

Abstract
Secreted Semaphorin 3E (Sema3E) promotes cancer cell invasiveness and metastatic spreading. The pro-metastatic activity of Sema3E is due to its proteolytic fragment p61, capable of transactivating the oncogenic tyrosine kinase ErbB2 that associates with the Sema3E receptor PlexinD1 in cancer cells. Here, we show that a mutated, uncleavable variant of Sema3E (Uncl-Sema3E) binds to PlexinD1 like p61-Sema3E, but does not promote the association of PlexinD1 with ErbB2 nor activates the ensuing signalling cascade leading to metastatic spreading. Furthermore, Uncl-Sema3E competes with endogenous p61-Sema3E produced by tumour cells, thereby hampering their metastatic ability. Uncl-Sema3E also acts independently as a potent anti-angiogenic factor. It activates a PlexinD1-mediated signalling cascade in endothelial cells that leads to the inhibition of adhesion to extracellular matrix, directional migration and cell survival. The putative therapeutic potential of Uncl-Sema3E was validated in multiple orthotopic or spontaneous tumour models in vivo, where either local or systemic delivery of Uncl-Sema3E-reduced angiogenesis, growth and metastasis, even in the case of tumours refractory to treatment with a soluble vascular endothelial growth factor trap. In summary, we conclude that Uncl-Sema3E is a novel inhibitor of tumour angiogenesis and growth that concomitantly hampers metastatic spreading.
Keywords: angiogenesis, metastasis, plexin, semaphorin, tumour growth
→ See accompanying article http://dx.doi.org/10.1002/emmm.201200206
INTRODUCTION
Semaphorins are evolutionarily conserved signalling molecules, initially identified as axonal guidance cues, and now thought to regulate a wide range of developmental and pathological processes including angiogenesis and cancer progression (Capparuccia & Tamagnone, 2009; Neufeld & Kessler, 2008; Zhou et al, 2008). Semaphorin receptors are found in the families of plexins and neuropilins. The associated signalling pathways start to be understood and often involve negative regulation of monomeric GTPases controlling cell-substrate adhesion and cell migration. However, in addition to this, certain semaphorins and plexins can trigger the activation of plexin-associated tyrosine kinases in a cell context-dependent manner, leading to activation of distinctive signalling cascades that promote cell migration and invasive growth (Franco & Tamagnone, 2008). Due to their emerging role in tumour angiogenesis, tumour growth and metastasis, semaphorins appear to be promising molecular targets controlling cancer progression. Intriguingly, certain semaphorins can regulate both cancer cells and stromal cells in the tumour microenvironment, leading to concomitant functional effects in vivo (reviewed by Capparuccia & Tamagnone, 2009).
Semaphorin 3E (Sema3E) has been found in metastatic tumour cells (Christensen et al, 1998, 2005) and it was then shown to be crucially involved in developmental and post-ischemic angiogenesis as an endothelial-repelling signal (Fukushima et al, 2011; Gu et al, 2005; Kim et al, 2011). Moreover, Sema3E can differentially regulate distinctive neuronal populations, mediating either axonal repulsion or attraction (Chauvet et al, 2007). Notably, unlike other class 3 secreted semaphorins, Sema3E binds directly to the PlexinD1 receptor and is not dependent on neuropilin co-receptors (Gu et al, 2005). We have recently shown that Sema3E-PlexinD1 signalling triggers two distinct pathways in cancer cells and in cells of the tumour microenvironment (Casazza et al, 2010). On one hand, endothelial cells and developing vessels are typically repelled by Sema3E, acting as an anti-angiogenic factor through PlexinD1 signalling (Casazza et al, 2010); this pathway seems to implicate an endothelial-specific regulation of the GTPases Arf6 (Sakurai et al, 2010) and RhoJ (Fukushima et al, 2011), affecting integrin-mediated adhesion and cytoskeletal remodelling. In cancer cells instead, Sema3E signalling promotes invasiveness and metastatic spreading in vivo, and this activity is mediated by the trans-activation of erythroblastic leukemia viral oncogene homolog 2 (ErbB2)-epidermal growth factor receptor (EGFR) oncogenic tyrosine kinase receptors (Casazza et al, 2010). In addition, Sema3E levels in human tumour samples were found to correlate with metastatic progression (Casazza et al, 2010). Therefore, although Sema3E could strongly inhibit angiogenesis in vivo, its potential application for the blockade of tumour progression is actually excluded by its intrinsic pro-metastatic activity.
Interestingly, it has been shown that the functional activity of class 3 semaphorins is subject to regulation by proteolytic cleavage due to furin-like pro-protein-convertases (Adams et al, 1997; Christensen et al, 2005; Kutschera et al, 2011; Varshavsky et al, 2008). While in most cases this processing was reported to blunt semaphorin signalling, in the case of Sema3E, it is actually required for revealing its pro-metastatic activity in cancer cells (Christensen et al, 2005). In fact, the proteolytic fragment p61 is the active and predominant form of Sema3E found in invasive and metastatic cancer cells (Casazza et al, 2010).
In this study, we have employed a point-mutated Sema3E isoform resistant to furin-mediated cleavage, named Uncl-Sema3E, in order to dissect the two distinctive signalling cascades mediated by this semaphorin in cancer progression. In fact, Uncl-Sema3E displayed potent endothelial-repelling and anti-angiogenic activities, thereby dramatically suppressing tumour growth; and yet, it was devoid of the pro-metastatic effect of p61-Sema3E. We found that Uncl-Sema3E competes with p61-Sema3E for binding to the receptor; thus, it can efficiently block the signalling of endogenous p61 released by cancer cells and blunt its autocrine pro-invasive and pro-metastatic activity. We have furthermore demonstrated the potential therapeutic efficacy of the systemic delivery of Uncl-Sema3E, acting as a dual anti-angiogenic and anti-metastatic molecule in multiple preclinical models of cancer progression in mice.
RESULTS
Full-length uncleaved Sema3E antagonizes mature p61-Sema3E and blocks its pro-metastatic signalling in cancer cells
Sema3E signalling in tumour cells promotes invasion and metastasis, but these activities crucially depend on its proteolytic maturation into the smaller fragment p61 (Christensen et al, 2005). In fact, a mutated ‘uncleavable’ full-length Sema3E, which cannot be targeted by furins and converted into p61 (Uncl-Sema3E; Fig 1A), did not promote metastasis formation in experiments based on direct cancer cell injection in the circulation (Christensen et al, 2005). Here, we also show that, in contrast to p61, Uncl-Sema3E does not induce cancer cell migration (Fig 1B). We have recently reported that the pro-invasive and pro-metastatic activity of p61 is due to the association between PlexinD1 and the oncogenic tyrosine-kinase receptor ErbB2 as well as the induction of ErbB2 tyrosine phosphorylation and intracellular signalling (Casazza et al, 2010). We therefore asked whether the proteolytic processing of Sema3E may be required for its ability to trigger ErbB2 signalling in cancer cells. Notably, Uncl-Sema3E was unable to induce the association of ErbB2 with PlexinD1 (Fig 1C, top), nor did it promote ErbB2 phosphorylation (Fig 1C, middle) or intracellular mitogen-activated protein kinase (MAPK) signalling (Fig 1D).
Figure 1. Uncl-Sema3E is unable to trigger ErbB2 activation and cancer cell migration.
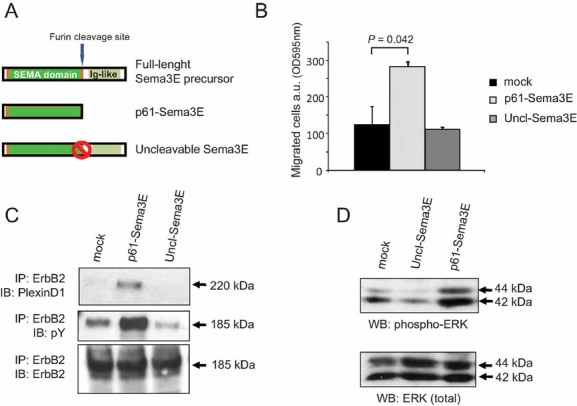
- Schematic diagram summarizing Sema3E isoforms: the full-length precursor of 87 kDa (p87), the proteolytically cleaved fragment p61 and the mutated uncleavable molecule (Uncl-Sema3E).
- Migration of MDA-MB435 cancer cells assayed in Transwell inserts in the presence of 7 nM Uncl-Sema3E, or 7 nM p61-Sema3E, or mock. Migrated cells were quantified by staining with crystal violet and measuring the absorbance at 595 nm (see Materials and Methods Section). Data are given as average ± SD of two independent experiments performed in duplicate (n = 4).
- Serum-starved A549 cancer cells were treated with either purified 7 nM p61-Sema3E, or 7 nM Uncl-Sema3E, or mock, for 10 min. The induced association between endogenous PlexinD1 receptor and ErbB2 tyrosine kinase was revealed by immunoblotting. The data are representative of three independent experiments yielding consistent results.
- A549 carcinoma cells were treated as above, and total protein lysates were analyzed by immunoblotting with phospho-specific antibodies to detect activated forms of MAPK, as well as with the respective non-phospho-specific antibody to provide loading controls.
Despite this apparent lack of activity, we found that Uncl-Sema3E is fully capable of binding the receptor PlexinD1 in analogy to p61-Sema3E isoform (Fig 2A). Moreover, we found that Uncl-Sema3E selectively competes with p61 for binding the receptor in the same nanomolar concentration range (Fig 2B). We then asked whether Uncl-Sema3E may potentially act as a dominant-negative molecule by interfering with p61 signalling. Indeed, ErbB2 transactivation elicited by p61 in A549 cells was strikingly reduced in the presence of Uncl-Sema3E (Fig 2C). Furthermore, data shown above in Fig 1C revealed that the basal level of ErbB2 tyrosine phosphorylation decreased in cells treated with Uncl-Sema3E, consistent with its functional competition with endogenous p61.
Figure 2. Uncl-Sema3E antagonizes p61 and blocks its pro-metastatic signalling in cancer cells.

- COS cells transfected to express PlexinD1 were probed with alkaline phosphatase (AP)-conjugated Uncl-Sema3E putative ligand. The binding was revealed by incubation with an AP substrate (see Supporting Information Methods). Scale bar: 30 µm.
- PlexinD1-expressing COS cells (as above) were incubated with AP-labelled p61-Sema3E (7 nM) in combination with increasing concentrations of Uncl-Sema3E (precisely: 3, 7, 15, 60 nM) (n = 6 for each experimental condition). Equimolar concentration of Uncl-Sema3E strongly reduced p61-Sema3E binding to PlexinD1, which was almost abrogated upon excess competition. Statistical significance was calculated by t-test with respect to p61-Sema3E binding in the absence of Uncl-Sema3E: *p = 1.3E−06; **p = 3.2E−08; §p = 1.4E−08; §§p = 1.5E−08. The binding of the independent semaphorin Sema4D to COS cells overexpressing its cognate receptor PlexinB1 provided a specificity control.
- Serum starved A549 cancer cells were incubated with 7 nM Uncl-Sema3E or mock for 1 h. Cells were then stimulated with mock, 7 nM p61, or 0.2 nM Heregulin-β1 (as positive control) in the presence of Uncl-Sema3E. ErbB2 tyrosine phosphorylation was revealed by immunoblotting.
- Migration of A549 and 4T1 cells expressing Uncl-Sema3E or control-EV, assayed in Transwell inserts (n = 4). Data shown indicate fold change versus respective controls.
- Fluorescently labelled A549 cells, either expressing Uncl-Sema3E or control-EV (same as above), were injected intravenously in nude mice. Forty-eight hours later the mice were sacrificed, and the lungs fixed and analyzed under a fluorescence microscope to reveal and quantify extravasated cancer cells colonizing the tissue (n = 5; see Materials and Methods Section for details). Representative images are shown on the right; scale bars: 100 µm.
- A549 and 4T1 cells expressing Uncl-Sema3E or control-EV were injected intravenously in nude mice (n = 5 and 6, respectively, for the two cell lines). Superficial metastatic colonies in the lungs were counted under a stereomicroscope; the graph indicates average values ± SD. Representative macroscopic images of the lungs are also shown, where red arrows indicate some of the metastatic lesions. Scale bars: 200 µm.
Altogether, these data suggested that Uncl-Sema3E, by blocking the transactivation of ErbB2, could interfere with the invasive-metastatic effect driven by p61-Sema3E. Intriguingly, earlier studies had tested the activity of full-length uncleavable Sema3E in non-metastatic 168-FARN mouse mammary carcinoma cells, which express very low levels of endogenous Sema3E (Christensen et al, 2005; and Supporting Information Fig 1). In contrast, we found that p61-Sema3E and the receptor PlexinD1 are well expressed in several cancer cells, establishing autocrine signalling circuits, which drive invasion and metastasis (Casazza et al, 2010). Therefore, in order to validate a potential dominant-negative effect of Uncl-Sema3E in vitro and in vivo, we ectopically expressed this mutant in two metastatic cancer cells with autocrine p61 signalling. First, we used 4T1 mouse mammary carcinoma cells, which are syngenic with (and have the same origin as) 168-FARN cells, but were selected for their strong metastatic behaviour (Aslakson & Miller, 1992); intriguingly, these cells carry high endogenous levels of Sema3E (Casazza et al, 2010; also see Supporting Information Fig 1). In addition, similar experiments were performed in A549 human lung carcinoma cells, which we also found to depend on endogenous p61 (Casazza et al, 2010). Uncl-Sema3E overexpression did not affect endogenous levels of p61-Sema3E secreted by cancer cells (Supporting Information Fig 2A) nor formed heterodimeric complexes with p61 (Supporting Information Fig 2B and C). Notably, although cancer cell viability was not impaired by Uncl-Sema3E overexpression (Supporting Information Fig 3A and B), we found that spontaneous cell migration was remarkably reduced in both models (Fig 2D). This was not associated with detectable changes in cellular phenotype or modified expression of the typical markers of epithelial–mesenchymal transition (Supporting Information Fig 3C–E), while it could be explained by the reduced activation of ErbB2 signalling demonstrated above. Moreover, we found that Uncl-Sema3E dramatically impaired the metastatic potential of tumour cells in vivo, as observed 48 h after injection of fluorescently labelled cells in the circulation (Fig 2E) as well as at later stages by counting macroscopic metastatic colonies in the lungs (Fig 2F). These results phenocopied those observed upon ribonucleic acid (RNA) interference (RNAi)-mediated Sema3E silencing in cancer cells (Casazza et al, 2010) and further suggested a dominant-negative function of Uncl-Sema3E, antagonizing p61-driven metastatic extravasation of tumour cells.
Uncl-Sema3E inhibits integrin-mediated adhesion and intracellular signalling in endothelial cells
Further investigating the functional relevance of uncleaved Sema3E, we observed that it could trigger the typical semaphorin-driven ‘collapsing’ response in COS cells expressing PlexinD1 (unpublished observation). Moreover, Uncl-Sema3E strongly and rapidly induced the retraction of cellular processes and the rounding of human umbilical vein-derived endothelial cells (HUVEC) (Figure 3A and Supporting Information Movies 1A–C). These effects have been reported for several semaphorins and are currently explained by the inhibitory activity mediated by plexins on integrin-based focal adhesive complexes (Barberis et al, 2004). Notably, a recent study showed that Sema3E triggers integrin-beta1 internalization leading to reduced cell-substrate adhesion (Sakurai et al, 2010). We therefore investigated the ability of Uncl-Sema3E to inhibit integrin function and intracellular signalling in endothelial cells. We found that as little as 5 min of treatment with Uncl-Sema3E were sufficient to trigger a dramatic disassembly of focal adhesion complexes (Fig 3B); these structures connect integrins with the cytoskeleton and are pivotally involved in the protrusion of cellular processes as well as in cell migration dynamics. Moreover, the presence of integrin-beta1 molecules in active conformation on the cell surface was significantly reduced upon treatment with Uncl-Sema3E (Fig 3C and Supporting Information Fig 4), further indicating reduced adhesion to the extracellular matrix. We also found that pre-treating endothelial cells with antibodies directed against the active conformation of integrin-beta1, which forcibly stabilizes cell-substrate adhesion, was sufficient to prevent cell contraction induced by Uncl-Sema3E (Supporting Information Fig 5A). This indicated that the loss of cell-substrate adhesion is crucial for the inhibitory activity of Uncl-Sema3E in endothelial cells. Notably, several studies underlined that substrate adhesion is a major requirement for the viability of endothelial cells, and its default leads to programmed cell death by anoikis, both in culture and in vivo (reviewed by Cheresh & Stupack, 2008). We found that focal adhesion disassembly triggered by Uncl-Sema3E further inhibited downstream intracellular signalling cascades, especially the activation of focal adhesion kinase (FAK) and the associated upregulation of phosphorylated MAPK/ERK (Fig 3D). It was shown previously that molecules interfering with FAK and subsequent MAPK activation in endothelial cells lead to an apoptotic response (Lu & Rounds, 2011). In fact, we observed an increased number of active-caspase 3-positive apoptotic endothelial cells upon treatment with Uncl-Sema3E as assessed by two independent methods (Fig 3E and F).
Figure 3. Uncl-Sema3E inhibits integrin-mediated adhesion and intracellular signalling in endothelial cells.

- HUVEC were stained with FITC-phalloidin to reveal F-actin (in green) and 4,6-diamidino-2-phenylindole (DAPI) to highlight nuclei. Upon incubation with 7 nM Uncl-Sema3E for 20 min, endothelial cells totally disassembled F-actin stress fibers, and cellular processes were retracted, leading to the typical collapsed phenotype (see Barberis et al, 2004). Scale bar: 50 µm.
- HUVEC were treated with 7 nM Uncl-Sema3E for the indicated times (or left untreated) and then subjected to immunofluorescence analysis by staining with anti-Paxillin antibodies (revealed in red) and with FITC-falloidin to detect F-actin (in green). On the right, images of cellular processes are magnified to reveal focal adhesions (FA) and the percent fraction of FA-containing cells is indicated aside. Scale bars: 20 and 10 µm in the magnified fields shown on the right.
- The graph summarizes the results of immunofluorescence analysis shown in Supporting Information Fig 4, and indicates the amount of integrin-β1 in active conformation on the surface of HUVEC, either untreated or treated with 7 nM Uncl-Sema3E for 5 min; n = 4 (each condition).
- HUVEC were treated with 7 nM Uncl-Sema3E for the indicated times (or left untreated) and their protein lysates were subjected to immuno-blotting analysis with antibodies directed against Y397 phosphorylated focal adhesion kinase (P-FAK), serine/threonine phosphorylated MAPK and AKT and vinculin (as protein loading control).
- HUVEC grown on glass coverslips were treated with 7 nM Uncl-Sema3E (n = 4) or with 200 nM staurosporin (STS; n = 4) for 1 h, or left untreated (n = 4). The early apoptotic marker activated-caspase 3 was revealed by immunofluorescence (shown in red); nuclei were counterstained with DAPI; scale bar: 50 µm. The graph indicates the average percent fraction of apoptotic cells in each condition ± SD; *p = 0.008; **p = 0.0002 (vs. control).
- HUVEC were treated with 7 nM Uncl-Sema3E for the indicated times (or left untreated) and then subjected to cytofluorimetric analysis with antibodies directed against activated-caspase 3. The experiment was repeated three times with consistent results.
Uncl-Sema3E inhibits endothelial cell migration and tube formation
Consistent with its potent regulatory activity on integrin function and cytoskeletal dynamics, we found that Uncl-Sema3E strongly inhibited endothelial cell migration in a PlexinD1-dependent manner (Fig 4A); notably, this activity paralleled that of the proteolytic fragment p61 (Casazza et al, 2011). Moreover, we found that this inhibitory effect of Sema3E in endothelial cells is dependent on the expression of the intracellular adaptor molecule Rnd2 (Supporting Information Fig 5B). We further assessed the activity of Uncl-Sema3E in HUVEC engaged in vascular tube formation. In fact, HUVEC grown in vitro on a basement membrane matrix (Matrigel) undergo spontaneous alignment into hollow tubes, forming capillary-like networks within 24 h (Grant et al, 1989). However, we found that endothelial tubule formation and stability was significantly impaired in the presence of Uncl-Sema3E compared to untreated cultures (Fig 4B). Moreover, when HUVEC were grown as spheroids in a three-dimensional collagen matrix in the presence of basic fibroblast growth factor (bFGF), they formed elongated sprouts, which were significantly reduced in the presence of either Uncl-Sema3E or the proteolytic fragment p61 (Fig 4C). These data further indicated that full-length uncleaved Sema3E is not an inactive precursor molecule, since it can elicit a potent PlexinD1-mediated inhibitory response in endothelial cells. Moreover, they demonstrated that the endothelial-repelling function of Sema3E pre-exists to, and it is not significantly blocked by, the proteolytic processing of the molecule. Taken together, these findings characterize Uncl-Sema3E as a partial agonist of PlexinD1 as compared to the cleaved mature p61 fragment. In fact, Uncl-Sema3E is able to bind to Plexin-D1 and elicits endothelial cell repulsion and anti-angiogenic activity in vitro, but unlike p61, it is unable to trigger the ErbB2-dependent pathway in cancer cells, and instead, it antagonizes this major signal promoting invasion and metastasis.
Figure 4. Uncl-Sema3E inhibits endothelial cell migration and tube formation.

- The haptotactic migration of HUVEC was assayed in Transwell inserts. In the presence of Uncl-Sema3E or p61-Sema3E (7 nM) endothelial cells migration was inhibited, in PlexinD1-dependent manner (PLXND1 knock-down was achieved by shRNA expression, see Materials and Methods Section). Data are given as average ± SD of two independent experiments performed in duplicate (n = 4).
- HUVEC were grown in Matrigel-coated wells for 24 h (see Materials and Methods Section) in the presence or absence of purified p61-Sema3E or Uncl-Sema3E. Representative phase-contrast microscopy images are shown on the left (scale bar: 200 µm). The assay was repeated several times (n = 4) and quantified by counting bifurcations of vessel-like tubular structures in multiple microscopic fields (the graph on the right shows mean values ± SD from at least five separate wells for each condition).
- HUVEC spheroids (containing approximately 400 cells each) were implanted in collagen gels, in the presence or absence of bFGF (see Materials and Methods Section for details). Moreover, either Uncl-Sema3E or p61 (1 µg/ml each) were included with bFGF in some of the wells (n = indicated in figure below respective bars). Spheroids were photographed after 24 h (representative images are shown; scale bar: 100 µm) and endothelial sprouts were quantified as described (Shraga-Heled et al, 2007).
Uncl-Sema3E inhibits tumour angiogenesis and tumour growth in vivo
A potential regulatory function of full-length uncleaved Sema3E in primary tumour development had never been investigated. We therefore assessed the activity of Uncl-Sema3E in multiple experimental models of tumour progression in mice. Initially, we exploited tumorigenic and weakly metastatic human MDA-MB-435 cancer cells that express PlexinD1 receptor but contain relatively low endogenous levels of Sema3E (see expression analysis in Supporting Information Fig 1). Notably, the over-expression of Uncl-Sema3E did not induce significant changes in the tumour cell proliferation rate in culture, even upon serum deprivation (Fig 5A and Supporting Information Fig 3A and B), consistent with our previous findings concerning p61-Sema3E isoform (Casazza et al, 2010). On the other hand, both Uncl-Sema3E and p61-Sema3E caused a striking inhibition of tumour growth upon subcutaneous transplantation of the overexpressing cells in immunodeficient mice (Fig 5B and C), putatively consistent with a paracrine effect in the tumour microenvironment. The histological analysis of explanted tumours actually revealed that Uncl-Sema3E had strongly reduced vessel density (Fig 5D). This was also confirmed in tumour samples explanted at an earlier stage when the size of control and Uncl-Sema3E-expressing xenografts was almost comparable (Supporting Information Fig 6A), consistent with the idea that the paracrine anti-angiogenic activity of this molecule was primarily responsible for tumour suppression. Moreover, we found that Uncl-Sema3E (and p61) impaired pericyte coverage of blood vessels (Fig 5E and Supporting Information Fig 6B), leading to reduced vessel functionality and perfusion (Fig 5F) as well as increased vessel permeability (Supporting Information Fig 6C). These data prompted us to investigate a potential regulatory activity of Uncl-Sema3E on vascular pericyte recruitment. In fact, we found that endothelial cells exposed to Uncl-Sema3E down-regulated the expression of PDGF-B, a major factor recruiting mural cells to vessels (Supporting Information Fig 7A). Moreover, the migration of primary microvascular pericytes in vitro was significantly inhibited by Uncl-Sema3E in a PlexinD1-dependent manner (Supporting Information Fig 7B). Thus, in addition to a direct inhibitory effect on endothelial cell adhesion, migration and survival, Uncl-Sema3E destabilized tumour vessels by interfering with pericyte recruitment.
Figure 5. Uncl-Sema3E inhibits tumour angiogenesis and tumour growth in vivo.
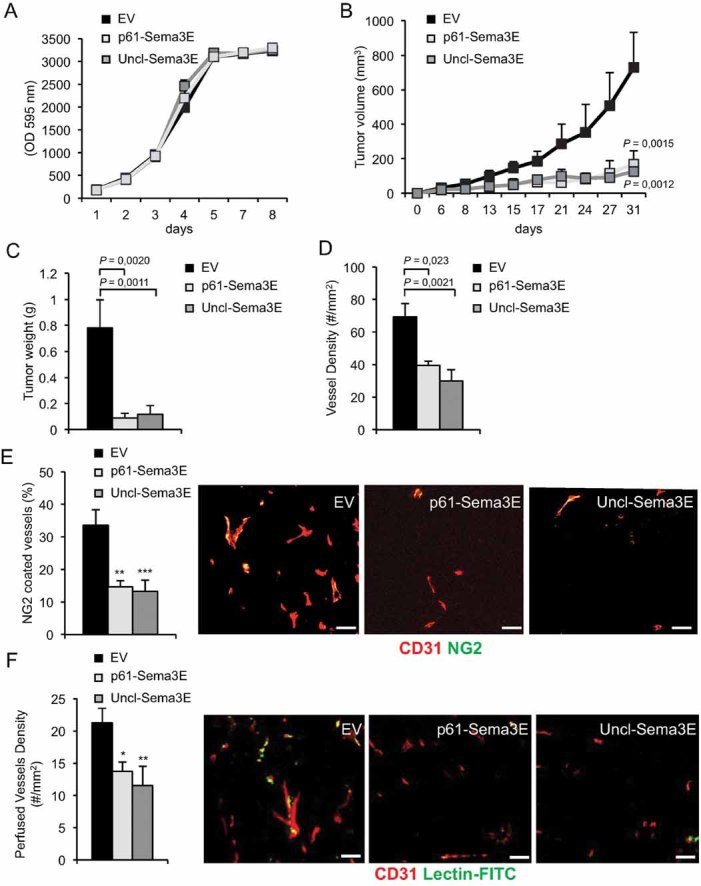
- A. MDA-MB435 cancer cells over-expressing Uncl-Sema3E, or p61-Sema3E, or carrying a non-coding empty vector (EV) were grown in culture for 3 days in 0.5% FBS. Cell growth was evaluated daily, by staining with crystal violet and quantifying absorbance at 595 nm. Indicated values are the average of two independent experiments performed in triplicates.
- B-C. MDA-MB435 cancer cells carrying Uncl-Sema3E, or p61-Sema3E, or control-EV (as above) were injected subcutaneously into nude mice (n = 5, per each experimental group). Panel B displays tumour volume growth over time, while panel C shows tumour weight at the end of the experiment. The graph shows the mean ± SD of six mice per each experimental condition.
- D. The tumours formed by engineered MDA-MB435 (described above) were explanted at the end of the experiment and tissue sections were immunostained for the endothelial cell marker CD31 to quantify vessel density.
- E. Double-staining for CD31 (red) and NG2 (green), endothelial and pericyte markers, respectively, revealed a reduced fraction of pericyte-covered tumour vessels in Uncl-Sema3E tumours compared to controls (scale bars: 100 µm); **p = 2.6E−05; ***p = 1.0E−05.
- F. Vessel perfusion was assessed upon intravenous injection of FITC-conjugated Lectin 10 min before mouse sacrifice and tumour excision. CD31 was counterstained in red to reveal endothelial cells (scale bars: 100 µm); *p = 0.018; **p = 0.0095.
The disruption of vessel networks usually correlates with deficient tissue oxygenation. In fact, we found that Uncl-Sema3E-expressing tumours were diffusely hypoxic as revealed by pimonidazole (PIMO) staining (Fig 6A) and heavily apoptotic compared to controls (Fig 6B); this likely accounted for the observed shrinkage of the tumours, as the mitotic index was not significantly affected by the treatment (Fig 6C). Notably, tumours expressing Uncl-Sema3E did not give rise to increased secondary metastatic foci with respect to controls (Fig 6D), whereas we have previously shown that either wild-type cleavable Sema3E or the mature fragment p61 are actively pro-metastatic in vivo (Casazza et al, 2010).
Figure 6. Uncl-Sema3E anti-angiogenic activity leads to tumour hypoxia and apoptosis.

- Tissue sections from tumours formed by MDA-MB435 control-EV cells, or cells overexpressing Uncl-Sema3E or p61-Sema3E, were stained with pimonidazole (PIMO, green) to reveal hypoxic areas (n = 5; see Materials and Methods Section) and for the endothelial marker CD31 (red); nuclei were stained with DAPI. Data are given as percent fraction of PIMO+ area/total area.
- Sections of the same tumours as above were immunostained for cleaved caspase 3 (green), CD31 (red) and DAPI. Uncl-Sema3E and p61 expression enhanced the apoptotic index (number of cleaved Casp3 positive cells/total cell number). The graph indicates mean values ± SD (n = 5); ***p = 5.9E−11; **p = 0.0046; §§§p = 1.9E−09; §§p = 0.0025.
- Sections of the same tumours as above were analyzed by immunohistochemistry to reveal the mitotic nuclear marker Ki67, followed by hematoxilin counterstaining. No significant difference was detected between samples. Scale bars (throughout this figure): 100 µm.
- Superficial metastatic colonies in the lungs of mice bearing the same tumours as above (n = 5 per each experimental group) were counted under a stereomicroscope after airways infusion with ink. Unlike what seen with p61, Uncl-Sema3E did not promote tumour metastatic spreading.
RIP-Tag2 (RT2) transgenic mice are a very well characterized and stereotyped model of pancreatic neuroendocrine spontaneous tumorigenesis (Hanahan, 1985). These mice develop islet carcinomas in a multi-step process characterized by the temporal appearance of distinctive lesions, including hyperplasia, followed by the so-called ‘angiogenic switch’ in the angiogenic islets, eventually accompanied by sporadic appearance of multiple tumours (Bergers et al, 2000). We therefore subjected tumour-bearing RT2 mice to the local delivery of affinity-purified Uncl-Sema3E (see Supporting Information Fig 8) into the pancreas by means of Alzet osmotic mini-pumps, in order to assess its activity in inhibiting angiogenesis and regressing tumour burden. The treatment with Uncl-Sema3E achieved a remarkable reduction of total tumour burden (Fig 7A) as well as of the average number of tumour foci (unpublished observation) compared to control-treated mice, while tumour cell proliferation was not significantly affected (Supporting Information Fig 9A). Importantly, the tumour vessel area was dramatically reduced (Fig 7B), vessel coverage was impaired (Fig 7C and Supporting Information Fig 9B) and endothelial cell apoptosis was increased (Supporting Information Fig 9C). As expected, the strong anti-angiogenic activity mediated by Uncl-Sema3E caused diffuse tumour hypoxia (Fig 7D). This effect on the primary tumours is comparable to that reported in the literature upon administration of validated vascular endothelial growth factor (VEGF)-targeted anti-angiogenic drugs in the same model (Bergers et al, 2003; Pàez-Ribes et al, 2009) as we confirmed in a series of mice treated in parallel with the tyrosine kinase inhibitor sunitinib (Fig 7A–D). Notably, it was shown that tumour shrinkage and tissue hypoxia generated by vessel disruption can trigger ‘escape’ mechanisms leading to drug resistance, increased local invasion and metastatic spreading (Casanovas et al, 2005; Ebos et al, 2009; Pàez-Ribes et al, 2009). This was confirmed in our RT2 mice treated with sunitinib, as in fact primary tumours were locally more invasive; in contrast, pancreatic tumours treated with Uncl-Sema3E not only were smaller, but also displayed a partial reduction of local invasiveness compared to controls (Fig 7E). Notably, tissue immunostaining revealed that the receptor PlexinD1 is expressed in both endothelial and tumour cells (Supporting Information Fig 9D) consistent with the idea that Uncl-Sema3E could regulate both cell types. Moreover, while the incidence of tumour-invaded peri-pancreatic lymph nodes as well as the volume of lymphonodal foci was dramatically increased in sunitinib-treated mice, in the presence of Uncl-Sema3E, tumour lymphatic infiltration was not significantly different from controls (Supporting Information Fig 10A). Notably, lymph node infiltration is a common finding in RT2 tumours, while distant liver metastases are observed in only 10–20% of cases (Pàez-Ribes et al, 2009). However, the increased primary tumour invasiveness caused by sunitinib further resulted into the formation of numerous distant metastasis in the liver, consistent with previous findings (Pàez-Ribes et al, 2009); in contrast, this was not observed upon treatment with Uncl-Sema3E, which was occasionally associated with smaller and isolated liver metastasis, as seen in controls (Supporting Information Fig 10B). Altogether, these data indicate that RT2 mice treatment with Uncl-Sema3E achieved striking tumour shrinkage consequent to vessel disruption, which was not associated with increased local invasion and metastatic spreading, as observed for sunitinib and other anti-angiogenic molecules.
Figure 7. Uncl-Sema3E inhibits angiogenesis and tumour growth in the RIP-Tag2 model.
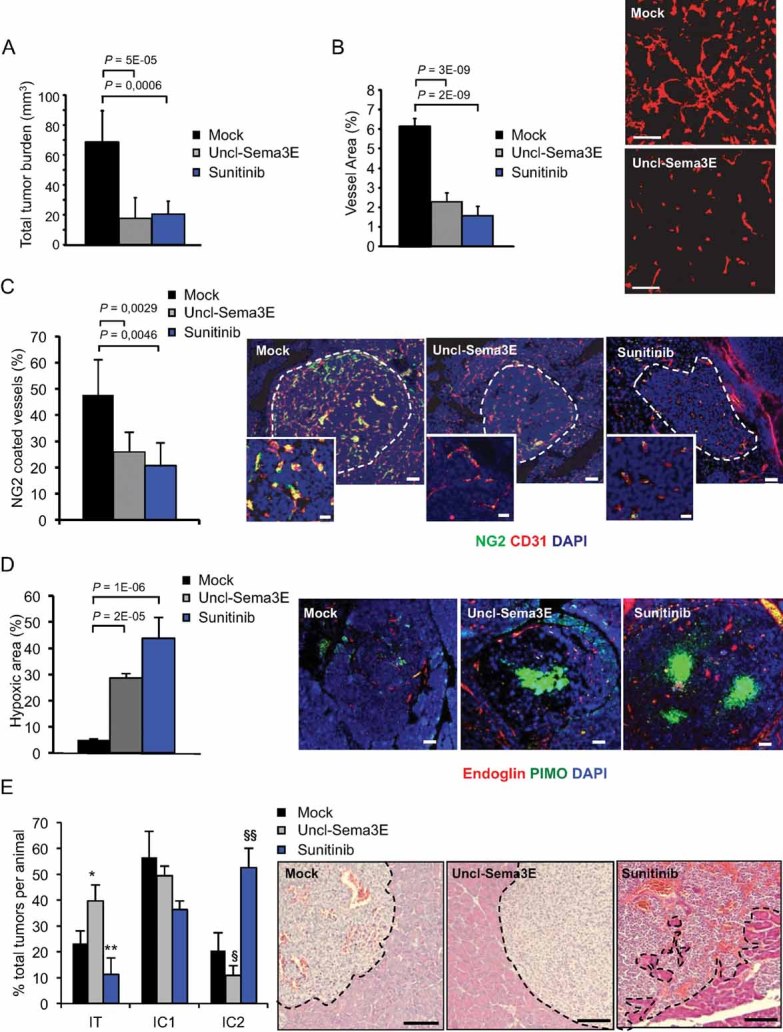
Alzet minipumps were implanted in 12-week-old RT2 mice to release locally in the pancreas Uncl-Sema3E (n = 14) or mock saline (n = 12) over a period of 2 weeks; other mice were treated orally with sunitinib (n = 8) for the same period (see Materials and Methods Section for details); two independent experiments were performed, yielding consistent results.
- Average global tumour burden per mice in different experimental conditions.
- Vessel area in treated tumours was estimated based on CD31 staining (n = 6, per each experimental group).
- Vessel coverage with mural cells was assessed by double staining for NG2 and CD31 (n = 6).
- Hypoxic areas in islet tumours were revealed by staining with pimonidazol (PIMO, see Materials and Methods Section; n = 6); the same sections were costained with the endothelial cell marker Endoglin-CD105, and with DAPI.
- Primary RT2 tumour invasiveness was assessed by morphometric analysis of H&E stained sections, as previously reported (Pàez-Ribes et al, 2009). Representative images of the different groups are shown on the right. Uncl-Sema3E treated tumours often displayed a non-invasive phenotype: i.e. the tumour was ‘encapsulated’ and well separated from the surrounding exocrine tissue (delimited by a dotted line). In contrast, in sunitinib-treated mice the tumour invasive front was often extensively intercalated with the surrounding normal exocrine tissue. The graph on the left indicates the average fraction of encapsulated (IT), microinvasive (IC1) and highly invasive (IC2) carcinomas per mice in each experimental condition (n = 8). We assessed the statistical significance of the different relative representation of each tumour histotype in drug-treated versus mock control mice; *p = 0.003; **p = 0.007; §p = 6.1E−05; §§p = 1.7E−06. Scale bars, throughout the entire figure: 100 µm (20 µm in magnified insets in panel C).
Systemic delivery of Uncl-Sema3E suppresses tumour growth and prevents metastatic spreading
We finally asked whether Uncl-Sema3E could be delivered systemically in mice as a therapeutic drug to interfere with tumour progression. To this end, we analyzed multiple overtly metastatic models and applied diverse strategies for the systemic delivery of Uncl-Sema3E in tumour-bearing mice: either based on gene transfer in vivo or on the administration of a purified recombinant protein. Hydrodynamic delivery of naked DNA expression plasmids in the circulation is commonly used to achieve transgene expression in mice, especially in the liver, and sustained plasmatic levels of secreted factors (Liu et al, 1999). We therefore compared the potential tumour suppressing activity of wild-type Sema3E that can be freely processed into p61 and its mutated counterpart Uncl-Sema3E by intravenously injecting immunodeficient mice with the expression constructs encoding either protein. The next day, we transplanted the mice with 4T1 mammary carcinoma cells. Moreover, we verified by enzyme-linked immunosorbent assay (ELISA) that the average blood concentration of Sema3E isoforms in the two groups of treated mice was comparable: i.e. 185 ± 88 µg/ml for Sema3E wt, and 179 ± 75 µg/ml for Uncl-Sema3E (vs. an endogenous level of approximately 6 ng/ml Sema3E in control mice). Primary tumours grew significantly less in the presence of either of the two Sema3E isoforms (Fig 8A and B) and vessel area was comparably reduced (Fig 8C), consistent with the fact that the proteolytic processing of this semaphorin does not impact on its anti-angiogenic activity. However, while metastases were increased with respect to controls following treatment with wild-type cleavable Sema3E, they were remarkably reduced in the presence of Uncl-Sema3E (Fig 8D) consistent with its ability to compete with endogenous p61. We further validated the tumour suppressing and anti-metastatic activity of systemic Uncl-Sema3E in another metastatic tumour model expressing endogenous Sema3E, i.e. lung carcinoma cells A549, achieving similar results (Supporting Information Fig 11). Notably, a histopathological analysis of liver, kidneys, heart and adrenal glands of mice treated with Uncl-Sema3E (delivered by different approaches) did not reveal significant signs of toxicity or any differences to tissues of mock-treated animals (Supporting Information Fig 12).
Figure 8. Systemic delivery of Uncl-Sema3E achieves a concomitant inhibition of tumour growth and metastatic spreading.

- A-D. cDNA constructs expressing either wild-type processable Sema3E, or Uncl-Sema3E (or a mock control) were hydroporated in nude mice (see Materials and Methods Section), achieving systemic delivery of the proteins in the circulation at comparable concentrations (see text). 4T1 carcinoma cells were then implanted subcutaneously in the engineered mice (n = 5, per each experimental group). Both Sema3E isoforms inhibited tumour growth (panels A and B) and tumour angiogenesis (revealed by CD31 staining, panel C; scale bar: 100 µm). However, while the occurrence of spontaneous metastasis in the lungs was significantly reduced in the presence of Uncl-Sema3E, wild-type Sema3E expression promoted metastatic spreading (panel D).
In order to compare the effect of Uncl-Sema3E with that of a validated anti-angiogenic molecule, we exploited two metastatic tumour models characterized by different responsiveness to VEGF-targeted anti-angiogenic therapy. In particular, 4T1 mammary carcinoma cells are highly responsive to anti-VEGF(R) treatments, which effectively leads to tumour shrinkage (Fischer et al, 2007); in contrast, it has been shown previously that tumours formed by Lewis Lung Carcinoma Cells (LLc) are refractory to this therapeutic approach (Shojaei et al, 2007). We therefore delivered complementary DNA (cDNA) constructs encoding either a soluble VEGF trap consisting of the extracellular portion of VEGFR2 (sFlk1), Uncl-Sema3E or mock control into immunodeficient mice. These mice were then randomized into two groups for each construct and transplanted with either 4T1 or LLc cells the day after hydroporation. The plasma levels of either molecule were comparable between mice carrying different models, as assessed by ELISA (Table 1), and the growth of 4T1 tumours was remarkably inhibited in the presence of both anti-angiogenic factors (Fig 9A and B). However, while LLc tumours were insensitive to anti-VEGF therapy (as expected), their growth was dramatically impaired by Uncl-Sema3E (Fig 9D and E), consistent with the direct inhibitory activity of this molecule in endothelial cells. Notably, vessel density was remarkably reduced in LLc tumours treated with Uncl-Sema3E, while it was comparable to controls in the presence of sFlk1 (Fig 9G). Upon analyzing the metastatic dissemination at the end of the experiment, we found that tumour shrinkage elicited by sFlk1 was accompanied by an increased number of lung metastasis (Fig 9C and F). In contrast, the tumour suppressing activity of Uncl-Sema3E was coupled with a reduction of metastatic dissemination compared to controls in both models (Fig 9C and F), indicating that it is a valuable therapeutic tool to treat tumours independently from their responsiveness to anti-VEGF drugs.
Table 1.
Protein concentration in the sera of tumor-bearing mice
| (µg/ml) | LLC | 4T1 |
|---|---|---|
| sFLK-1 | 315.2 ± 72.7 | 271.2 ± 37.2 |
| Uncl-Sema3E | 212.4 ± 22.8 | 179.8 ± 8.7 |
Protein concentration of sFLK-1 and Uncl-Sema3E in the sera of (LLc or 4T1 tumor-bearing) mice subjected to gene hydroporation (mean values ±SD, assessed by ELISA).
Figure 9. Tumours refractory to VEGF-blockade are efficiently inhibited by systemic Uncl-Sema3E, further reducing distant metastasis.

- A-F. The expression of either the validated VEGF-trap molecule sFlk1 or Uncl-Sema3E (or mock control) was achieved in immunodeficient mice by naked cDNA hydroporation (see Materials and Methods Section). The concentration of proteins delivered in the systemic circulation was assessed by ELISA (see Table 1). Mice treated with either of the two anti-angiogenic molecules were randomized into two experimental groups (n = 10) and transplanted with either 4T1 (VEGF-inhibitor responsive) or LLc (VEGF-inhibitor non-responsive) cancer cells (n = 5 per each experimental condition). Panels A and D show the volumetric growth of 4T1 and LLc tumours over time, respectively (statistical significance was calculated vs. respective control tumours). Panels B and E show endpoint 4T1 and LLc tumour burden, respectively. Vessel density is shown in panel G for both models (based on CD31 staining). The load of spontaneous lung metastasis is shown in panels C and F.
Finally, we tested the therapeutic efficacy of affinity-purified Uncl-Sema3E molecule administered systemically to tumour-bearing mice. Mice were transplanted orthotopically with 4T1 highly metastatic tumour cells and, as soon as developing palpable tumours (4–5 days after transplantation), they were treated i.p. with 30 mg/kg of purified Uncl-Sema3E three times/week for 18 days; the same amount of albumin was administered in control mice. Importantly, blood counts as well as liver and kidney markers were not altered in tumour-bearing mice treated with Uncl-Sema3E compared to controls (Supporting Information Fig 13A). Moreover, experiments performed in young healthy mice revealed that the systemic delivery of Uncl-Sema3E did not interfere with body growth or mouse behaviour (Supporting Information Fig 13B and unpublished observations), further suggesting that this treatment does not produce major adverse side effects. The systemic delivery of purified Uncl-Sema3E strikingly inhibited tumour growth (Fig 10A and B). Moreover, this treatment efficiently reduced metastasis formation (Fig 10C). Notably, Uncl-Sema3E inhibited tumour growth in vivo independently from PlexinD1 expression in cancer cells, as demonstrated by gene knock down experiments (Supporting Information Fig 14). This indicates that the activity of Uncl-Sema3E in the microenvironment is pivotal for the inhibition of tumour growth. Notably, primary tumours formed by PlexinD1-depleted cells are basally less metastatic due to the impairment of autocrine p61 signalling (Casazza et al, 2010); thus, the dominant-negative anti-metastatic effect of Uncl-Sema3E cannot be seen in tumours lacking expression of the implicated receptor PlexinD1 in cancer cells. In sum, either the removal of PlexinD1 receptor in tumour cells or the systemic delivery of the p61-competing molecule Uncl-Sema3E similarly achieved a significant reduction of the metastatic spreading in vivo.
Figure 10. Systemic delivery of purified Uncl-Sema3E suppresses tumour growth and prevents metastatic spreading.

- A-C. 4T1 cancer cell were orthotopically transplanted into the mammary fat pad of nude mice. Four days after transplant, mice bearing palpable tumours were randomized and divided into two groups (n = 8, each group); control group was treated with 30 mg/kg of purified Uncl-Sema3E, while the other was treated with the same concentration of bovine albumin (mock negative control). The treatment with purified Uncl-Sema3E impaired the growth of orthotopic tumours (A and B) and the formation of spontaneous lung metastasis (C).
In conclusion, our data show that the systemic delivery of an uncleavable full-length isoform of Sema3E inhibits tumour angiogenesis and tumour growth, while blocking the metastatic dissemination of cancer cells.
DISCUSSION
Due to their emerging role in tumour angiogenesis, tumour growth and metastasis, semaphorins appear as promising targets for strategies aimed at interfering with cancer progression. In particular, we have recently demonstrated that Sema3E expression in human tumours correlates with the metastatic progression (Casazza et al, 2010). This is explained by an autocrine signalling mechanism in cancer cells, implicating the receptor PlexinD1 and the associated oncogenic tyrosine kinase ErbB2. Notably, Sema3E was furthermore described as endothelial-repelling cue regulating angiogenesis (Casazza et al, 2010; Fukushima et al, 2011; Gu et al, 2005; Kigel et al, 2008; Sakurai et al, 2010). These multiple functional activities of Sema3E are currently explained by cell context-specific signalling cascades. In addition, Sema3E is subject to regulation by furin-like pro-protein convertases. These proteolytic enzymes, particularly abundant in advanced invasive and metastatic cancers (Bassi et al, 2005), are responsible for converting full-length Sema3E into the smaller fragment p61. We have shown here that this process reveals an otherwise concealed signalling function of Sema3E, i.e. the ability to induce association of PlexinD1 with the oncogenic tyrosine kinase ErbB2 and to trigger its intracellular signalling cascade. In fact, while full-length uncleaved Sema3E and the p61 fragment can both bind to the extracellular domain of PlexinD1, the former cannot induce the formation of a complex with ErbB2, likely due to steric hindrance by the large immunoglobulin-like domain that is removed upon proteolytic cleavage. As a consequence, Uncl-Sema3E cannot induce cancer cell migration and is devoid of pro-metastatic effects. We have reported previously that in tumours, the balance between precursor full-length Sema3E and p61 is commonly tilted in favour of the pro-metastatic fragment (Casazza et al, 2010). Thus, while cancer cells may convert a larger fraction of precursor-Sema3E into pro-metastatic p61, the treatment with Uncl-Sema3E could tilt the balance in the opposite direction by competing for receptor binding.
In addition and contrary to previous assumptions, here we showed that uncleaved full-length Sema3E is not at all an inactive molecule. In fact, endothelial cells treated with Uncl-Sema3E undergo cell-substrate detachment, cell repulsion and the typical collapsing response, depending on the PlexinD1 signalling cascade. This means that, unexpectedly, the proteolytic processing of Sema3E has no role in regulating its inhibitory activity in endothelial cells. Thus, in this study, for the first time, we could dissect the two distinctive signalling functions mediated by Sema3E in cancer cells and endothelial cells, by exploiting a mutated isoform resistant to furin-mediated cleavage. The functional relevance in physiology of non-cleaved full-length Sema3E precursor versus the p61 fragment is not addressed in this study, but it would deserve future investigation, e.g. by using molecular genetics to knock-in an uncleavable Sema3E allele in the mouse genome.
It is well established that angiogenesis plays a pivotal role for tumour growth, and disrupting the tumour vasculature has become a common strategy to interfere with cancer progression. For instance, inhibitors of the prominent VEGF signalling pathway supporting angiogenesis are applied in the treatment of human cancers; yet resistance to anti-VEGF therapy has been observed in both preclinical and clinical trials (Rapisarda & Melillo, 2009), seemingly due to the involvement of additional/alternate pro-angiogenic signals. Notably, we found that Uncl-Sema3E not only inhibited endothelial cells in culture, but its ectopic expression strongly impaired tumour angiogenesis and tumour growth in multiple preclinical tumour models in mice, either spontaneously or generated by cancer cell transplantation. In fact, upon treatment with Uncl-Sema3E, tumour vasculature was reduced and lacked appropriate coverage with mural cells; moreover, tumours became diffusely hypoxic and apoptotic. Notably, we found that Uncl-Sema3E inhibited tumour growth in vivo even upon PlexinD1 knockdown in cancer cells, consistent with that its activity in the microenvironment is essentially responsible for this effect.
The anti-angiogenic and tumour growth inhibitory effect attained by Uncl-Sema3E in mice seemed to be comparable or superior to that achieved by therapeutic concentrations of a validated VEGF trap or a VEGFR2 inhibitor. Moreover, Uncl-Sema3E effectively suppressed the growth of a tumour model non-responsive to VEGF-targeted anti-angiogenic therapy. This is consistent with the fact that this molecule carries an intrinsic inhibitory activity in endothelial cells, mediated by PlexinD1, rather than acting by blocking the pro-angiogenic function of VEGF or another specific factor. Interestingly, we found that integrin-mediated adhesion to the extracellular matrix was rapidly impaired in endothelial cells treated with Uncl-Sema3E; thereby, focal adhesions were disassembled and the actin cytoskeleton rearranged leading to the typical ‘collapsed’ phenotype. These findings are consistent with previous data indicating that inhibitory signals mediated by semaphorins (including Sema3E) depend on plexin-mediated inactivation of the monomeric GTPase R-Ras, stabilizing integrin active conformation, and on the expression of the regulatory molecule Rnd2 (Casazza et al, 2010; Oinuma et al, 2004). Moreover, a recent report indicated that PlexinD1 activation by Sema3E elicits integrin-beta1 endocytosis mediated by the monomeric GTPase Arf6 (Sakurai et al, 2010). Now we found that the impairment of integrin function elicited by Uncl-Sema3E in endothelial cells correlated with a reduced activation of the signal transducer FAK and of its downstream effector MAPK. Interestingly, endothelial cells are strictly dependent on these consensus signals mediated by the extracellular matrix for adhesion, migration and survival (Cheresh & Stupack, 2008), which may explain their overt collapsing response to Uncl-Sema3E stimulation. Notably, reduced substrate adhesion and FAK inactivation have been reported to induce endothelial cell apoptosis (reviewed by Lu & Rounds, 2011). In line with these data, we found that endothelial cells treated with Uncl-Sema3E show an increased tendency to undergo programmed cell death, likely due to impaired cell-substrate adhesion (anoikis).
It was demonstrated that tumour shrinkage induced by anti-angiogenic drugs blocking VEGF signalling correlates with a severely hypoxic environment, which leads to increased cancer cell invasiveness and distant metastasis (Ebos et al, 2009; Pàez-Ribes et al, 2009). In contrast, the anti-angiogenic activity of Uncl-Sema3E (even when associated with tumour hypoxia) did not induce invasion and metastasis, suggesting that this molecule might concomitantly control cancer cell behaviour. Notably, a wide range of human tumour cells express PlexinD1 (Casazza et al, 2010; Roodink et al, 2005, 2009) and depend on autocrine p61-Sema3E signalling for invasiveness and metastatic spreading in vivo (Casazza et al, 2010). Here, we showed that p61/PlexinD1 signalling in tumour cells can be conveniently blocked by the competitor molecule Uncl-Sema3E, hindering invasiveness and metastasis in preclinical models. In fact, Uncl-Sema3E not only inhibited tumour growth without inducing metastasis formation, but its activity effectively reduced the spreading of multiple highly metastatic cells to distant organs. Uncl-Sema3E could not block proximal lymph node infiltration by tumour cells in RT2 mice. This is consistent with our analysis of gene expression in human primary colorectal tumours indicating that elevated Sema3E levels did not correlate with the colonization of loco-regional lymph nodes, but they significantly correlated with the metastatic spreading to the liver and with patient's survival (Casazza et al, 2010). Moreover, this is in keeping with a major role of p61-PlexinD1 signalling in cancer cell transmigration through endothelial layers and hematogenous metastatic dissemination in experimental models (Casazza et al, 2010).
We have validated the tumour-suppressing activity of Uncl-Sema3E through multiple delivery approaches in preclinical models in mice including the systemic administration of a purified molecule. The effective Uncl-Sema3E concentrations achieved in the blood of mice subject to systemic delivery are in the low-micromolar range, similar to those applied for the validated VEGF-trap sFlk1, and they did not induce appreciable adverse effects. Notably, while PlexinD1 is widely distributed in human tumours, it is expressed at low levels in normal adult tissues (Roodink et al, 2005). In fact, several lines of evidence indicate that plexin-D1 expression is dynamically regulated during development and in the adult, and most prominently associated with actively growing vessels. For instance, a very recent study nicely showed the specific expression of PlexinD1 in tip cells of developing vessels in the retina and proposed that its expression is induced in these cells by VEGF-VEGFR2 signalling (Kim et al, 2011). Moreover, PlexinD1 expression was shown to increase in endothelial cells upon ischemia-induced neovascularization (Fukushima et al, 2011), and it was typically found expressed in tumour vessels but not in normal quiescent adult vasculature (Roodink et al, 2005).
In sum, our study demonstrates in vitro as well as in preclinical models in mice, that the genetically modified semaphorin isoform Uncl-Sema3E is usefully endowed with a double-tiered activity against cancer: on one side it strongly impairs tumour angiogenesis suppressing tumour growth, on the other it efficiently inhibits the invasive and metastatic behaviour of cancer cells mediated by Sema3E. Since tumour hypoxia can promote invasion and metastasis, anti-angiogenic drugs may not be the risk-free stand-alone therapeutic approach they were hoped to be. Thus, scientists are now interested in finding drug combinations, which can associate anti-angiogenic molecules with others capable of blocking any ensuing invasive and metastatic spreading of cancer cells. In this perspective, Uncl-Sema3E appears to be ideally endowed with both anti-angiogenic and anti-metastatic activities, since it can concomitantly act in the tumour vasculature and in cancer cells. In conclusion, our data suggest that both cancer cells and the supporting vasculature could be targeted in several tumours by using Uncl-Sema3E, a novel potent tumour suppressor molecule, inhibiting neo-angiogenesis while preventing invasion and metastasis.
MATERIALS AND METHODS
Fluorescent labelling of living cells
A549 cells were incubated with 10 µM CFDA SE Vybrant® cell tracer (Molecular Probes) for 15′ at 37°C, then the loading solution was replaced with fresh prewarmed medium and the cells incubated for further 30 min at 37°C before harvesting.
Endothelial tube formation assay
This assay was previously described (Grant et al, 1989). Briefly, HUVEC were seeded in 48-well tissue culture plates (2 × 104 cells per well) coated with 150 µl Matrigel in HUVEC growth medium. After 24 h in the presence or absence of either purified p61-Sema3E or Uncl-Sema3E (2 µg/ml each), the wells were photographed using phase-contrast microscopy, and the assay was quantified by counting tube bifurcations in seven independent fields per each experimental condition.
Endothelial spheroids sprouting assay
Spheroids containing 400 cells each were prepared by the hanging drop method (Laib et al, 2009) from cultured HUVEC. Spheroids were harvested and implanted in collagen gels in the presence or absence of bFGF (10 ng/ml). Moreover, either Uncl-Sema3E or p61 (1 µg/ml each) were included with bFGF in some of the wells. Spheroids were photographed after 24 h and sprouts formation assessed and quantified as previously described (Shraga-Heled et al, 2007).
The paper explained
PROBLEM
Angiogenesis plays an important role in the growth and spreading of cancer. New blood vessels ‘feed’ the cancer cells with oxygen and nutrients, allowing the tumour to grow, invade nearby tissues, spread to other organs, and form new metastatic colonies. Ideally, anti-angiogenic therapies are aimed at starving cancer cells and induce tumour regression. However, recent studies have shown that current anti-angiogenic drugs seem to promote a rebound effect, leading to even more invasive cancer growth and increased metastasis. In fact, these molecules attack the vasculature without controlling the invasiveness of cancer cells.
RESULTS
We studied a mutated form of Semaphorin 3E (Uncl-Sema3E), which acts as a partial agonist of the specific receptor PlexinD1. On one hand, Uncl-Sema3E inhibits endothelial cells and efficiently hampers tumour angiogenesis and tumour growth. At the same time, this molecule competes with a metastasis-promoting isoform of Sema3E active in cancer cells, and inhibits invasiveness and metastatic dissemination. We demonstrated in multiple preclinical mouse models that the systemic delivery of Uncl-Sema3E induces tumour shrinkage (comparable to VEGF inhibitors or possibly better) and concomitantly prevents cancer cell invasion and metastatic spreading.
IMPACT
This study shows for the first time that the molecule Uncl-Sema 3E could be considered a new-generation anti-cancer drug, capable to target both endothelial and tumour cells, concomitantly blocking vessel-borne nutrient supply and the ability of cancer cells to escape and form metastasis.
Transplanted tumour models in mice
In vivo studies were conducted in 6–8 weeks old immunodeficient mice (Charles River Laboratory Lecco, Milan, Italy). Unless otherwise indicated, in each experiment, we analyzed six tumours per each condition. For subcutaneous tumour models, depending on cell lines, 2 × 106 MDA-MB-435, 8 × 106 A549, 5 × 105 4T1 cells or 2 × 106 LLc cells were injected into the right posterior flank of anaesthetized animals. Tumour size was measured externally every 2 days using a caliper, and tumour volume was estimated using the equation: V = 4/3π × (d/2)2 × D/2, where d is the minor tumour axis and D is the major tumour axis. Mice were sacrificed after 4–6 weeks from transplant, as indicated, and tumours were weighted after dissection. Superficial pulmonary metastases were contrasted by black India-ink airways infusion, and counted on dissected lung lobes under a stereoscopic microscope. For orthotopic transplant of breast carcinoma cells, 1 × 106 4T1 cells resuspended in 40 ml phosphate-buffered saline were injected into the second mammary fat pad of anesthetized female mice. Tumour growth and spontaneous lung metastasis were analyzed as above. For experimental metastasis assays, depending on cell lines, 4.5 × 106 A549 or 2 × 105 4T1 cells were injected into lateral mice tail vein; cell colonies were scored in the lung by black India-ink airways infusion. Short-term metastatic extravasation of fluorescent cancer cells in the lungs was quantified by analyzing five pairs of lungs for each experimental group (at least five independent microscopic fields each) with ImageJ software. The statistical significance of results was verified by calculating p values with Student's t-test. Animal procedures had been approved by the Ethical Commission of the University of Torino, Italy, and by the Italian Ministry of Health.
Systemic protein delivery by naked DNA hydroporation in vivo
In vivo gene transfer of expression constructs leading to secretion of wild-type Sema3E, Uncl-Sema3E or sFlk1 (Fischer et al, 2007; Van de Veire et al, 2010) proteins in mice was achieved by naked DNA hydrodynamic delivery of naked DNA, as previously described (Liu et al, 1999). Briefly, immunodeficient mice were injected in the tail vein with 50 µg of the indicated plasmid DNA in 2.5 ml of Ringer Solution, over a time of 7 s. One day after hydroporation, tumour cells were subcutaneously implanted in mice. Proteins concentration in the circulating blood was determined by ELISA using anti-VEGFR-2 (for sFlk1) and anti-Sema3E specific immunoassays (R&D Systems), according to manufacturer's protocols.
Experiments in RIP-Tag2 mouse model
Generation of RT2 mice as a model of pancreatic β-cell carcinogenesis has been reported previously (Hanahan, 1985), and exploited for other studies in our lab (Maione et al, 2009). Alzet osmotic minipumps (2002 model, Charles River Laboratories) were used to accomplish local delivery of Uncl-Sema3E in the pancreas of experimental mice between 12 and 14 weeks of age (see Supporting Information Methods for details). Sunitinib l-malate (Axon Medchem BV) was administered to mice daily by oral gavage, at a dose of 40 mg/kg. Total tumour burden was quantified by measuring with a caliper and estimating the volume of individually excised macroscopic tumours (>1 mm3) with the formula: V = a × b2 × 0.52, where a and b represent the longer and shorter diameter of the tumour, respectively.
Statistical analysis
Data are reported in graphs as mean values ± SD (indicated by scale bars). The number of independent samples analyzed (N) is indicated in figure legends. Statistical significance was evaluated by unpaired Student's t-test (or by Anova-2way test for line graphs), with reference to respective controls (unless otherwise indicated) and expressed as p values within figures or in figure legends.
Acknowledgments
We gratefully acknowledge the help and support of all members of the Tamagnone Lab and other colleagues at IRCC (Candiolo, Italy). In particular, we thank Dr. Francesco Sassi for providing his expert opinion on mouse tissue histo-pathology, Dr. Guido Serini for help in confocal cell imaging, Dr. Antonino Sottile for blood tests in mice, Dr. Elisa Vigna for support on gene transfer experiments in vivo. We thank Claus Christensen and Eugene Lukanidin (Danish Cancer Society, Copenhagen) for generously providing Sema3E expression constructs and for useful discussion of the data. We thank S. Giove, L. Palmas, M. Accardo, L. Fontani, F. Maina, T. Werdiningsih and Y. Jonsson for excellent technical support. The work was supported by grants from Italian Association for Cancer Research-AIRC and Regione Piemonte (to L.T. and E.G.), from the Association for International Cancer Research (AICR-UK; Grant 11-0274 to L.T.), and by grants from the Israel Science Foundation (ISF) and from the Rappaport Family Institute for Research in the Medical Sciences of Technion (to G.N.). A.C. and F.M. were supported by a FIRC Fellowship. A.C. was also recipient of an EMBO long term fellowship. Boaz Kigel was supported by a Gutwirth Fellowship.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
Experiments were conceived and designed by AC, MM, EG, GN and LT. Experiments were performed by AC, BK, LC and OK; FM performed experiments on RT2 model. The paper was written by GN and LT.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Adams RH, Lohrum M, Klostermann A, Betz H, Püschel AW. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J. 1997;16:6077–6086. doi: 10.1093/emboj/16.20.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- Barberis D, Artigiani S, Casazza A, Corso S, Giordano S, Love CA, Jones EY, Comoglio PM, Tamagnone L. Plexin signaling hampers integrin-based adhesion, leading to Rho-kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. FASEB J. 2004;18:592–594. doi: 10.1096/fj.03-0957fje. [DOI] [PubMed] [Google Scholar]
- Bassi DE, Fu J, Lopez de Cicco R, Klein-Szanto AJ. Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog. 2005;44:151–161. doi: 10.1002/mc.20134. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparuccia L, Tamagnone L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment—two sides of a coin. J Cell Sci. 2009;122:1723–1736. doi: 10.1242/jcs.030197. [DOI] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Casazza A, Finisguerra V, Capparuccia L, Camperi A, Swiercz JM, Rizzolio S, Rolny C, Christensen C, Bertotti A, Sarotto I, et al. Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J Clin Invest. 2010;120:2684–2698. doi: 10.1172/JCI42118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A, Fu X, Johansson I, Capparuccia L, Andersson F, Giustacchini A, Squadrito ML, Venneri MA, Mazzone M, Larsson E, et al. Systemic and targeted delivery of Semaphorin 3A inhibits tumor angiogenesis and progression in mouse tumor models. Arterioscler Thromb Vasc Biol. 2011;31:741–749. doi: 10.1161/ATVBAHA.110.211920. [DOI] [PubMed] [Google Scholar]
- Chauvet S, Cohen S, Yoshida Y, Fekrane L, Livet J, Gayet O, Segu L, Buhot MC, Jessell TM, Henderson CE, et al. Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron. 2007;56:807–822. doi: 10.1016/j.neuron.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh DA, Stupack DG. Regulation of angiogenesis: apoptotic cues from the ECM. Oncogene. 2008;27:6285–6298. doi: 10.1038/onc.2008.304. [DOI] [PubMed] [Google Scholar]
- Christensen CR, Klingelhöfer J, Tarabykina S, Hulgaard EF, Kramerov D, Lukanidin E. Transcription of a novel mouse semaphorin gene, M-semaH, correlates with the metastatic ability of mouse tumor cell lines. Cancer Res. 1998;58:1238–1244. [PubMed] [Google Scholar]
- Christensen C, Ambartsumian N, Gilestro G, Thomsen B, Comoglio P, Tamagnone L, Guldberg P, Lukanidin E. Proteolytic processing converts the repelling signal Sema3E into an inducer of invasive growth and lung metastasis. Cancer Res. 2005;65:6167–6177. doi: 10.1158/0008-5472.CAN-04-4309. [DOI] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Franco M, Tamagnone L. Tyrosine phosphorylation in semaphorin signaling: shifting into overdrive. EMBO Rep. 2008;9:865–871. doi: 10.1038/embor.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima Y, Okada M, Kataoka H, Hirashima M, Yoshida Y, Mann F, Gomi F, Nishida K, Nishikawa S, Uemura A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest. 2011;121:1974–1985. doi: 10.1172/JCI44900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant DS, Tashiro K, Segui-Real B, Yamada Y, Martin GR, Kleinman HK. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell. 1989;58:933–943. doi: 10.1016/0092-8674(89)90945-8. [DOI] [PubMed] [Google Scholar]
- Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin AL, Ginty DD. Semaphorin 3E and plexinD1 control vascular pattern independently of neuropilins. Science. 2005;307:265–268. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic b-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Kigel B, Varshavsky A, Kessler O, Neufeld G. Successful inhibition of tumor development by specific class-3 semaphorins is associated with expression of appropriate semaphorin receptors by tumor cells. PLoS One. 2008;3:e3287. doi: 10.1371/journal.pone.0003287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Oh WJ, Gaiano N, Yoshida Y, Gu C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011;25:1399–1411. doi: 10.1101/gad.2042011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutschera S, Weber H, Weick A, De Smet F, Genove G, Takemoto M, Prahst C, Riedel M, Mikelis C, Baulande S, et al. Differential endothelial transcriptomics identifies semaphorin 3G as a vascular class 3 semaphorin. Arterioscler Thromb Vasc Biol. 2011;31:151–159. doi: 10.1161/ATVBAHA.110.215871. [DOI] [PubMed] [Google Scholar]
- Laib AM, Bartol A, Alajati A, Korff T, Weber H, Augustin HG. Spheroid-based human endothelial cell microvessel formation in vivo. Nat Protoc. 2009;4:1202–1215. doi: 10.1038/nprot.2009.96. [DOI] [PubMed] [Google Scholar]
- Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- Lu Q, Rounds S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc Res. 2012;83:56–63. doi: 10.1016/j.mvr.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione F, Molla F, Meda C, Latini R, Zentilin L, Giacca M, Seano G, Serini G, Bussolino F, Giraudo E. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest. 2009;119:3356–3372. doi: 10.1172/JCI36308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer. 2008;8:632–645. doi: 10.1038/nrc2404. [DOI] [PubMed] [Google Scholar]
- Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science. 2004;305:862–865. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat. 2009;12:74–80. doi: 10.1016/j.drup.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodink I, Raats J, van der Zwaag B, Verrijp K, Kusters B, van Bokhoven H, Linkels M, de Waal RM, Leenders WP. Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel target for diagnosis and therapy. Cancer Res. 2005;65:8317–8323. doi: 10.1158/0008-5472.CAN-04-4366. [DOI] [PubMed] [Google Scholar]
- Roodink I, Verrijp K, Raats J, Leenders WP. Plexin D1 is ubiquitously expressed on tumor vessels and tumor cells in solid malignancies. BMC Cancer. 2009;9:297. doi: 10.1186/1471-2407-9-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Gavard J, Annas-Linhares Y, Basile JR, Amornphimoltham P, Palmby TR, Yagi H, Zhang F, Randazzo PA, Li X, et al. Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R-Ras. Mol Cell Biol. 2010;30:3086–398. doi: 10.1128/MCB.01652-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- Shraga-Heled N, Kessler O, Prahst C, Kroll J, Augustin H, Neufeld G. Neuropilin-1 and neuropilin-2 enhance VEGF121 stimulated signal transduction by the VEGFR-2 receptor. FASEB J. 2007;21:915–926. doi: 10.1096/fj.06-6277com. [DOI] [PubMed] [Google Scholar]
- Van de Veire S, Stalmans I, Heindryckx F, Oura H, Tijeras-Raballand A, Schmidt T, Loges S, Albrecht I, Jonckx B, Vinckier S, et al. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell. 2010;141:178–190. doi: 10.1016/j.cell.2010.02.039. [DOI] [PubMed] [Google Scholar]
- Varshavsky A, Kessler O, Abramovitch S, Kigel B, Zaffryar S, Akiri G, Neufeld G. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res. 2008;68:6922–6931. doi: 10.1158/0008-5472.CAN-07-5408. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gunput RA, Pasterkamp RJ. Semaphorin signaling: progress made and promises ahead. Trends Biochem Sci. 2008;33:161–170. doi: 10.1016/j.tibs.2008.01.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.