stress (Scheinman & Lam, 2006). Although the very high mortality rate (30–35% by the age of 35 years) calls for effective preventive and therapeutic measures, current clinical management of CPVT is based on the symptomatic treatment with β-blockers to reduce the frequency of arrhythmias and the implantation of automated defibrillators (ICDs) to terminate fatal arrhythmias (Kaufman, 2009). Two genetic forms of the disease have been described: one accounting for at least 50% of all cases and associated with autosomal dominant mutations in the cardiac ryanodine receptor, RYR2 (CPVT1; Priori et al, 2001) and a very rare one linked to recessive mutations in calsequestrin (CPVT2; Postma et al, 2002). Both proteins belong to the multimolecular calcium (Ca2+) release channel complex of the sarcoplasmic reticulum (SR) which supports myocyte Ca2+ cycling and contractile activity (Berridge, 2003; Bers, 2004; Gyorke & Terentyev, 2008; Kaye et al, 2008; Lanner et al, 2010). Accumulating evidence from animal models suggests that CPVT mutations result in Ca2+ diastolic leak from the SR causing arrhythmogenic delayed after depolarizations (DADs) in cardiac myocytes (Liu et al, 2009). However, the molecular mechanisms underlying the pathogenesis of these mutations are still controversial.
Dantrolene, a hydantoin derivative that acts as muscle relaxant, is currently the only specific and most effective treatment for malignant hyperthermia, a rare life-threatening familial disorder caused by mutations in the skeletal ryanodine receptor (RYR1) (Kobayashi et al, 2009). It is also used in the management of other disorders, such as neuroleptic malignant syndrome and muscle spasticity (Krause et al, 2004). The therapeutic action of dantrolene seems to be due to its binding to an amino-terminal sequence of RYR1, which restores inter-domain interactions critical for the closed state of the channel (Paul-Pletzer et al, 2002). Recently, dantrolene has been shown to target a corresponding sequence in RYR2 (Paul-Pletzer et al, 2005) and to improve intracellular Ca2+ handling in failing cardiomyocytes from a canine model of heart failure (Kobayashi et al, 2009) and arrhythmias in a mouse model of CPVT1 (Kobayashi et al, 2010; Uchinoumi et al, 2010). Although experimental animals have been extremely valuable for investigating cardiac function and pathogenesis as well as for drug assessment and development, they cannot completely model human cardiomyocytes. Induced pluripotent stem cells (iPSCs) offer the possibility to obtain human myocytes in vitro from patients with cardiac abnormalities (Carvajal-Vergara et al, 2010; Moretti et al, 2010b; Takahashi et al, 2007; Yazawa et al, 2011). Here, we report the generation of the first human patient-specific iPSC-based system of CPVT1 and tested whether dantrolene can rescue the disease phenotype and thus represent a potential novel drug compound for the causal treatment of CPVT.
RESULTS
Derivation of iPSC lines and their differentiation into the cardiac lineage
Dermal fibroblasts were obtained from a 24-year-old woman with a diagnosis of familial CPVT, who underwent cardiac arrest at the age of 23 years and received an ICD after cardiac resuscitation. Genetic screening showed that she carried a novel autosomal dominant S406L missense mutation in the RYR2 gene, caused by a C → T nucleotide substitution in exon 14 at position 1217 of the coding region (Fig 1A and B). The mutation is located in the N-terminal domain (amino acids 1–600) of the RYR2 Ca2+ release channel, which represents, together with the central domain (amino acids 2000–2500) and the carboxy-terminal transmembrane domain, one of the three hotspots for CPVT-associated RYR2 mutations (George et al, 2007; Thomas et al, 2010; Fig 1C). Fibroblast transduction with retroviral vectors encoding for SOX2, OCT4, KLF4 and c-MYC generated several CPVT patient-specific iPSC clones, three of which were further characterized and used for cardiomyocyte differentiation. Similarly, control iPSCs were created using fibroblasts from a 32-year-old healthy female (Moretti et al, 2010b). The S406L heterozygous mutation was identified exclusively in CPVT-iPSCs. All iPSC lines showed human embryonic stem cell morphology, expression of the pluripotency markers NANOG and TRA1-81, alkaline phosphatase activity, reactivation of endogenous pluripotency genes (OCT4, SOX2, NANOG, REX1 and TDGF1), silencing of the four retroviral transgenes and normal karyotype (Fig 1D–F; Fig S1A and B of Supporting information). Pluripotency of each iPSC line was assessed by upregulation of genes specific of all three germ layers in in vitro-differentiating embryoid bodies (EBs) (Fig S1C of Supporting information).
Figure 1. Generation of CPVT-iPSCs.

- Pedigree of the CPVT-affected patient (III-1) showing autosomal dominant inheritance in the family.
- Sequence analysis of RYR2 gene in fibroblasts from control and CPVT patient, revealing a novel heterozygous missense mutation in exon 14 (position 1217C > T of the coding sequence). Same results were obtained from all analysed control and CPVT-iPSC clones.
- Schematic representation of RYR2 channel and localization of the S406L mutation (red circle) at the N-terminal domain. Yellow circles indicate reported putative pathogenic mutations.
- Representative images of CPVT-iPSC colonies in bright field (top, clone a) and after staining for alkaline phosphatase (AP) activity (bottom, clone c). Scale bars, 100 µm.
- Representative images of a CPVT-iPSC colony (clone b) after immunostaining for the pluripotency markers NANOG (red) and TRA1-81 (green). Merged image is the magnified area marked by the white box. Scale bars, 100 µm.
- Karyogram of CPVT-iPSC clone a.
To direct iPSCs into the cardiac lineage, we used the EB differentiation system as previously described (Moretti et al, 2010a, b). Spontaneously beating areas, which started to appear within 10–12 days, were manually explanted and allowed to further mature for additional 2–4 months. Quantitative real-time PCR (qRT-PCR) in cardiac explants revealed that, after 2 months maturation, expression of most genes involved in myocytic Ca2+ handling and excitation–contraction (EC) coupling reaches similar levels to those of fetal human heart and is comparable among different control and CPVT-iPSC clones (Fig 2A). Consistent with already reported transcriptional profile data on human iPSC-/ESC-derived cardiac explants (Gupta et al, 2010), calsequestrin (CASQ2) expression is almost undetectable in all iPSC-derived cardiac explants at this maturation stage, while RYR2 is already expressed. However, protein analysis by western blotting at 3–4 months maturation demonstrated similar expression levels of pivotal Ca2+ handling proteins, such as RYR2, CASQ2, triadin (TRDN), junctin (JCTN) and phospholamban (PLN), among control-, CPVT-iPSC-derived cardiomyocytes and adult human heart tissue, suggesting further development of Ca2+ cycling molecular components (Fig 2B). Moreover, we evaluated the expression and subcellular localization of RYR2 by confocal immunofluorescence analysis on single control and diseased iPSC-derived cardiomyocytes (Fig 2C). In both cells, RYR2 was similarly distributed in the cytosol and perinuclear region, and partially co-localized with the myofilaments (Fig 2C). Importantly, RYR2 spatial cluster density was also comparable in control and CPVT myocytes (Fig 2D), suggesting that the S406L mutation does not interfere with trafficking of the homotetrameric channel.
Figure 2. Expression analysis of genes involved in myocytic Ca2+ handling and excitation–contraction coupling in iPSC-derived cardiomyocytes.

- Comparison of transcriptional profile of 2-month-old iPSC-derived cardiac explants, human adult (AH) and fetal (FH) heart tissue. qRT-PCR analysis was performed on 28 key genes involved in cardiomyocyte EC-coupling. All values are normalized for TNNT2 and relative to AH tissue.
- Western blot of whole cell extracts from 3 to 4-month-old iPSC-derived cardiac explants and human adult heart tissue (AH). Cardiac troponin T (cTNT) and β-actin were used as loading controls.
- Confocal immunofluorescence images of RYR2 (green) and actin (red) in human cardiomyocytes generated from control (top) and CPVT-iPSCs (bottom). Actin is marked by phalloidin. From left to right, the third panels display the merged image of the first two panels and the last panels depict RYR2 and actin expression patterns in a optical section at the nuclear plane. N indicates the cell nucleus. Scale bars, 15 µm.
- RYR2 cluster density in cardiomyocytes derived from control (black) and CPVT-iPSCs (red) (n = 17 in each group).
Stress-induced Ca2+ cycling abnormalities in CPVT-iPSC-derived cardiomyocytes
To assess whether CPVT-iPSC-derived cardiomyocytes recapitulate the disease phenotype, we analysed Ca2+ handling properties in single cells at 3–4 months maturation. We first examined whether CPVT myocytes display altered control of Ca2+ release during excitation–contraction (EC) by measuring electrically evoked Ca2+ transients at different pacing rates in absence and in presence of isoproterenol to mimic catecholaminergic stress (Fig 3 and Fig S2 of Supporting information). Increasing stimulation frequencies from 0.5 to 1.5 Hz correlated with a higher percentage of cells with abnormal Ca2+ handling in both control and CPVT myocytes (Fig 3A). However, this effect was significantly more pronounced in the diseased cells and was comparable among different CPVT-iPSC lines (Fig 3A and Fig S3 of Supporting information). We could observe three types of stress-induced Ca2+ cycling abnormalities, which associated with different severities of arrhythmogenicity: Ca2+ alternans, in which Ca2+ transients alternate between large and small on successive beats (AR1); Ca2+ transient fusion, characterized by absence of triggered Ca2+ transients at every second stimulation (AR2); and very irregular Ca2+ oscillations (AR3). Thus, frequency-induced stress appears to be one major arrhythmic trigger in CPVT-iPSC-derived myocytes. Deeper analysis of Ca2+ cycling properties in rhythmic cells revealed that, under basal conditions, control and CPVT myocytes presented comparable resting Ca2+ levels, similar systolic and diastolic Ca2+ concentration during electrical stimulation at different rates and equal SR Ca2+ content, determined by caffeine application (Fig 3B–E and Fig S4 of Supporting information). However, in presence of isoproterenol diastolic Ca2+ was significantly elevated in CPVT compared to control cells, while systolic Ca2+ levels remained similar (Fig 3C and D). Moreover, in contrast to control myocytes, SR Ca2+ load was not increased by isoproterenol treatment in CPVT cells (Fig 3E). These data suggest that in situations of catecholamine-induced elevated luminal Ca2+ the S406L-mutation in the RYR2 channels results in diastolic Ca2+ leak from the SR. This effect may be attributable to an increased S406L-RYR2 Ca2+ sensitivity, which lowers the release threshold to produce spontaneous activity during the diastolic period (Eisner et al, 2009; Priori & Chen, 2011). To investigate whether CPVT-iPSC-derived myocytes indeed possess an enhanced spontaneous Ca2+ release during adrenergic stimulation, we measured Ca2+ sparks in single cells during rest (Fig 4 and Movies S1-S4 of Supporting information). Ca2+ sparks are the elementary release events in cardiac EC coupling and derive from the local activity of RYR2 channel clusters (Cheng et al, 1993). Under basal conditions, Ca2+ spark frequency did not differ between control and CPVT myocytes, although Ca2+ spark amplitude, full width at 50% peak amplitude and decay time were significantly higher in diseased cells (Fig 4A–C). Moreover, only in CPVT myocytes, abnormal Ca2+ sparks with a prolonged plateau phase were observed (Fig 4B, ii). Under catecholaminergic stress, Ca2+ spark frequency considerably increased in CPVT compared to control cells, and associated with a greater decay time constant and even longer abnormal sparks (Fig 4B and C and Movies S3 and S4 of Supporting information). These results indicate that elevated diastolic Ca2+ and reduced SR Ca2+ load during catecholaminergic challenge in CPVT-iPSC-derived myocytes are caused by hyperactivity of individual Ca2+ release units.
Figure 3. Intracellular Ca2+ signalling in control and CPVT-iPSC-derived cardiomyocytes.

- A. Images of Fura-2 Ca2+ recordings depicting normal (R) and aberrant (AR1, AR2 and AR3) Ca2+ cycling in electrically stimulated iPSC-derived myocytes (top, from CPVT cells) and their percentage occurrence during pacing at either 0.5, 1.0 or 1.5 Hz (bottom). Red lines indicate electric stimulation and n the number of cells analysed.
- B. Bar graphs comparing the average resting intracellular Ca2+ ([Ca2+]i) before electrical stimulation started in control (black, n = 191) and CPVT (red, n = 211) myocytes from three different iPCS lines per group. Data are means ± SEM from four independent differentiation experiments.
- C,D. Average of diastolic and systolic [Ca2+]i in control (black) and CPVT (red) rhythmic myocytes during sequential pacing at 0.5, 1.0 and 1.5 Hz in absence (circles) and in presence (squares) of 10 µM isoproterenol. Between 4 and 42 cells were analysed per group; no rhythmic cells were observed with isoproterenol at 1.5 Hz. Data are means ± SEM. ***p < 0.001 versus CPVT and Control + Iso, ###p = 0.001 versus Control in C; *p = 0.04, ***p < 0.001 versus same group without isoproterenol in D; two-tailed t-test.
- E. Average (±SEM) of maximum caffeine-induced [Ca2+]i as measurement of SR Ca2+ content, in control (black) and CPVT (red) myocytes in absence (basal, n = 33 vs. n = 8 cells) and in presence of isoproterenol (n = 24 vs. n = 17 cells); *p = 0.03 versus control basal and CPVT + Iso, two-tailed t-test.
Figure 4. Ca2+ spark properties in control and CPVT-iPSC-derived myocytes.

- A,B. Panel (i) shows representative pseudo-coloured images of fluo-4-AM loaded control (A) and CPVT (B) myocytes in the absence (left) or in the presence (right) of 1 µM isoproterenol. Below, typical Ca2+ traces, recorded at 105 images/s, corresponding to each of the five individual regions of interest marked in the top images. Panel (ii) displays original line-scan images of Ca2+ sparks at a higher temporal resolution (1000 lines/s, top), a portion of the corresponding Ca2+ traces (middle), and 3D surface plots of representative Ca2+ sparks (bottom), highlighting the extended time course of CPVT-sparks.
- C. Summary of Ca2+ spark characteristics from control (black) and CPVT (red) myocytes in absence (basal) or presence of 1 µM isoproterenol. FWHM, full width at half maximum. Between 38 and 142 cells from three iPSC lines were analysed per group. Data are means ± SEM from four independent experiments; p-values from two-tailed t-test.
Since in CPVT patients tachycardia is restricted to the ventricles under stress condition, it might be expected that CPVT is a disease of ventricular cardiomyocytes. Our cardiac differentiation protocol leads to the generation of all three subtypes of cardiomyocytes, namely ventricular-like, atrial-like and nodal-like cells, which can be distinguished by the expression of specific myocytic lineage markers and by the shape of the action potential (Moretti et al, 2010b). Immunohistochemical analysis for ventricular and atrial myosin light chain 2 (MLC2v and MLC2a) proteins and electrophysiological measurements of action potentials in single iPSC-derived myocytes demonstrated that the ventricular subtype is largely predominant, accounting for 70–80% of all myocytes similarly in both control and CPVT groups (Fig 5A). To assess whether Ca2+ spark properties were specifically altered only in ventricular cells, we stained the cells with an antibody against MLC2v retrospectively and analysed spark data from ventricular (MLC2v+ cells) and non-ventricular (MLC2v− cells) myocytes separately. The observed differences in Ca2+ spark properties between control and CPVT cells persisted in the ventricular and non-ventricular subpopulations (Fig 5B), indicating that the mutated S406L-RYR2 channels are dysfunctional in all myocytes.
Figure 5. Myocytic subtypes of iPSC-derived myocytes and their Ca2+ spark properties.

- Percentage of ventricular-, atrial- and nodal-like myocytes after 3–4 month cardiac iPSC differentiation based on single cell electrophysiological measurements of action potentials (n = 47–50 cells) and expression of specific myocytic lineage markers (MLC2v, for ventricular cells, and MLC2a, for atrial cells) by immunohistochemistry (n = 100 cells). Scale bars, 10 µm. Dotted lines in the action potential traces indicate 0 mV.
- Summary of Ca2+ spark characteristics from control (black) and CPVT (red) cells under basal conditions when all myocytes or specifically ventricular (MLC2v+) and non-ventricular (MLC2v−) subtypes are analysed. Fold changes are relative to all myocytes control. Staining for MLC2v was performed after Ca2+ spark imaging. Between 21 and 113 cells from three iPSC lines were analysed per group. Data are means ± SEM from three independent experiments; p-values from two-tailed t-test.
Rescue of S406L-RYR2 malfunction by dantrolene in CPVT-iPSC-derived cardiomyocytes
Two mechanisms have been proposed to explain how CPVT-RYR2 mutations alter the sensitivity of the channel to luminal and/or cytosolic Ca2+ activation, leading to enhanced stress-induced diastolic Ca2+ release: (a) weakening of the interdomain interactions within the RYR2 channels, which destabilizes the closed state (‘domain unzipping’; George et al, 2007; Ikemoto & Yamamoto, 2000) and (b) disruption of critical interaction between the RYR2 channels and their modulating proteins (Priori & Chen, 2011; Wehrens et al, 2003). The N-terminal and central regions, although separated by ∼2000 residues in the linear sequence, interact with each other to form a “domain switch” that stabilizes the closed state of RYR channels (Liu et al, 2010; Yamamoto et al, 2000). Disturbance of this interaction leads to a prolongation of Ca2+ sparks (Uchinoumi et al, 2010), as observed in the CPVT myocytes. Docking of the recent crystal structure of RYR1 amino-terminal residues 1–559 into 3D reconstructions from cryo-electron microscopy of RYR1 has suggested that indeed multiple domain-domain interfaces are involved in disruption of Ca2+ regulation by various disease-causing mutations in RYR1 and RYR2 (Tung et al, 2010). Similar modelling for the N-terminal region of RYR2 has revealed that the S406L mutation is indeed located at the interface between two domains (Fig S5 of Supporting information). Thus, ‘domain unzipping’ is likely to be the pathomechanism of this mutation. To further verify this hypothesis, we investigated whether dantrolene, which is believed to stabilize the ‘domain switch’ by binding to a N-terminal sequence of skeletal and cardiac RYRs (Kobayashi et al, 2005, 2009, 2010; Paul-Pletzer et al, 2002, 2005; Wang et al, 2011), could suppress the impact of the S406L mutation in CPVT-iPSC-derived myocytes. Treatment with dantrolene restored normal Ca2+ spark properties in CPVT myocytes under basal conditions and corrected S406L-RYR2 hyperactivity induced by adrenergic stimulation, with minimal effects in control cells (Fig 6A). It has been demonstrated that elevated spontaneous Ca2+ release during diastole can be arrhythmogenic by activation of the Na+/Ca2+ exchanger, which generates a transient depolarizing current leading to DADs and triggered activity (TA; Schlotthauer & Bers, 2000). Therefore, we finally examined the incidence of spontaneous DADs/TA in iPSC-derived myocytes by measuring membrane potentials in single ventricular and atrial cells following electrical stimulation (Fig 6B–D and Fig S6 of Supporting information). All investigated cells presented no spontaneous activity before pacing and we did not observe any differences in resting membrane potential nor in the duration of electrically induced action potentials between equivalent subtype of myocytes in control and CPVT groups (Fig 6B and Fig S6C of Supporting information). When stimulated at 1 Hz, 56% of the control ventricular-like myocytes (5:9 cells) and 11% of the control atrial-like cells (1:9 cells) showed no spontaneous after-potentials and maintained stable resting voltage after electric pacing ended (Fig 6C, left, and Fig S6A of Supporting information). However, we observed DADs and TA in 88 and 89% of the ventricular-like and atrial-like CPVT cells, respectively (14:16 ventricular cells, Fig 6C, right; 8:9 atrial cells, Fig S6B of Supporting information). Moreover, when compared to control cells, diseased myocytes exhibited a much higher incidence of spontaneous action potentials after termination of pacing (1.4 ± 0.3/s versus 0.5 ± 0.3/s for ventricular and 1.1 ± 0.3/s versus 0.2 ± 0.1/s for the atrial cells). Interestingly, DADs and triggered arrhythmias were completely abolished by dantrolene treatment in all investigated CPVT cells (Fig 6D and Fig S6B of Supporting information), suggesting that a defective inter-domain interaction within the RYR2 is the underlying arrhythmogenic mechanism of the S406L mutation. Moreover, the rescue of the CPVT-disease phenotype in a patient-specific iPSC-based system by dantrolene provides the first evidence that, as in the case of malignant hyperthermia, correction of defective inter-domain interaction within mutated human RYR2 may represent an effective novel causal therapy for CPVT1.
Figure 6. Dantrolene corrects the disease phenotype in CPVT-iPSC-derived myocytes.
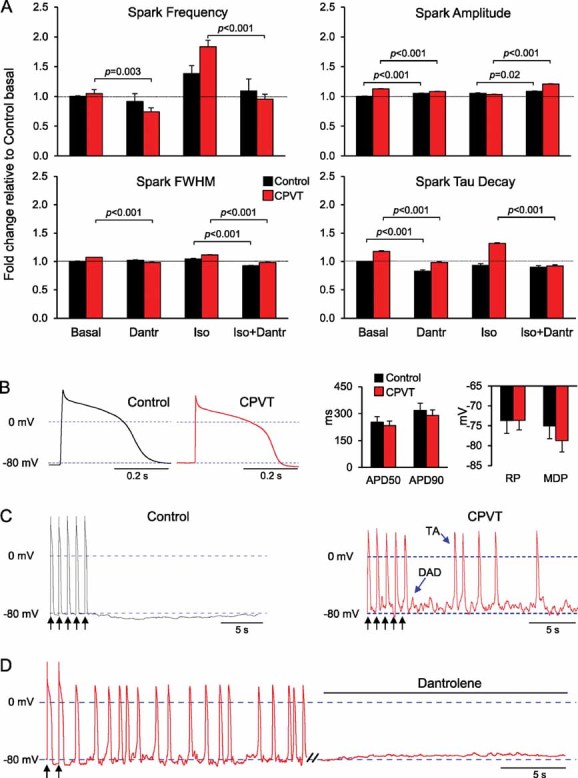
- Fold change of Ca2+ spark characteristics relative to control cells under basal conditions in control (black) and CPVT (red) myocytes after treatment with 10 µM dantrolene alone, 1 µM isoproterenol alone or both drugs combined. Between 32 and 142 cells from three iPSC lines were analysed per group. Data are means ± SEM from four independent experiments; p-values from one-way ANOVA followed by Tukey's test.
- Representative traces of electrically evoked action potentials from control (black) and CPVT (red) ventricular myocytes (left) and bar graphs of the averaged action potential duration at 50% (APD50) and 90% (APD90) repolarization, the maximum diastolic potential and the resting potential (right) during stimulation at 1 Hz.
- Typical action potential recordings from a control (black) and a CPVT (red) ventricular myocyte. Black arrows indicate the last five paced action potentials at 1 Hz stimulation; blue arrows mark an example of DAD and triggered activity.
- Representative action potential recording from a CPVT ventricular cell showing that superfusion with 10 µM dantrolene completely abolished DADs and TA. Black arrows indicate the last two paced action potentials at 1 Hz stimulation.
DISCUSSION
We have developed the first human stem cell-based model for CPVT1, bearing a novel S406L missense mutation in RYR2, and demonstrated its suitability to recapitulate molecular and physiological aspects of the disease phenotype. Until now, only heterologous expression systems and genetic mouse models have been used to study the cellular and molecular aspects of CPVT-linked RYR2 mutations. Based on these studies, two mechanisms for RYR2-mediated CPVT have been proposed. The first mechanism suggests that RYR2 mutants reduce the binding affinity of the channel for its auxiliary stabilizing protein FKBP12.6 and this is further aggravated in situation of catecholamine-induced hyperphosphorylation of RYR2 with consequential dissociation of FKBP12.6 and Ca2+ leakage from the SR (Lehnart et al, 2008; Marx et al, 2000; Wehrens et al, 2003). Although it may be possible that selected mutations alter FKBP12.6 binding to RYR2, this hypothesis has been recently challenged and increasing body of evidence clearly demonstrates that alterations in FKBP12.6-RYR2 interaction are unlikely to be the common cause of CPVT1 (George et al, 2003; Guo et al, 2010; Jiang et al, 2005; Liu et al, 2006; Xiao et al, 2007). Alternatively, it has been proposed that RYR2 mutations in the N-terminal and central regions of the protein weaken interactions between these two domains that are critical in stabilizing the closed state of the channel, resulting in an increased open probability and enhanced spontaneous Ca2+ release during stress-induced SR Ca2+ overload (Ikemoto & Yamamoto, 2000; Tateishi et al, 2009). In support of this ‘domain unzipping’ mechanism, dantrolene has been shown to suppress abnormal Ca2+ leak from mutated RYR1 and RYR2 by binding to a N-terminal sequence and stabilizing domain–domain contacts within the N-terminal and central regulatory regions (Kobayashi et al, 2005, 2009; Paul-Pletzer et al, 2002, 2005). Interestingly, dantrolene binding to RYR2 seems to be dependent on a particular conformational state of the channel that takes place only in disease conditions (Kobayashi et al, 2009; Paul-Pletzer et al, 2005). Therefore, the results that this drug rescues the disease phenotype in our patient-specific iPSC-based CPVT model would indicate that ‘domain unzipping’ is likely to be the pathomechanism of the novel N-terminal S406L-RYR2 mutation. Evidence for unzipping of the interaction between N-terminal and central domains raises the question of whether ‘domain unzipping’ is present in other regions of RYR2 in which CPVT mutations are located. Thus, it would be of great interest to investigate dantrolene effects in other patient-specific iPSC-CPVT models bearing different RYR2 mutations situated throughout the molecule. Our study on a human model of CPVT provides valuable insights into the pathophysiology of the disease and suggests dantrolene as a potential novel drug for the causal treatment of cardiac arrhythmias in CPVT1 patients carrying N-terminal mutations. Yet, clinical trials demonstrated that current treatments with β-blockers and ICDs are not fully protective in all CPVT patients and showed that these regimens are less attractive in CPVT compared to other forms of inherited ventricular tachycardia, such as long QT or Brugada syndrome (Kaufman, 2009). Although the precise mechanistic basis of CPVT most likely depends on the hotspot in which mutations are residing and hence might call for the development of location-specific drugs to address the functional heterogeneity, our work highlights the potential of human iPSCs in the emerging field of personalized medicine (Zhu et al, 2011) by demonstrating the ability to screen the effects of potential disease aggravators and novel customized treatment options.
MATERIALS AND METHODS
Human iPSC generation and cardiomyocyte differentiation
After approval by the institutional review board, we recruited a 24-year-old caucasian female CPVT patient and a 32-year-old female caucasian control without history of cardiac disease scheduled for plastic surgery to undergo dermal biopsy after obtaining written informed consent. Reprogramming of primary skin fibroblasts and differentiation into cardiomyocytes were performed as described previously (Moretti et al, 2010a, b). Briefly, fibroblasts were infected with retroviruses encoding OCT4, SOX2, KLF4 and c-MYC and cultured on murine embryonic feeder cells until iPSC colonies could be picked. EB differentiation was achieved by aggregating the cells on low-attachment plates and EBs were plated on gelatin-coated dishes at day 7. Spontaneously contracting areas were manually dissected and cultured further until day 90–130 of differentiation. Cells for physiological experiments were collagenase-dissociated into single cells, plated on fibronectin-coated glass coverslips, and analysed within 3–6 days.
Genomic sequencing and karyotype analysis
The presence of the RYR2-S406L mutation in the patient and its absence in the control and in blood samples of 100 coronary artery disease patients without CPVT was verified by polymerase chain reaction-based sequencing of genomic DNA isolated from skin fibroblasts, from iPSCs and from blood using a Genomic DNA Purification Kit (Gentra Systems). Karyotyping of the iPSC lines was performed at the Institute of Human Genetics of the Technical University Munich using standard methodology.
Quantitative real-time PCR
Total mRNA was isolated from fibroblasts, iPSC clones, EBs, and myocytic explants using the Stratagene Absolutely RNA kit. One microgram of total RNA was used to synthesize cDNA from fibroblasts, iPSC clones and EBs, using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). RNA from cardiomyocyte explants and from human adult and fetal heart (Clontech) was linearly amplified using the RNA Amplification RampUP Kit (Genisphere) and subsequently 1 µg of amplified RNA was used to synthetize cDNA. Gene expression was quantified by qRT-PCR using 1 µl of the RT reaction and the Power SYBR Green PCR Master Mix (Applied Biosystems). Gene expression levels were normalized to GAPDH or to TNNT2, as indicated in the dedicated figure legends. Primer sequences are provided in Table S1 of Supporting information.
Phenotypic characterization of iPSC lines and their differentiated progeny
Differential gene expression was assessed by qRT-PCR reaction as described previously (Moretti et al, 2010a, b). For histochemistry, cells were fixed with 3.7% v/v formaldehyde. Nuclei were visualized with Hoechst-33528 (1 µg/ml), F-actin with Phalloidin-Alexa-Fluor-594-conjugate (Invitrogen, 1:40), and alkaline phosphatase activity with NBT/BCIP substrate (Roche). Immunostaining was performed with standard protocols using the following primary antibodies: human NANOG (rabbit polyclonal, Abcam, 1:500), TRA1-81-Alexa-Fluor-488-conjugated (mouse monoclonal, BD Pharmingen, 1:20), RYR2 (mouse monoclonal, Thermo Scientific, 4 µg/ml), MLC2a (mouse monoclonal, Synaptic Systems, 5 µg/ml) and MLC2v (mouse monoclonal, Synaptic Systems, 5 µg/ml). Bright-field and fluorescence microscopy were performed using imaging systems (DMI6000-AF6000), filters and software from Leica microsystems. Confocal imaging (Leica SP5-II LSCM) was used to analyse expression of sarcomeric proteins and to assess RYR2 subcellular distribution. RYR2 cluster density was calculated based on particle counting after thresholding with ImageJ PlugIns (Wayne Rasant, NIH, Bethesda, USA). Western blotting on whole cell lysate from iPSC-derived cardiac explants and human adult heart (Imgenex) was performed with standard protocols using 20 µg proteins and the following primary antibodies: RYR2 (mouse monoclonal, Thermo Scientific, 0.4 µg/ml), PLB (mouse monoclonal, Thermo Scientific, 2 µg/ml), TRDN (goat polyclonal, Santa Cruz, 1 µg/ml), JCTN (goat polyclonal, Santa Cruz, 1 µg/ml), CASQ2 (rabbit polyclonal, Abcam, 0.08 µg/ml), β-actin (rabbit polyclonal, Abcam, 1:1000) and cTNT (mouse monoclonal, NeoMarkers, 0.2 µg/ml).
The paper explained
PROBLEM
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited cardiac disease that, under physical and emotional stress, leads to life-threatening arrhythmia followed by syncopes and sudden cardiac death at a young age in patients with structurally normal heart. Despite the very high mortality rate, no causative treatment exists and the development of new drugs is hampered by the difficulty of obtaining patient cardiac myocytes and maintaining them in culture without loss of their physiological properties.
RESULTS
Taking dermal fibroblasts from a 24-year-old woman with a diagnosis of familial CPVT, we generated iPSC lines that were subsequently differentiated into cardiomyocytes. These cardiomyocytes recapitulated, under catecholaminergic stress, all major hallmarks of the disease, such as elevated diastolic Ca2+ concentrations, a reduced SR Ca2+ content, and an increased susceptibility to arrhythmias. Additionally, we found the drug dantrolene to be protective and efficient in suppressing stress-induced arrhythmic events in CPVT cardiac myocytes.
IMPACT
In this study, we generated the first human model of CPVT. Our findings indicate that cardiomyocytes derived from CPVT patient-specific iPSCs can be used as an in vitro model system to study disease mechanisms, screen drug compounds for individual risk stratification and develop patient-specific therapies.
Physiological characterization of iPSC-derived cardiomyocytes
Intracellular free Ca2+ was measured at 20–22°C in cells loaded with fura-2-AM (5 µM for 20 min; Fluka, Buchs, Switzerland) in HEPES-buffered saline (in mM: NaCl (140), KCl (5.4), MgCl2 (1), CaCl2 (2), glucose (10), HEPES (10), pH 7.4) containing 0.1% w/v bovine serum albumin on a monochromator-equipped (Polychrome-V, TILL-Photonics, Gräfelfing, Germany) inverted microscope (Olympus-IX 71 with an UPlanSApo 20×/0.85 oil immersion objective). Fluorescence was excited at 340 and 380 nm, emission recorded at 22–24 Hz with a 14-bit EMCCD camera (iXON3 885, Andor, Belfast, UK), and Ca2+ concentrations calculated as described previously (Grynkiewicz et al, 1985). For field stimulation, 5 ms depolarizing voltage pulses at 90 V (Stimulator Type 201, Hugo Sachs Elektronik, March-Hugstetten, Germany) were applied using platinum electrodes (RC-37FS, Warner Instruments, Hamden, USA). Drugs were applied by solution exchange via continuous perfusion. Caffeine (100 mM) was applied 25 s after the stimulation period.
Spontaneous Ca2+ sparks were imaged at 20–22°C in cells loaded with fluo-4-AM (0.6 µM for 30 min, Invitrogen) in extracellular solution (in mM: NaCl (135), KCl (5.4), MgCl2 (2), CaCl2 (1.8), HEPES (10), glucose (10), pH 7.35) on an inverted confocal microscope (Leica SP5-II LSCM) through a 63x oil immersion objective (HCX PL APO, 1.4, Leica), exciting with the 488 nm line of an Arg/Kr laser (Lasos, Jena, Germany) and collecting emission at 495–600 nm, acquiring 512 × 120 pixel frames at 105 Hz using the resonant scanner, keeping laser, spectral and gain settings constant throughout all experiments. Time series (each 1000 images) were recorded at a 1-min interval. Drug effects were assessed 10–15 min after manual solution exchange, which was verified not to alter Ca2+ spark properties by a mock solution exchange. Images stored in a database (OMERO, http://www.openmicroscopy.org) were analysed off-line using a custom-designed algorithm performing automatic cell- and spark-detection and subsequent fitting of the single spark fluorescence distributions with a 2D Gauss over time approach.
Myocyte action potentials were recorded at 35 ± 0.5°C in the current clamp mode of the perforated patch-clamp technique using 300 µg/ml water-soluble amphotericin B (Sigma–Aldrich, Deisenhofen, Germany) in the pipette solution (in mM: KCl (30), K-aspartate (110), MgCl2 (1), HEPES (10), EGTA (0.1), pH 7.2), sampling at 10 kHz with an EPC10 patch-clamp amplifier (HEKA, Lambrecht, Germany). Cells were superfused with bath solution (in mM: NaCl (135), KCl (5), MgCl2 (1), CaCl2 (2), glucose (10), HEPES (10), pH 7.4), additionally containing 10 µM dantrolene where indicated. The liquid junction potential was +13.8 mV and offset corrections were made by the Patchmaster software. Pipette series resistance ranged from 5.5 to 19 MΩ. Perforation started shortly after seal formation and reached steady state within 3–5 min.
All experiments and analysis were performed by investigators blinded to the genotype of the cells.
Statistical analysis
Data that passed tests for normality and equal variance were analysed with the use of Student's t-test or one-way analysis of variance followed by Tukey's test, when appropriate. Two-sided p-values of <0.05 were considered statistically significant. All data are shown as means ± SEM.
For more detailed Materials and Methods see the Supporting information.
Author contributions
KLL, DS, TG and PL conceived the experiments; CBJ, AM, and MB generated and characterized control and CPVT iPSCs; Generation and characterization of human cardiomyocytes was performed by CBJ, LI, MB, TD, AG and JTL; CBJ performed histochemistry/immunostaining with LI and SR and western blotting with TD; MMS, US and SP conducted Ca2+ imaging and electrophysiological measurements and PL Ca2+ spark experiments; RJD and MS recruited the CPVT patient family; DS performed docking structure modelling; KLL, DS and AM wrote the manuscript.
Acknowledgments
We thank T. Kitamura and S. Yamanaka for providing viral vectors through Addgene, and especially the member of the CPVT family and the healthy volunteers who provided us with skin biopsies for the reprogramming. We would like to acknowledge Diana Grewe and Christina Scherb for their technical assistance in cell culture and Gabi Lederer (Cytogenetic Department, TUM) for karyotyping. This work was supported by grants from the European Research Council (Marie Curie Excellence Team Grant MEXT-23208; ERC 261053–CHD-iPS), the German Research Foundation (Research Unit 923, Mo 2217/1-1 and La 1238 3-1/4-1) and the German Ministry for Education and Research (01 GN 0826). CBJ was supported by a scholarship (AFR-PHD-09-169) granted by the National Research Fund, Luxembourg.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Berridge MJ. Cardiac calcium signalling. Biochem Soc Trans. 2003;31:930–933. doi: 10.1042/bst0310930. [DOI] [PubMed] [Google Scholar]
- Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation–contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. 2009;73:1561–1567. doi: 10.1253/circj.cj-09-0478. [DOI] [PubMed] [Google Scholar]
- George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- George CH, Jundi H, Thomas NL, Fry DL, Lai FA. Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol. 2007;42:34–50. doi: 10.1016/j.yjmcc.2006.08.115. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MK, Illich DJ, Gaarz A, Matzkies M, Nguemo F, Pfannkuche K, Liang H, Classen S, Reppel M, Schultze JL, et al. Global transcriptional profiles of beating clusters derived from human induced pluripotent stem cells and embryonic stem cells are highly similar. BMC Dev Biol. 2010;10:98. doi: 10.1186/1471-213X-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- Ikemoto N, Yamamoto T. Postulated role of inter-domain interaction within the ryanodine receptor in Ca(2+) channel regulation. Trends Cardiovasc Med. 2000;10:310–316. doi: 10.1016/s1050-1738(01)00067-6. [DOI] [PubMed] [Google Scholar]
- Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- Kaufman ES. Mechanisms and clinical management of inherited channelopathies: long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm. 2009;6:S51–S55. doi: 10.1016/j.hrthm.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Hoshijima M, Chien KR. Reversing advanced heart failure by targeting Ca2+ cycling. Annu Rev Med. 2008;59:13–28. doi: 10.1146/annurev.med.59.052407.103237. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Bannister ML, Gangopadhyay JP, Hamada T, Parness J, Ikemoto N. Dantrolene stabilizes domain interactions within the ryanodine receptor. J Biol Chem. 2005;280:6580–6587. doi: 10.1074/jbc.M408375200. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, Xu X, Uchinoumi H, Okuda S, Yamamoto T, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993–2005. doi: 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010;74:2579–2584. doi: 10.1253/circj.cj-10-0680. [DOI] [PubMed] [Google Scholar]
- Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F. Dantrolene—a review of its pharmacology, therapeutic use and new developments. Anaesthesia. 2004;59:364–373. doi: 10.1111/j.1365-2044.2004.03658.x. [DOI] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J Mol Cell Cardiol. 2009;46:149–159. doi: 10.1016/j.yjmcc.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang R, Tian X, Zhong X, Gangopadhyay J, Cole R, Ikemoto N, Chen SR, Wagenknecht T. Dynamic, inter-subunit interactions between the N-terminal and central mutation regions of cardiac ryanodine receptor. J Cell Sci. 2010;123:1775–1784. doi: 10.1242/jcs.064071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Jung CB, Thies TM, Takashima Y, Bernshausen A, Schiemann M, Fischer S, Moosmang S, Smith AG, et al. Mouse and human induced pluripotent stem cells as a source for multipotent Isl1+ cardiovascular progenitors. FASEB J. 2010a;24:700–711. doi: 10.1096/fj.09-139477. [DOI] [PubMed] [Google Scholar]
- Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flugel L, Dorn T, Goedel A, Hohnke C, Hofmann F, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010b;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Paul-Pletzer K, Yamamoto T, Bhat MB, Ma J, Ikemoto N, Jimenez LS, Morimoto H, Williams PG, Parness J. Identification of a dantrolene-binding sequence on the skeletal muscle ryanodine receptor. J Biol Chem. 2002;277:34918–34923. doi: 10.1074/jbc.M205487200. [DOI] [PubMed] [Google Scholar]
- Paul-Pletzer K, Yamamoto T, Ikemoto N, Jimenez LS, Morimoto H, Williams PG, Ma J, Parness J. Probing a putative dantrolene-binding site on the cardiac ryanodine receptor. Biochem J. 2005;387:905–909. doi: 10.1042/BJ20041336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–e26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- Scheinman MM, Lam J. Exercise-induced ventricular arrhythmias in patients with no structural cardiac disease. Annu Rev Med. 2006;57:473–484. doi: 10.1146/annurev.med.57.081704.090151. [DOI] [PubMed] [Google Scholar]
- Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tateishi H, Yano M, Mochizuki M, Suetomi T, Ono M, Xu X, Uchinoumi H, Okuda S, Oda T, Kobayashi S, et al. Defective domain–domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NL, Maxwell C, Mukherjee S, Williams AJ. Ryanodine receptor mutations in arrhythmia: the continuing mystery of channel dysfunction. FEBS Lett. 2010;584:2153–2160. doi: 10.1016/j.febslet.2010.01.057. [DOI] [PubMed] [Google Scholar]
- Tung CC, Lobo PA, Kimlicka L, Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhong X, Meng X, Koop A, Tian X, Jones PP, Fruen BR, Wagenknecht T, Liu Z, Chen SR. Localization of the dantrolene-binding sequence near the FK506-binding protein-binding site in the three-dimensional structure of the ryanodine receptor. J Biol Chem. 2011;286:12202–12212. doi: 10.1074/jbc.M110.194316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, Zhang L, Duff HJ, Gillis AM, Fleischer S, et al. Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Lensch MW, Cahan P, Daley GQ. Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011;12:266–275. doi: 10.1038/nrg2951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.